Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
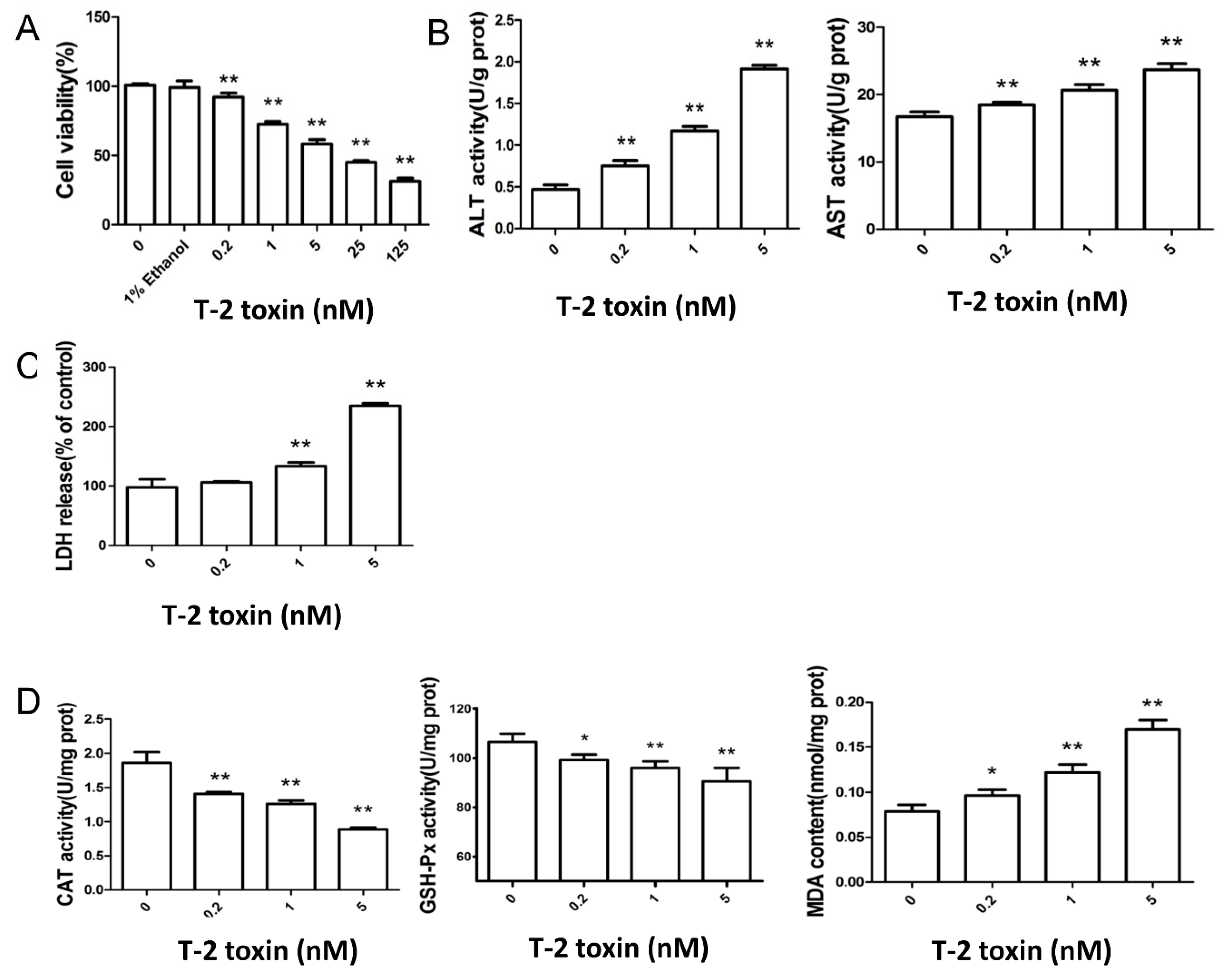
2.1. T-2 Toxin Led to Cytotoxicity and Oxidative Damage in L02 Cells
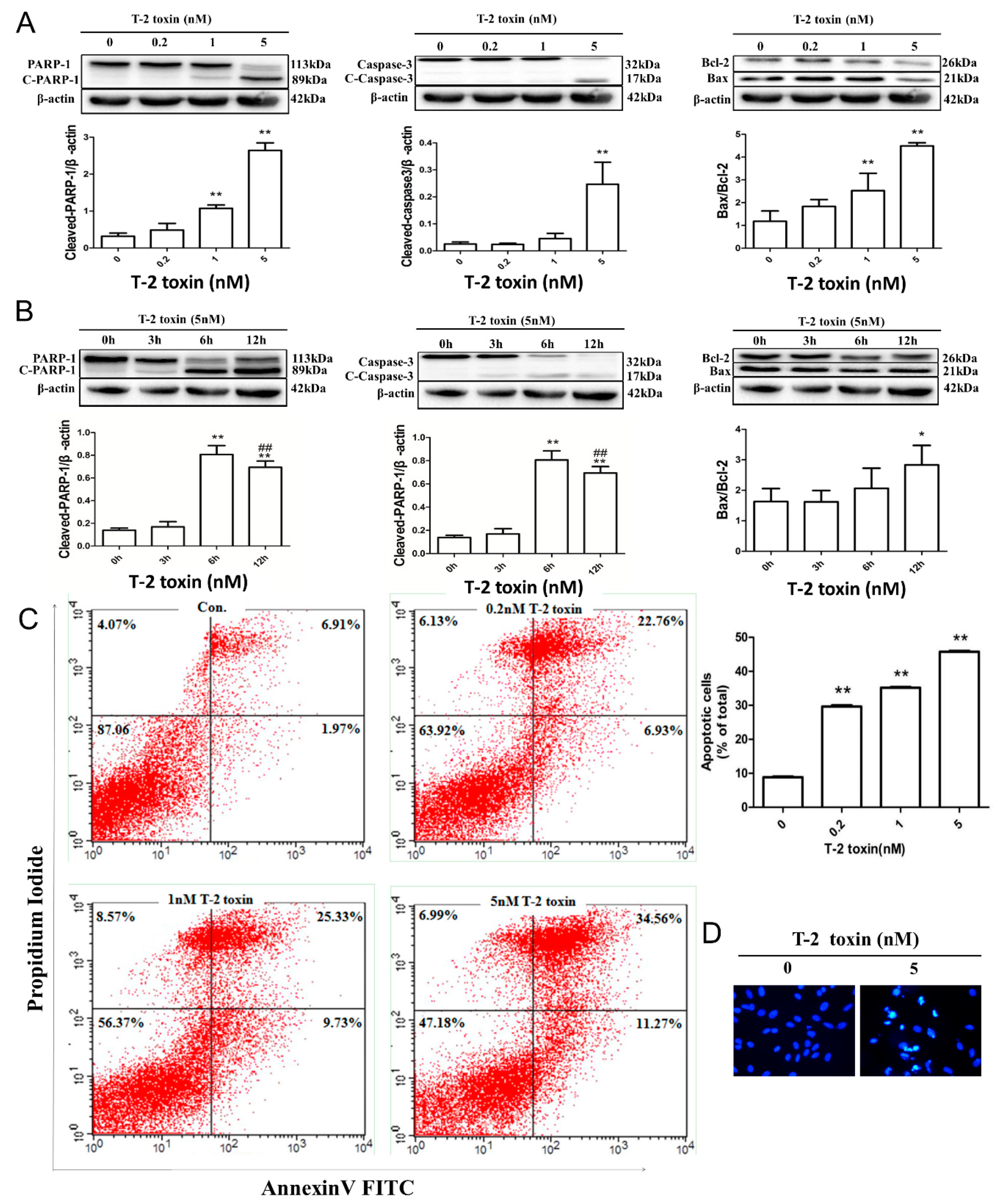
2.2. T-2 Toxin-Mediated Apoptotic Effects in L02 Cells
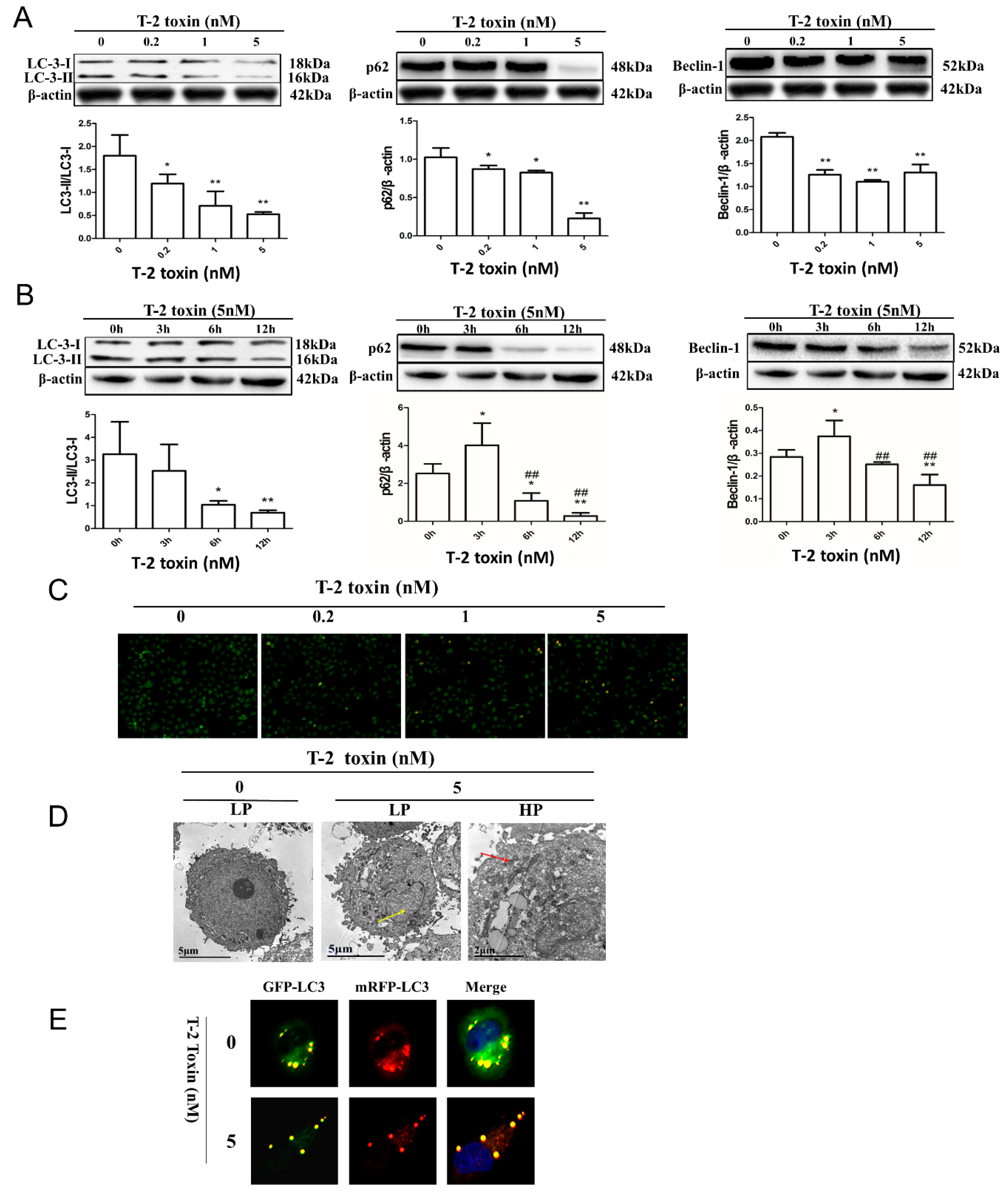
2.3. T-2 Toxin-Mediated Autophagy in L02 Cells
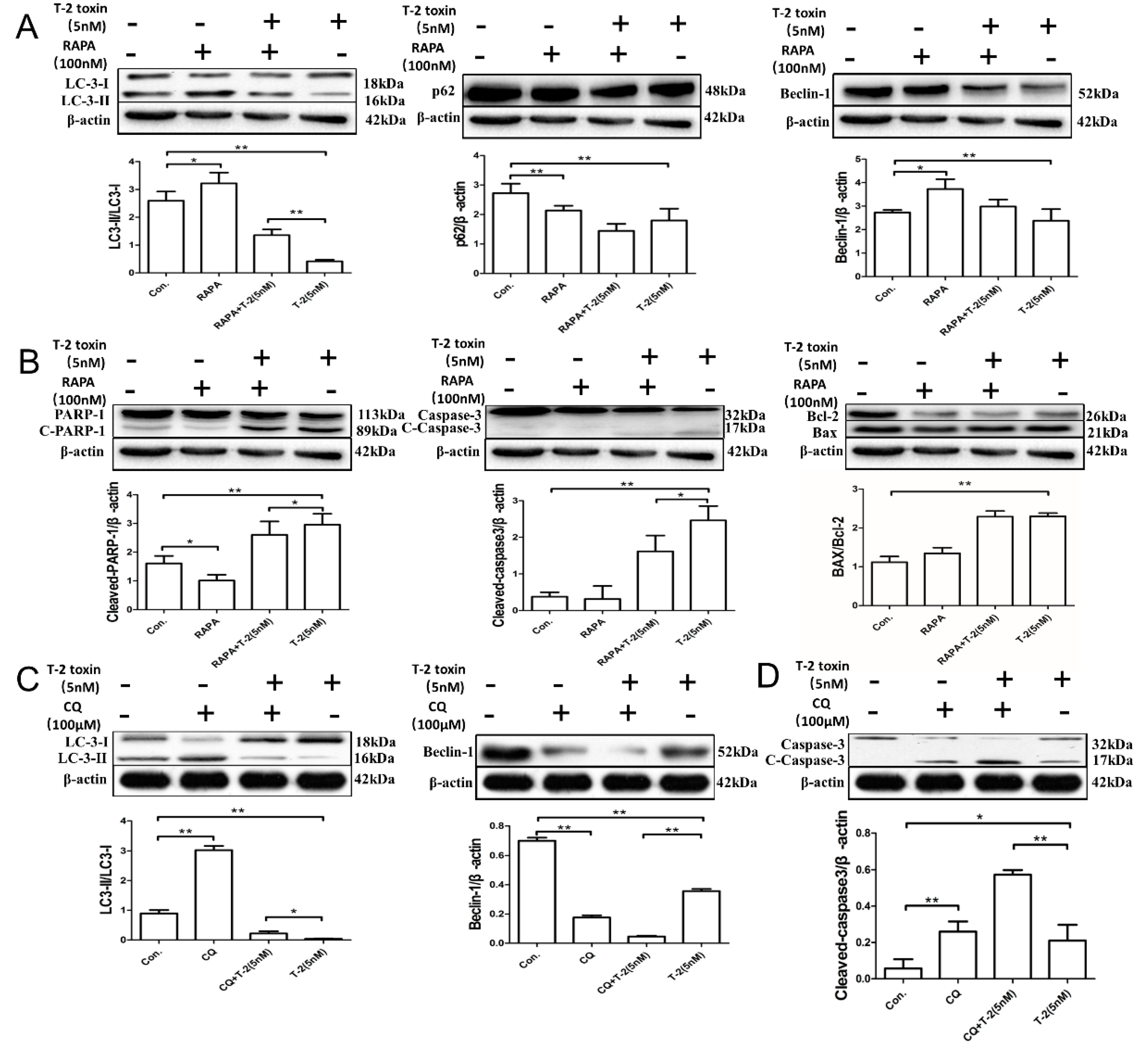
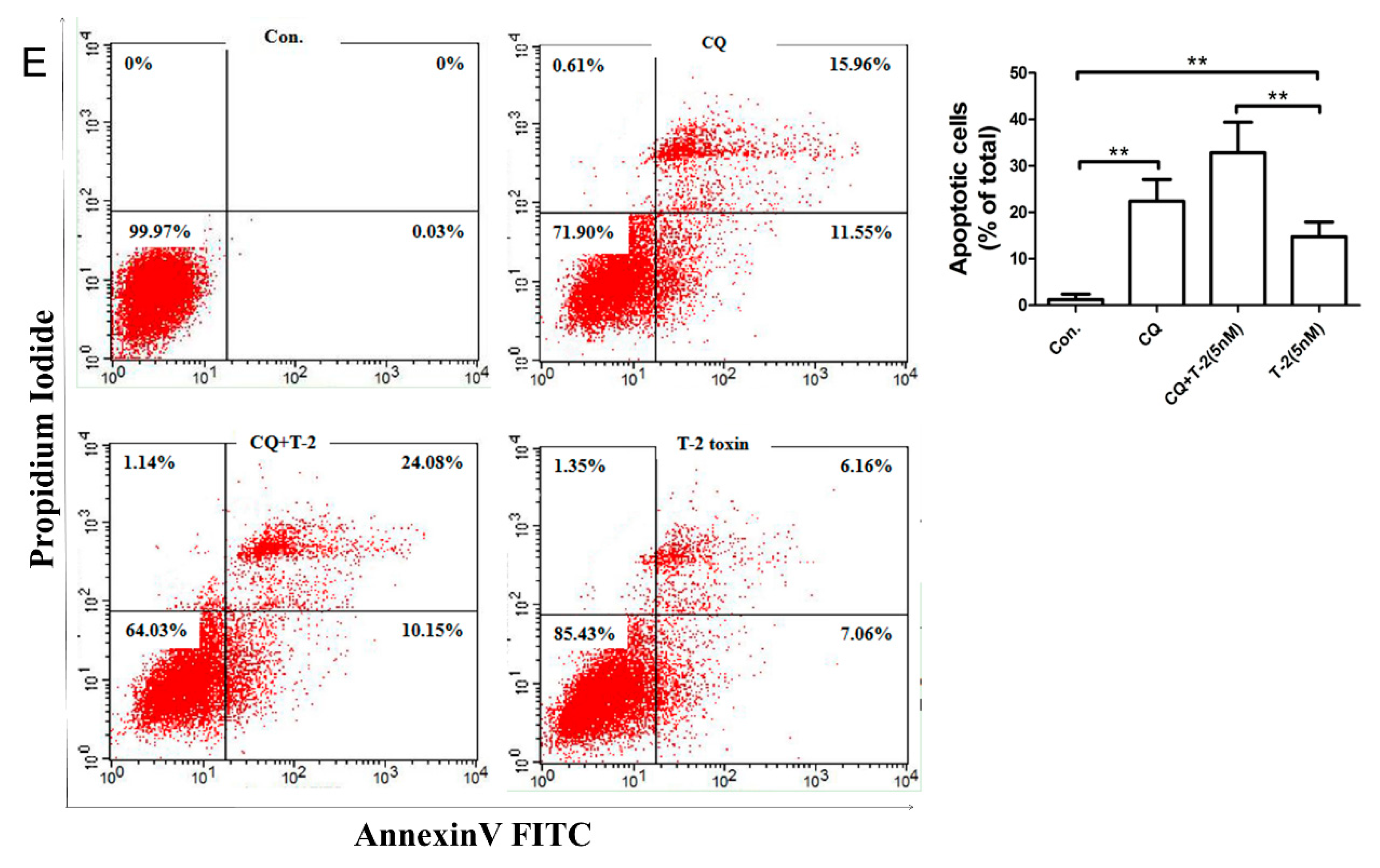
2.4. Autophagy Blocks T-2 Toxin-Mediated Apoptosis
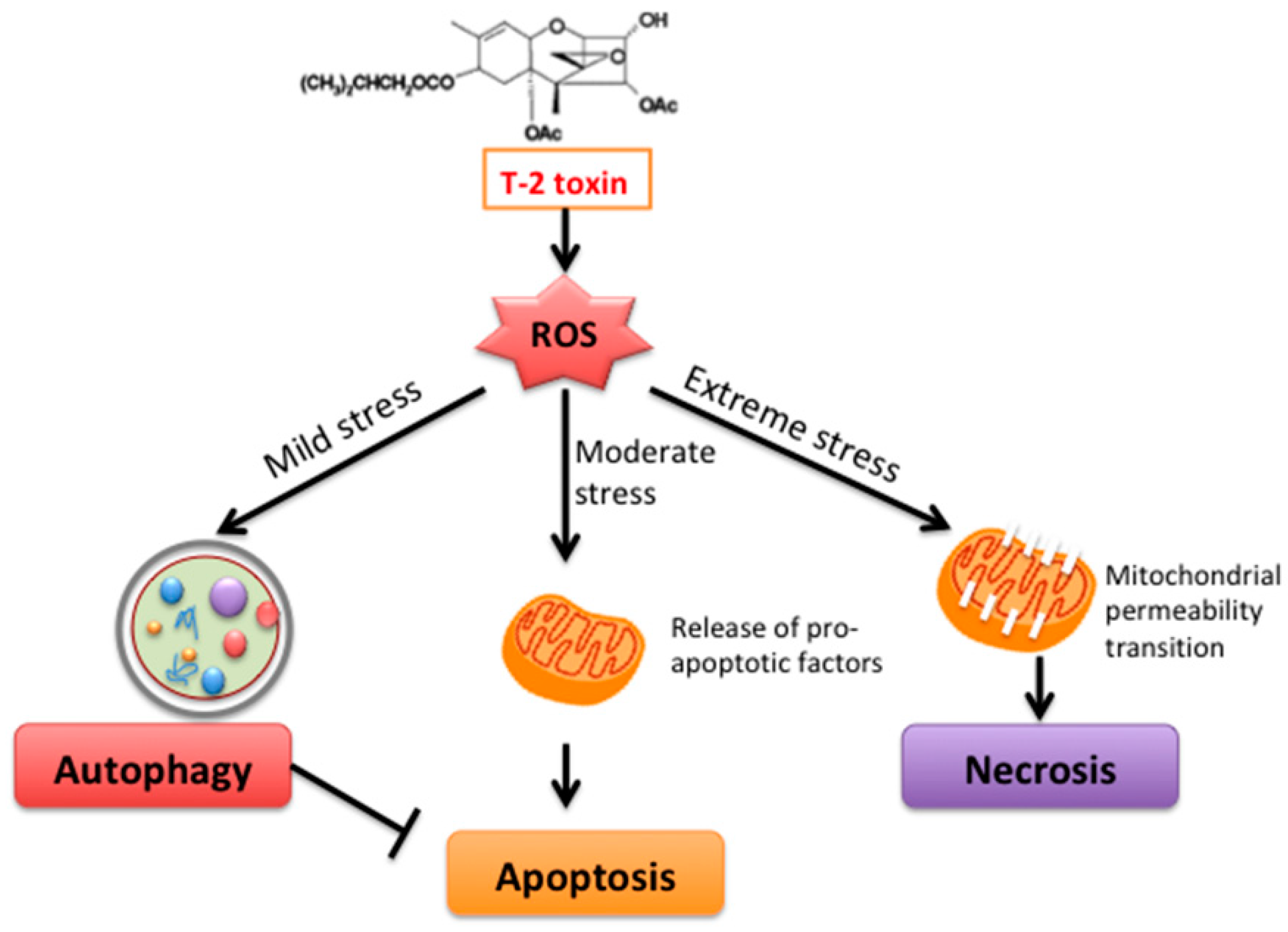
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cells Culture
4.3. Determination of Cell Viability
4.4. Evaluation of Cytotoxicity Induced by T-2 Toxin
4.5. Observation of Oxidative Stress Caused by T-2 Toxin
4.6. Determination of Apoptotic Effects Induced by T-2 Toxin
4.7. Determination of Autophagic Effects
4.8. Determination of Autophagy Flux
4.9. Protein Extraction and Western Blot
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Creppy, E.E. Update of survey, regulation and toxic effects of mycotoxins in Europe. Toxicol. Lett. 2002, 127, 19–28. [Google Scholar] [CrossRef]
- De Ruyck, K.; De Boevre, M.; Huybrechts, I.; De Saeger, S. Dietary mycotoxins, co-exposure, and carcinogenesis in humans: Short review. Mutat. Res. Rev. Mutat. Res. 2015, 766, 32–41. [Google Scholar] [CrossRef]
- World Health Organization. Selected Mycotoxins: Ochratoxins, Trichothecenes, Ergot; World Health Organization: Geneva, Switzerland, 1990. [Google Scholar]
- Yao, Y.-F.; Li, X.-B.; Yang, J.; Shen, B.; Zhou, Z.-K.; Pei, F.-X. Study on the effect of T-2 toxin combined with low nutrition diet on rat epiphyseal plate growth and development. Int. Orthop. 2010, 34, 1351–1356. [Google Scholar] [CrossRef]
- Chen, J.; Chu, Y.; Cao, J.; Yang, Z.; Guo, X.; Wang, Z. T-2 toxin induces apoptosis, and selenium partly blocks, T-2 toxin induced apoptosis in chondrocytes through modulation of the Bax/Bcl-2 ratio. Food Chem. Toxicol. 2006, 44, 567–573. [Google Scholar] [CrossRef]
- Nagase, M.; Alam, M.M.; Tsushima, A.; Yoshizawa, T.; Sakato, N. Apoptosis induction by T-2 toxin: Activation of caspase-9, caspase-3, and DFF-40/CAD through cytosolic release of cytochrome c in HL-60 cells. Biosci. Biotechnol. Biochem. 2001, 65, 1741–1747. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, D.; Huang, Y.; Wang, S. Study on the apoptosis mechanism induced by T-2 toxin. PLoS ONE 2013, 8, e83105. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Ihsan, A.; Huang, L.; Dai, M.; Hao, H.; Cheng, G.; Liu, Z.; Wang, Y.; Yuan, Z. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264. 7 cells induced by trichothecenes as DON and T-2 toxin. Toxicol. Sci. 2012, 127, 412–424. [Google Scholar] [CrossRef]
- Sehata, S.; Kiyosawa, N.; Makino, T.; Atsumi, F.; Ito, K.; Yamoto, T.; Teranishi, M.; Baba, Y.; Uetsuka, K.; Nakayama, H. Morphological and microarray analysis of T-2 toxin-induced rat fetal brain lesion. Food Chem. Toxicol. 2004, 42, 1727–1736. [Google Scholar] [CrossRef]
- Doi, K.; Ishigami, N.; Sehata, S. T-2 toxin-induced toxicity in pregnant mice and rats. Int. J. Mol. Sci. 2008, 9, 2146–2158. [Google Scholar] [CrossRef]
- Chen, J.-H.; Cao, J.-L.; Chu, Y.-L.; Wang, Z.-L.; Yang, Z.-T.; Wang, H.-L. T-2 toxin-induced apoptosis involving Fas, p53, Bcl-xL, Bcl-2, Bax and caspase-3 signaling pathways in human chondrocytes. J. Zhejiang Univ. Sci. B 2008, 9, 455–463. [Google Scholar] [CrossRef]
- Han, J.; Guo, X.; Wu, C.; Li, C.; He, S.; Duan, C.; Ning, Y. Nano-Se-chondroitin sulfate inhibits T-2 toxin-induced apoptosis of cultured chondrocytes from patients with Kashin-Beck disease. Nan Fang Yi Ke Da Xue Xue Bao 2013, 33, 225–229. [Google Scholar]
- Weidner, M.; Lenczyk, M.; Schwerdt, G.; Gekle, M.; Humpf, H.-U. Neurotoxic potential and cellular uptake of T-2 toxin in human astrocytes in primary culture. Chem. Res. Toxicol. 2013, 26, 347–355. [Google Scholar] [CrossRef]
- Fang, H.; Wu, Y.; Guo, J.; Rong, J.; Ma, L.; Zhao, Z.; Zuo, D.; Peng, S. T-2 toxin induces apoptosis in differentiated murine embryonic stem cells through reactive oxygen species-mediated mitochondrial pathway. Apoptosis 2012, 17, 895–907. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, J.; Zhang, H.; Wang, J.; Cai, H.; Li, C.; Li, K.; Liu, J.; Guo, X.; Zou, G. Integrated transcriptional and proteomic analysis with in vitro biochemical assay reveal the important role of CYP3A46 in T-2 toxin hydroxylation in porcine primary hepatocytes. Mol. Cell. Proteom. 2011. [Google Scholar] [CrossRef]
- Shinozuka, J.; Suzuki, M.; Noguchi, N.; Sugimoto, T.; Uetsuka, K.; Nakayama, H.; Doi, K. T-2 toxin-induced apoptosis in hematopoietic tissues of mice. Toxicol. Pathol. 1998, 26, 674–681. [Google Scholar] [CrossRef]
- Albarenque, S.M.; Shinozuka, J.; Iwamoto, S.; Nakayama, H.; Doi, K. T-2 toxin-induced acute skin lesions in Wistar-derived hypotrichotic WBNIILA-Ht rats. Histol. Histopathol. 1999, 14, 337–342. [Google Scholar]
- Wu, J.; Tu, D.; Yuan, L.-Y.; Yi, J.-E.; Tian, Y. T-2 toxin regulates steroid hormone secretion of rat ovarian granulosa cells through cAMP-PKA pathway. Toxicol. Lett. 2015, 232, 573–579. [Google Scholar] [CrossRef]
- Kiessling, K.-H. Biochemical mechanism of action of mycotoxins. Pure Appl. Chem. 1986, 58, 327–338. [Google Scholar] [CrossRef]
- Wu, J.; Tu, D.; Yuan, L.-Y.; Yuan, H.; Wen, L.-X. T-2 toxin exposure induces apoptosis in rat ovarian granulosa cells through oxidative stress. Environ. Toxicol. Pharmacol. 2013, 36, 493–500. [Google Scholar] [CrossRef]
- Yuan, Z.; Matias, F.B.; Yi, J.-E.; Wu, J. T-2 toxin-induced cytotoxicity and damage on TM3 Leydig cells. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2016, 181, 47–54. [Google Scholar] [CrossRef]
- Wu, J.; Huang, W.; Xiao, H.; Xie, Y.; Yuan, Z.; Yi, J.; Chen, J.; Tu, D.; Tian, Y. Procyanidins B2 reverses the T-2 toxin-induced mitochondrial apoptosis in TM3 Leydig cells. J. Funct. Foods 2018, 45, 118–128. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Park, K.-I.; Kim, S.-H.; Yu, S.-N.; Park, S.-G.; Kim, Y.; Seo, Y.-K.; Ma, J.-Y.; Ahn, S.-C. Inhibition of autophagy promotes salinomycin-induced apoptosis via reactive oxygen species-mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer cells. Int. J. Mol. Sci. 2017, 18, 1088. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.; Vandenabeele, P.; Corazzari, M. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef]
- Wirawan, E.; Walle, L.V.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2011, 1, e18. [Google Scholar] [CrossRef]
- Velmurugan, B.; Selvanayagam, M.; Cengiz, E.I.; Unlu, E. Histopathology of lambda-cyhalothrin on tissues (gill, kidney, liver and intestine) of Cirrhinus mrigala. Environ. Toxicol. Pharmacol. 2007, 24, 286–291. [Google Scholar] [CrossRef]
- Bouaziz, C.; El Golli, E.; Abid-Essefi, S.; Brenner, C.; Lemaire, C.; Bacha, H. Different apoptotic pathways induced by zearalenone, T-2 toxin and ochratoxin A in human hepatoma cells. Toxicology 2008, 254, 19–28. [Google Scholar] [CrossRef]
- Caloni, F.; Ranzenigo, G.; Cremonesi, F.; Spicer, L.J. Effects of a trichothecene, T-2 toxin, on proliferation and steroid production by porcine granulosa cells. Toxicon 2009, 54, 337–344. [Google Scholar] [CrossRef]
- El Golli, E.; Hassen, W.; Bouslimi, A.; Bouaziz, C.; Ladjimi, M.M.; Bacha, H. Induction of HSP 70 in Vero cells in response to mycotoxins: Cytoprotection by sub-lethal heat shock and by Vitamin E. Toxicol. Lett. 2006, 166, 122–130. [Google Scholar] [CrossRef]
- Wu, J.; Jing, L.; Yuan, H.; Peng, S.-Q. T-2 toxin induces apoptosis in ovarian granulosa cells of rats through reactive oxygen species-mediated mitochondrial pathway. Toxicol. Lett. 2011, 202, 168–177. [Google Scholar] [CrossRef]
- Wu, Q.-H.; Wang, X.; Yang, W.; Nüssler, A.K.; Xiong, L.-Y.; Kuča, K.; Dohnal, V.; Zhang, X.-J.; Yuan, Z.-H. Oxidative stress-mediated cytotoxicity and metabolism of T-2 toxin and deoxynivalenol in animals and humans: An update. Arch. Toxicol. 2014, 88, 1309–1326. [Google Scholar] [CrossRef]
- Landmesser, U.; Harrison, D.G. Oxidative stress and vascular damage in hypertension. Coronary Artery Dis. 2001, 12, 455–461. [Google Scholar] [CrossRef]
- Ozguner, F.; Koyu, A.; Cesur, G. Active smoking causes oxidative stress and decreases blood melatonin levels. Toxicol. Ind. Health 2005, 21, 21–26. [Google Scholar] [CrossRef]
- Antonsson, B. Bax and other pro-apoptotic Bcl-2 family “killer-proteins” and their victim the mitochondrion. Cell Tissue Res. 2001, 306, 347–361. [Google Scholar] [CrossRef]
- Borner, C. The Bcl-2 protein family: Sensors and checkpoints for life-or-death decisions. Mol. Immunol. 2003, 39, 615–647. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, H.; An, M. mTORC1 regulates apoptosis and cell proliferation in pterygium via targeting autophagy and FGFR3. Sci. Rep. 2017, 7, 7339. [Google Scholar] [CrossRef]
- Stanton, M.J.; Dutta, S.; Zhang, H.; Polavaram, N.S.; Leontovich, A.A.; Hönscheid, P.; Sinicrope, F.A.; Tindall, D.J.; Muders, M.H.; Datta, K. Autophagy control by the VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Du, D.; Hu, L.; Wu, J.; Wu, Q.; Cheng, W.; Guo, Y.; Guan, R.; Wang, Y.; Chen, X.; Yan, X. Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J. Neuroinflamm. 2017, 14, 169. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Fu, L.-L.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, Y.; Long, M.; Luo, T.; Bian, J.; Liu, X.; Gu, J.; Zou, H.; Song, R.; Wang, Y. Beclin-1-mediated autophagy protects against cadmium-activated apoptosis via the Fas/FasL pathway in primary rat proximal tubular cell culture. Sci. Rep. 2017, 7, 977. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Zhou, Y.; Yuan, Z.; Yi, J.; Chen, J.; Wang, N.; Tian, Y. Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells. Toxins 2019, 11, 45. https://doi.org/10.3390/toxins11010045
Wu J, Zhou Y, Yuan Z, Yi J, Chen J, Wang N, Tian Y. Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells. Toxins. 2019; 11(1):45. https://doi.org/10.3390/toxins11010045
Chicago/Turabian StyleWu, Jing, Yu Zhou, Zhihang Yuan, Jine Yi, Jingshu Chen, Naidong Wang, and Yanan Tian. 2019. "Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells" Toxins 11, no. 1: 45. https://doi.org/10.3390/toxins11010045
APA StyleWu, J., Zhou, Y., Yuan, Z., Yi, J., Chen, J., Wang, N., & Tian, Y. (2019). Autophagy and Apoptosis Interact to Modulate T-2 Toxin-Induced Toxicity in Liver Cells. Toxins, 11(1), 45. https://doi.org/10.3390/toxins11010045