Insecticidal Activity of a Vip3Ab1 Chimera Is Conferred by Improved Protein Stability in the Midgut of Spodoptera eridania

Abstract
1. Introduction
2. Results
2.1. Protein Characterization by Midgut Fluid Digestion and Analytical Size Exclusion Chromatography
2.2. Characterization of Proteases Present in S. frugiperda and S. eridania Midgut Fluids
2.3. Effect of Protease Inhibitors on In-Vivo Diet Bioassay
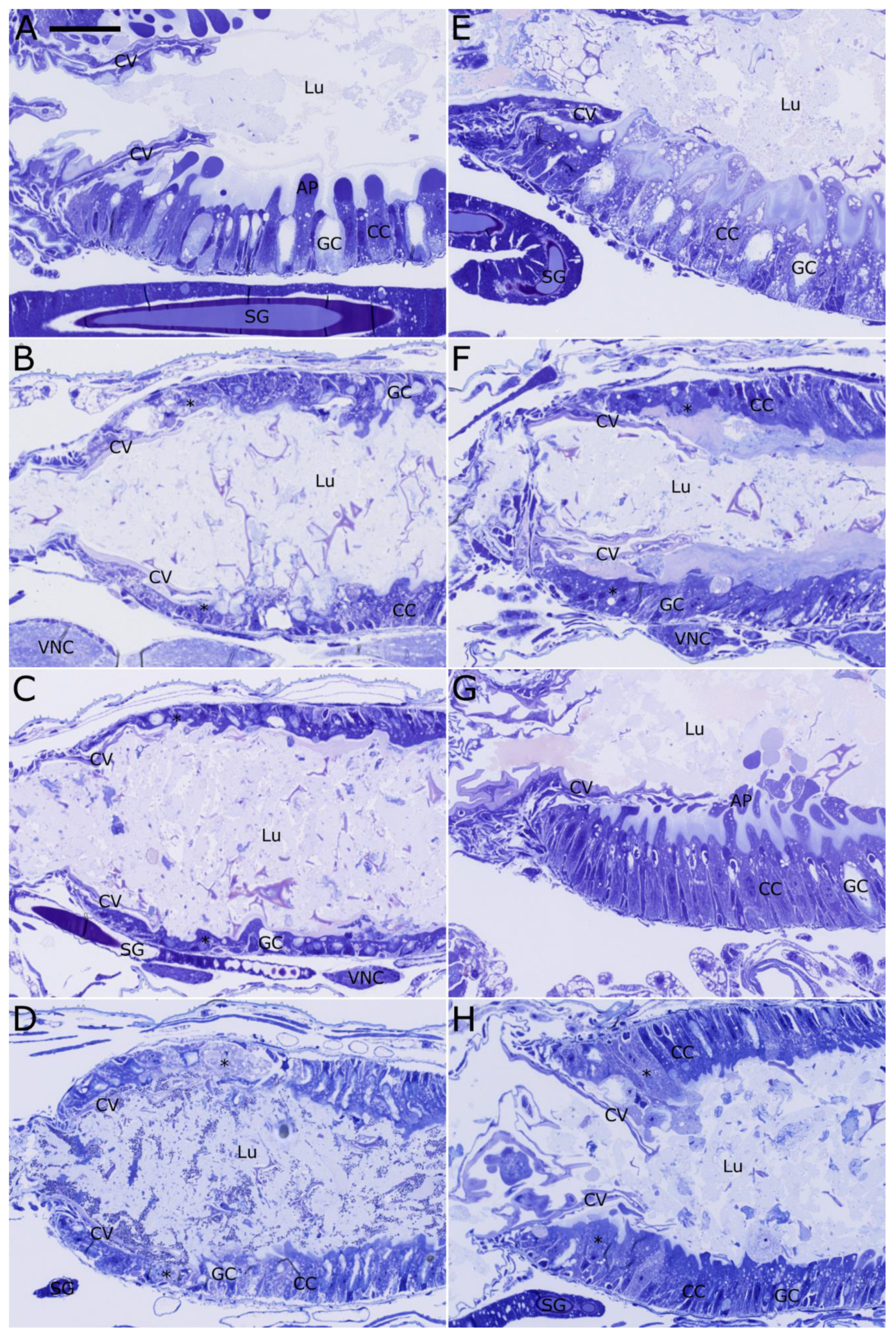
2.4. Histopathological Evaluation of Vip3Ab1 and VipAb1-740 Effects on S. eridania and S. frugiperda
2.5. Immunolocalization of Vip3Ab1 and VipAb1-740 in S. eridania and S. frugiperda
3. Discussion
4. Materials and Methods
4.1. Gene and Protein Sequences
4.2. Construct Design
4.3. Protein Expression and Purification
4.4. SDS-PAGE Analysis
4.5. Lepidopteran Midgut Fluid Protein Digestion and Analytical Size Exclusion Chromatography
4.6. Protease Inhibitors Used in Midgut Fluid Digestions and Insect Bioassay
4.6.1. Zymogram Analysis
4.6.2. Insect Diet Overlay Bioassays
4.7. Histopathology
4.8. Immunolocalization
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chakroun, M.; Banyuls, N.; Bel, Y.; Escriche, B.; Ferre, J. Bacterial Vegetative Insecticidal Proteins (Vip) from Entomopathogenic Bacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 329–350. [Google Scholar] [CrossRef]
- Estruch, J.J.; Warren, G.W.; Mullins, M.A.; Nye, G.J.; Craig, J.A.; Koziel, M.G. Vip3A, a novel Bacillus thuringiensis vegetative insecticidal protein with a wide spectrum of activities against lepidopteran insects. Proc. Natl. Acad. Sci. USA 1996, 93, 5389–5394. [Google Scholar] [CrossRef]
- Kunthic, T.; Watanabe, H.; Kawano, R.; Tanaka, Y.; Promdonkoy, B.; Yao, M.; Boonserm, P. pH regulates pore formation of a protease activated Vip3Aa from Bacillus thuringiensis. Biochim. Biophys. Acta 2017, 1859, 2234–2241. [Google Scholar] [CrossRef]
- Palma, L.; Scott, D.J.; Harris, G.; Din, S.U.; Williams, T.L.; Roberts, O.J.; Young, M.T.; Caballero, P.; Berry, C. The Vip3Ag4 Insecticidal Protoxin from Bacillus thuringiensis Adopts A Tetrameric Configuration That Is Maintained on Proteolysis. Toxins (Basel) 2017, 9, 165. [Google Scholar] [CrossRef]
- Zack, M.D.; Sopko, M.S.; Frey, M.L.; Wang, X.; Tan, S.Y.; Arruda, J.M.; Letherer, T.T.; Narva, K.E. Functional characterization of Vip3Ab1 and Vip3Bc1: Two novel insecticidal proteins with differential activity against lepidopteran pests. Sci. Rep. 2017, 7, 11112. [Google Scholar] [CrossRef]
- Anilkumar, K.J.; Rodrigo-Simon, A.; Ferre, J.; Pusztai-Carey, M.; Sivasupramaniam, S.; Moar, W.J. Production and characterization of Bacillus thuringiensis Cry1Ac-resistant cotton bollworm Helicoverpa zea (Boddie). Appl. Environ. Microbiol. 2008, 74, 462–469. [Google Scholar] [CrossRef]
- Ben Hamadou-Charfi, D.; Boukedi, H.; Abdelkefi-Mesrati, L.; Tounsi, S.; Jaoua, S. Agrotis segetum midgut putative receptor of Bacillus thuringiensis vegetative insecticidal protein Vip3Aa16 differs from that of Cry1Ac toxin. J. Invertebr. Pathol. 2013, 114, 139–143. [Google Scholar] [CrossRef]
- Chakroun, M.; Ferre, J. In vivo and in vitro binding of Vip3Aa to Spodoptera frugiperda midgut and characterization of binding sites by (125)I radiolabeling. Appl. Environ. Microbiol. 2014, 80, 6258–6265. [Google Scholar] [CrossRef]
- Gouffon, C.; Van Vliet, A.; Van Rie, J.; Jansens, S.; Jurat-Fuentes, J.L. Binding Sites for Bacillus thuringiensis Cry2Ae Toxin on Heliothine Brush Border Membrane Vesicles Are Not Shared with Cry1A, Cry1F, or Vip3A Toxin. Appl. Environ. Microb. 2011, 77, 3182–3188. [Google Scholar] [CrossRef]
- Lee, M.K.; Miles, P.; Chen, J.S. Brush border membrane binding properties of Bacillus thuringiensis Vip3A toxin to Heliothis virescens and Helicoverpa zea midguts. Biochem. Biophys. Res. Commun. 2006, 339, 1043–1047. [Google Scholar] [CrossRef]
- Welch, K.L.; Unnithan, G.C.; Degain, B.A.; Wei, J.; Zhang, J.; Li, X.; Tabashnik, B.E.; Carriere, Y. Cross-resistance to toxins used in pyramided Bt crops and resistance to Bt sprays in Helicoverpa zea. J. Invertebr. Pathol. 2015, 132, 149–156. [Google Scholar] [CrossRef]
- Horikoshi, R.J.; Bernardi, D.; Bernardi, O.; Malaquias, J.B.; Okuma, D.M.; Miraldo, L.L.; Amaral, F.S.; Omoto, C. Effective dominance of resistance of Spodoptera frugiperda to Bt maize and cotton varieties: implications for resistance management. Sci. Rep. 2016, 6, 34864. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Qureshi, J.A.; Meagher, R.L., Jr.; Reisig, D.D.; Head, G.P.; Andow, D.A.; Ni, X.; Kerns, D.; Buntin, G.D.; Niu, Y.; et al. Cry1F resistance in fall armyworm Spodoptera frugiperda: Single gene versus pyramided Bt maize. PLoS ONE 2014, 9, e112958. [Google Scholar] [CrossRef]
- Lee, M.K.; Walters, F.S.; Hart, H.; Palekar, N.; Chen, J.S. The mode of action of the Bacillus thuringiensis vegetative insecticidal protein Vip3A differs from that of Cry1Ab delta-endotoxin. Appl. Environ. Microbiol. 2003, 69, 4648–4657. [Google Scholar] [CrossRef]
- Martinelli, S.; Barata, R.M.; Zucchi, M.I.; Silva-Filho Mde, C.; Omoto, C. Molecular variability of Spodoptera frugiperda (Lepidoptera: Noctuidae) populations associated to maize and cotton crops in Brazil. J. Econ. Entomol. 2006, 99, 519–526. [Google Scholar] [CrossRef]
- Omoto, C.; Bernardi, O.; Salmeron, E.; Sorgatto, R.J.; Dourado, P.M.; Crivellari, A.; Carvalho, R.A.; Willse, A.; Martinelli, S.; Head, G.P. Field-evolved resistance to Cry1Ab maize by Spodoptera frugiperda in Brazil. Pest Manag. Sci. 2016, 72, 1727–1736. [Google Scholar] [CrossRef]
- Storer, N.P.; Babcock, J.M.; Schlenz, M.; Meade, T.; Thompson, G.D.; Bing, J.W.; Huckaba, R.M. Discovery and characterization of field resistance to Bt maize: Spodoptera frugiperda (Lepidoptera: Noctuidae) in Puerto Rico. J. Econ. Entomol. 2010, 103, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, M.; Banyuls, N.; Walsh, T.; Downes, S.; James, B.; Ferre, J. Characterization of the resistance to Vip3Aa in Helicoverpa armigera from Australia and the role of midgut processing and receptor binding. Sci. Rep. 2016, 6, 24311. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, F.; Bai, J.; Deng, G.; Qin, S.; Bao, Q. Evidence for positive Darwinian selection of Vip gene in Bacillus thuringiensis. J. Genet. Genomics. 2007, 34, 649–660. [Google Scholar] [CrossRef]
- Sopko, M.S.; Narva, K.E.; Bowling, A.J.; Pence, H.E.; Hasler, J.M.; Letherer, T.J.; Larsen, C.L.; Zack, M.D. Modification of Vip3Ab1 C-terminus confers broadened plant protection from lepidopteran pests. Toxins 2019. (under review). [Google Scholar]
- Murua, M.G.; Vera, M.A.; Herrero, M.I.; Fogliata, S.V.; Michel, A. Defoliation of Soybean Expressing Cry1Ac by Lepidopteran Pests. Insects 2018, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Bergamasco, V.B.; Mendes, D.R.; Fernandes, O.A.; Desiderio, J.A.; Lemos, M.V. Bacillus thuringiensis Cry1Ia10 and Vip3Aa protein interactions and their toxicity in Spodoptera spp. (Lepidoptera). J. Invertebr. Pathol. 2013, 112, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, M.; Bel, Y.; Caccia, S.; Abdelkefi-Mesrati, L.; Escriche, B.; Ferre, J. Susceptibility of Spodoptera frugiperda and S. exigua to Bacillus thuringiensis Vip3Aa insecticidal protein. J. Invertebr. Pathol. 2012, 110, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, J.; Tang, L.; Tang, M.; Shi, Y.; Pang, Y. Comparison of the expression of Bacillus thuringiensis full-length and N-terminally truncated vip3A gene in Escherichia coli. J. Appl. Microbiol. 2003, 95, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Palma, L.; de Escuder, I.R.; Maeztu, M.; Caballero, P.; Munoz, D. Screening of vip genes from a Spanish Bacillus thuringiensis collection and characterization of two Vip3 proteins highly toxic to five lepidopteran crop pests. Biol. Control. 2013, 66, 141–149. [Google Scholar] [CrossRef]
- Ruiz de Escudero, I.; Banyuls, N.; Bel, Y.; Maeztu, M.; Escriche, B.; Munoz, D.; Caballero, P.; Ferre, J. A screening of five Bacillus thuringiensis Vip3A proteins for their activity against lepidopteran pests. J. Invertebr. Pathol. 2014, 117, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Abdelgaffar, H.M.; Oppert, C.; Sun, X.; Monserrate, J.; Jurat-Fuentes, J.L. Differential heliothine susceptibility to Cry1Ac associated with gut proteolytic activity. Pestic. Biochem. Physiol. 2019, 153, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Banyuls, N.; Hernandez-Rodriguez, C.S.; Van Rie, J.; Ferre, J. Critical amino acids for the insecticidal activity of Vip3Af from Bacillus thuringiensis: Inference on structural aspects. Sci. Rep. 2018, 8, 7539. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Chen, R.R.; Ren, X.L.; Wan, P.J.; Mu, L.L.; Li, G.Q. Combined effects of three crystalline toxins from Bacillus thuringiensis with seven proteinase inhibitors on beet armyworm, Spodoptera exigua Hubner (Lepidoptera: Noctuidae). Pestic. Biochem. Phys. 2013, 105, 169–176. [Google Scholar] [CrossRef]
- Purcell, J.P.; Greenplate, J.T.; Sammons, R.D. Examination of Midgut Luminal Proteinase Activities in 6 Economically Important Insects. Insect Biochem. Molec. 1992, 22, 41–47. [Google Scholar] [CrossRef]
- Shao, Z.Z.; Cui, Y.L.; Liu, X.L.; Yi, H.Q.; Ji, J.H.; Yu, Z.N. Processing of delta-endotoxin of Bacillus thuringiensis subsp. kurstaki HD-1 in Heliothis armigera midgut juice and the effects of protease inhibitors. J. Invertebr. Pathol. 1998, 72, 73–81. [Google Scholar] [CrossRef]
- Fortier, M.; Vachon, V.; Frutos, R.; Schwartz, J.L.; Laprade, R. Effect of insect larval midgut proteases on the activity of Bacillus thuringiensis Cry toxins. Appl. Environ. Microbiol. 2007, 73, 6208–6213. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Lopez, L.; Munoz-Garay, C.; Porta, H.; Rodriguez-Almazan, C.; Soberon, M.; Bravo, A. Strategies to improve the insecticidal activity of Cry toxins from Bacillus thuringiensis. Peptides 2009, 30, 589–595. [Google Scholar] [CrossRef]
- Zhu, Y.C.; Guo, Z.B.; Abel, C. Cloning eleven midgut trypsin cDNAs and evaluating the interaction of proteinase inhibitors with Cry1Ac against the tobacco budworm, Heliothis virescens (F.) (Lepidoptera: Noctuidae). J. Invertebr. Pathol. 2012, 111, 111–120. [Google Scholar] [CrossRef]
- Zhu, Y.C.; West, S.; Liu, F.X.; He, Y.P. Interaction of proteinase inhibitors with Cry1Ac toxicity and the presence of 15 chymotrypsin cDNAs in the midgut of the tobacco budworm, Heliothis virescens (F.) (Lepidoptera: Noctuidae). Pest Manag. Sci. 2012, 68, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M.; Kay, L.M.; Stroud, R.M. Structure and specific binding of trypsin: comparison of inhibited derivatives and a model for substrate binding. J. Mol. Biol. 1974, 83, 209–230. [Google Scholar] [CrossRef]
- Abdelkefi-Mesrati, L.; Boukedi, H.; Chakroun, M.; Kamoun, F.; Azzouz, H.; Tounsi, S.; Rouis, S.; Jaoua, S. Investigation of the steps involved in the difference of susceptibility of Ephestia kuehniella and Spodoptera littoralis to the Bacillus thuringiensis Vip3Aa16 toxin. J. Invertebr. Pathol. 2011, 107, 198–201. [Google Scholar] [CrossRef]
- Abdelkefi-Mesrati, L.; Boukedi, H.; Dammak-Karray, M.; Sellami-Boudawara, T.; Jaoua, S.; Tounsi, S. Study of the Bacillus thuringiensis Vip3Aa16 histopathological effects and determination of its putative binding proteins in the midgut of Spodoptera littoralis. J. Invertebr. Pathol. 2011, 106, 250–254. [Google Scholar] [CrossRef]
- Sellami, S.; Cherif, M.; Jamoussi, K. Effect of adding amino acids residues in N- and C-terminus of Vip3Aa16 (L121I) toxin. J. Basic. Microbiol. 2016, 56, 654–661. [Google Scholar] [CrossRef]
- Yu, C.G.; Mullins, M.A.; Warren, G.W.; Koziel, M.G.; Estruch, J.J. The Bacillus thuringiensis vegetative insecticidal protein Vip3A lyses midgut epithelium cells of susceptible insects. Appl. Environ. Microbiol. 1997, 63, 532–536. [Google Scholar]
- Bel, Y.; Sheets, J.J.; Tan, S.Y.; Narva, K.E.; Escriche, B. Toxicity and Binding Studies of Bacillus thuringiensis Cry1Ac, Cry1F, Cry1C, and Cry2A Proteins in the Soybean Pests Anticarsia gemmatalis and Chrysodeixis (Pseudoplusia) includens. Appl. Environ. Microbiol. 2017, 83, e00326. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Vip3Ab1 | Vip3Ab1-740 | ||||||
|---|---|---|---|---|---|---|---|---|
| LC50 (ng/cm2) | Lower CI (95%) | Upper CI (95%) | N | LC50 (ng/cm2) | Lower CI (95%) | Upper CI (95%) | N | |
| Full length | 177.9 | 138.6 | 230.7 | 362 | 80.8 | 69.0 | 94.6 | 1056 |
| Processed | 590.2 | 468.4 | 739.0 | 224 | 435.0 | 368.6 | 552.4 | 224 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowling, A.J.; Sopko, M.S.; Tan, S.Y.; Larsen, C.M.; Pence, H.E.; Zack, M.D. Insecticidal Activity of a Vip3Ab1 Chimera Is Conferred by Improved Protein Stability in the Midgut of Spodoptera eridania. Toxins 2019, 11, 276. https://doi.org/10.3390/toxins11050276
Bowling AJ, Sopko MS, Tan SY, Larsen CM, Pence HE, Zack MD. Insecticidal Activity of a Vip3Ab1 Chimera Is Conferred by Improved Protein Stability in the Midgut of Spodoptera eridania. Toxins. 2019; 11(5):276. https://doi.org/10.3390/toxins11050276
Chicago/Turabian StyleBowling, Andrew J., Megan S. Sopko, Sek Yee Tan, Cory M. Larsen, Heather E. Pence, and Marc D. Zack. 2019. "Insecticidal Activity of a Vip3Ab1 Chimera Is Conferred by Improved Protein Stability in the Midgut of Spodoptera eridania" Toxins 11, no. 5: 276. https://doi.org/10.3390/toxins11050276
APA StyleBowling, A. J., Sopko, M. S., Tan, S. Y., Larsen, C. M., Pence, H. E., & Zack, M. D. (2019). Insecticidal Activity of a Vip3Ab1 Chimera Is Conferred by Improved Protein Stability in the Midgut of Spodoptera eridania. Toxins, 11(5), 276. https://doi.org/10.3390/toxins11050276