Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of Delta-Toxin on Fluid Accumulation in Mouse Intestinal Loops
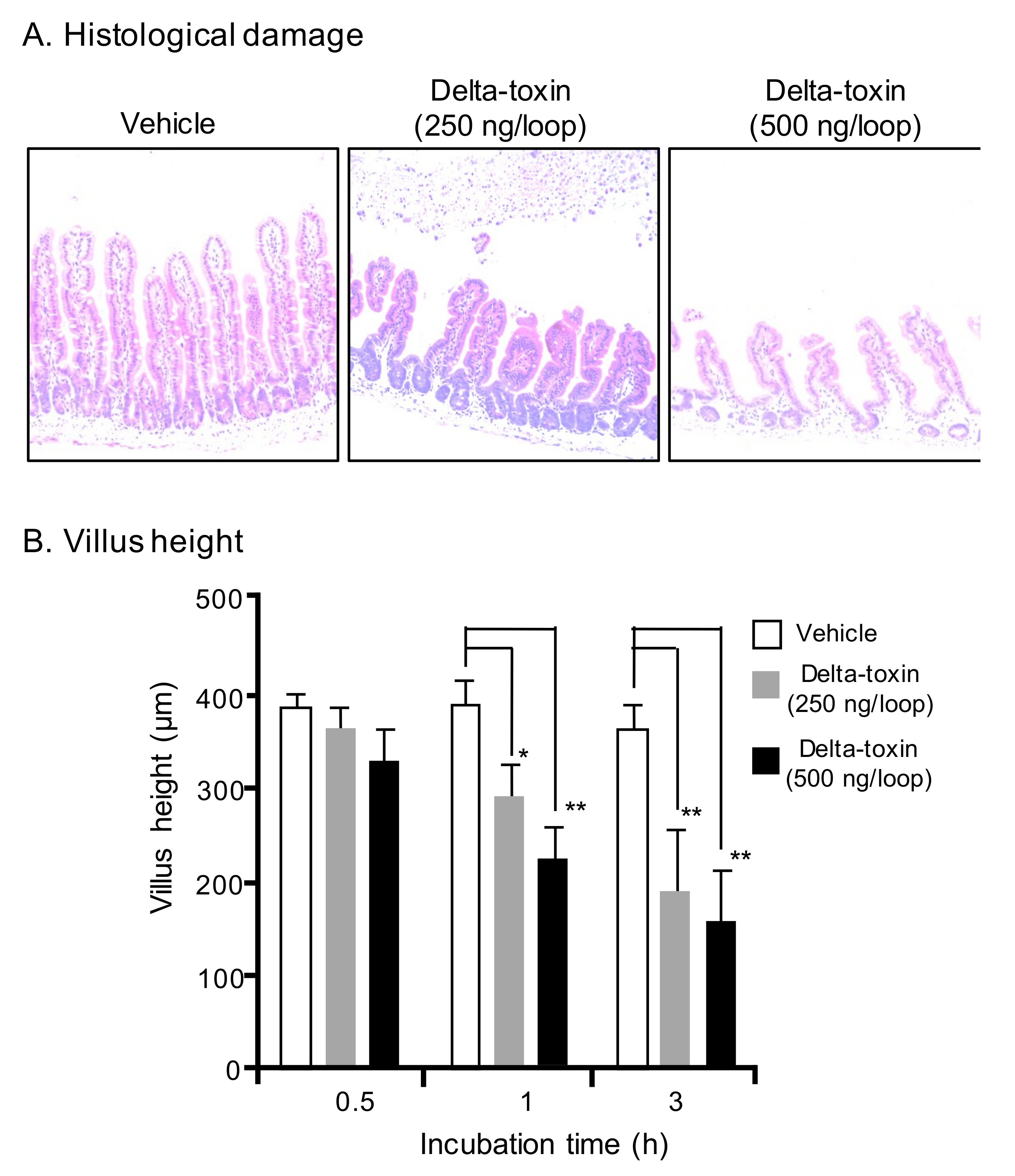
2.2. Histopathological Damage Caused by Delta-Toxin in Mouse Intestinal Loops
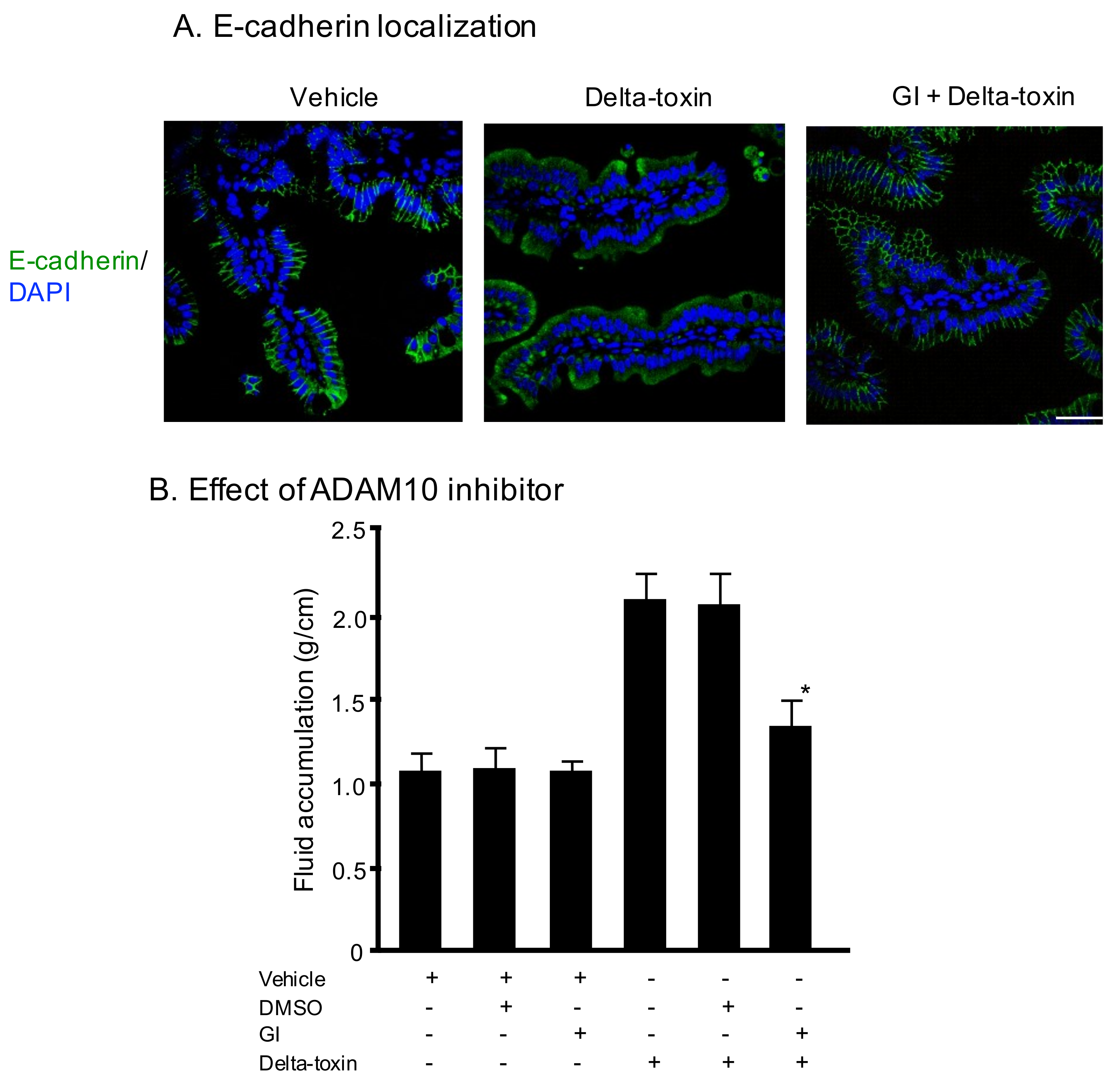
2.3. E-Cadherin Degradation Induced by Delta-Toxin in Mouse Intestinal Loops
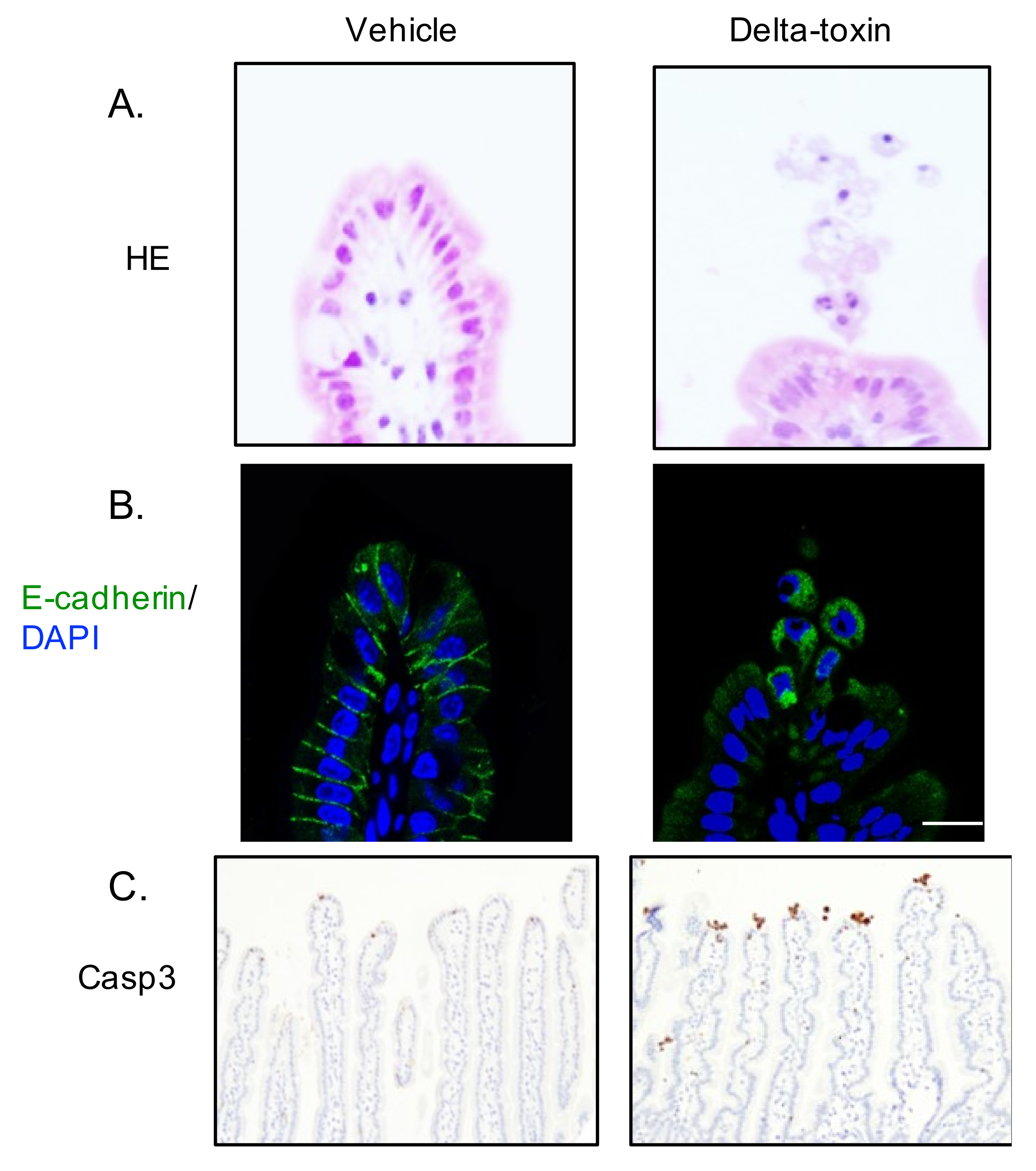
2.4. Delta-Toxin-Induced Cell Shedding from Small Intestinal Villi
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Mice
5.3. Mouse Loop Assay
5.4. Paracellular Permeability
5.5. Histological Analysis
5.6. Immunohistochemistry
5.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Popoff, M.R.; Bouvet, P. Clostridial toxins. Future Microbiol. 2009, 4, 1021–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Adams, V.; Bannam, T.L.; Miyamoto, K.; Garcia, J.P.; Uzal, F.A.; Rood, J.I.; McClane, B.A. Toxin Plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 2013, 77, 208–233. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; McClane, B.A. Recent progress in understanding the pathogenesis of Clostridium perfringens type C infections. Vet. Microbiol. 2011, 153, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Ochi, S.; Oda, M.; Miyamoto, K.; Takehara, M.; Kobayashi, K. Recent Insights into Clostridium perfringens Beta-Toxin. Toxins 2015, 7, 396–406. [Google Scholar] [CrossRef]
- Alouf, J.E.; Jolivet-Reynaud, C. Purification and characterization of Clostridium perfringens delta-toxin. Infect. Immun. 1981, 31, 536–546. [Google Scholar] [PubMed]
- Jolivet-Reynaud, C.; Hauttecoeur, B.; Alouf, J.E. Interaction of Clostridium perfringens delta toxin with erythrocyte and liposome membranes and relation with the specific binding to the ganglioside GM2. Toxicon 1989, 27, 1113–1126. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Jolivet-Reynaud, C.; Fitting, C.; David, B.; Alouf, J.E. Ganglioside Identification on Human Monocyte Membrane with Clostridium perfringens Delta-Toxin. J. Leukoc. Biol. 1986, 40, 65–72. [Google Scholar] [CrossRef]
- Jolivet-Reynaud, C.; Cavaillon, J.M.; Alouf, J.E. Selective cytotoxicity of Clostridium perfringens delta toxin on rabbit leukocytes. Infect. Immun. 1982, 38, 860–864. [Google Scholar]
- Manich, M.; Knapp, O.; Gibert, M.; Maier, E.; Jolivet-Reynaud, C.; Geny, B.; Benz, R.; Popoff, M.R. Clostridium perfringens delta toxin is sequence related to beta toxin, netB, and Staphylococcus pore-forming toxins, but shows functional differences. PLoS ONE 2008, 3, e3764. [Google Scholar] [CrossRef]
- Popoff, M.R. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe 2014, 30, 220–238. [Google Scholar] [CrossRef]
- Huyet, J.; Naylor, C.E.; Savva, C.G.; Gibert, M.; Popoff, M.R.; Basak, A.K. Structural insights into Clostridium perfringens delta toxin pore formation. PLoS ONE 2013, 8, e66673. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Miyamoto, K.; Kobayashi, K.; Takehara, M.; Nagahama, M. Clostridium perfringens Delta-Toxin Induces Rapid Cell Necrosis. PLoS ONE 2016, 11, e0147957. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Uzal, F.A.; Fisher, D.J.; Saputo, J.; Vidal, J.E.; Chen, Y.; Gupta, P.; Rood, J.I.; McClane, B.A. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 2008, 67, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Bücker, R.; Krug, S.M.; Rosenthal, R.; Günzel, D.; Fromm, A.; Zeitz, M.; Chakraborty, T.; Fromm, M.; Epple, H.-J.; Schulzke, J.-D. Aerolysin from Aeromonas hydrophila Perturbs Tight Junction Integrity and Cell Lesion Repair in Intestinal Epithelial HT-29/B6 Cells. J. Infect. Dis. 2011, 204, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.-K.; Vikström, E.; Magnusson, K.-E.; Vécsey-Semjén, B.; Colque-Navarro, P.; Möllby, R. The Staphylococcus aureus Alpha-Toxin Perturbs the Barrier Function in Caco-2 Epithelial Cell Monolayers by Altering Junctional Integrity. Infect. Immun. 2012, 80, 1670–1680. [Google Scholar] [CrossRef]
- Cajnko, M.M.; Marusic, M.; Kisovec, M.; Rojko, N.; Bencina, M.; Caserman, S.; Anderluh, G. Listeriolysin O affects the permeability of Caco-2 monolayer in a pore-dependent and Ca2+-independent manner. PLoS ONE 2015, 10, e0130471. [Google Scholar] [CrossRef]
- Wilke, G.A.; Wardenburg, J.B. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Wardenburg, J.B. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef]
- von Hoven, G.; Rivas, A.J.; Neukirch, C.; Klein, S.; Hamm, C.; Qin, Q.; Meyenburg, M.; Fuser, S.; Saftig, P.; Hellmann, N.; et al. Dissecting the role of ADAM10 as a mediator of Staphylococcus aureus alpha-toxin action. Biochem. J. 2016, 473, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Takehara, M.; Takagishi, T.; Miyamoto, K.; Kobayashi, K.; Nagahama, M. Delta-toxin from Clostridium perfringens perturbs intestinal epithelial barrier function in Caco-2 cell monolayers. Biochim. Biophys. Acta Biomembr. 2018, 1860, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Dahlhoff, M.; Horst, D.; Hirschi, B.; Trülzsch, K.; Müller-Höcker, J.; Vogelmann, R.; Allgäuer, M.; Gerhard, M.; Steininger, S.; et al. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS ONE 2010, 5, e14325. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Martínez, V.-H.; Petit, C.S.; Fouquet, S.; Le Beyec, J.; Chambaz, J.; Pinçon-Raymond, M.; Cardot, P.; Thenet, S. Epidermal growth factor receptor is involved in enterocyte anoikis through the dismantling of E-cadherin-mediated junctions. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G235–G244. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Watson, A.J.M.; Frey, M.R.; Miguel, J.C.; Burkitt, M.D.; Sutton, R.; Hughes, K.R.; Hall, L.J.; Caamaño, J.H.; et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis. Model. Mech. 2013, 6, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, F.; Zou, Z.; Liu, D.; Wang, J.; Su, Y. Active deformation of apoptotic intestinal epithelial cells with adhesion-restricted polarity contributes to apoptotic clearance. Lab. Invest. 2011, 91, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, P.; Su, Q.; Wang, S.; Wang, F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE 2012, 7, e34946. [Google Scholar]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seike, S.; Takehara, M.; Kobayashi, K.; Nagahama, M. Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine. Toxins 2019, 11, 232. https://doi.org/10.3390/toxins11040232
Seike S, Takehara M, Kobayashi K, Nagahama M. Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine. Toxins. 2019; 11(4):232. https://doi.org/10.3390/toxins11040232
Chicago/Turabian StyleSeike, Soshi, Masaya Takehara, Keiko Kobayashi, and Masahiro Nagahama. 2019. "Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine" Toxins 11, no. 4: 232. https://doi.org/10.3390/toxins11040232
APA StyleSeike, S., Takehara, M., Kobayashi, K., & Nagahama, M. (2019). Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine. Toxins, 11(4), 232. https://doi.org/10.3390/toxins11040232