Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide


Abstract
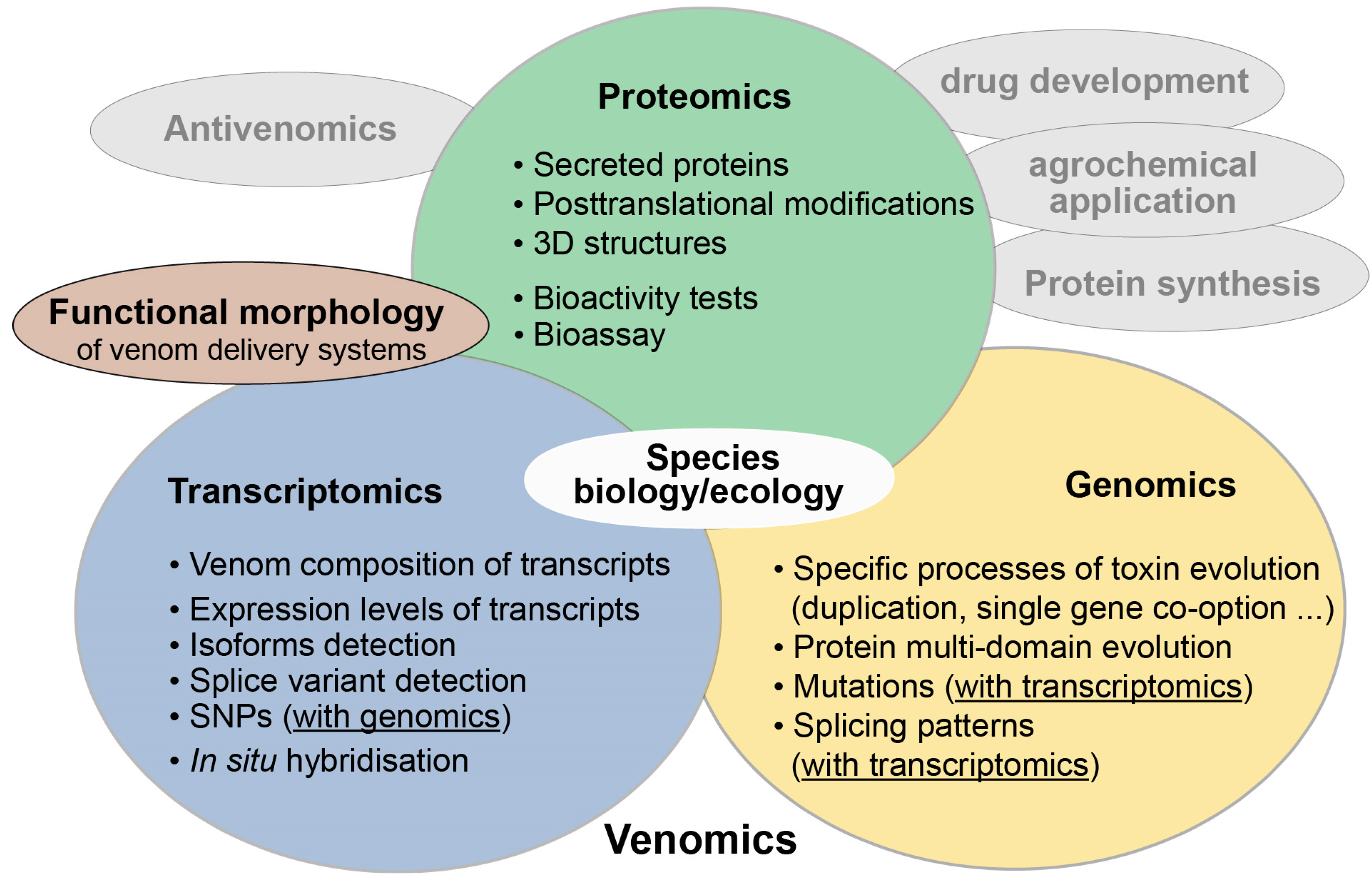
1. What is Modern Evolutionary Venomics?
2. Transcriptomics—One Major Pillar in Evolutionary Venomics
3. Theoretical Considerations from Collection to Sequencing
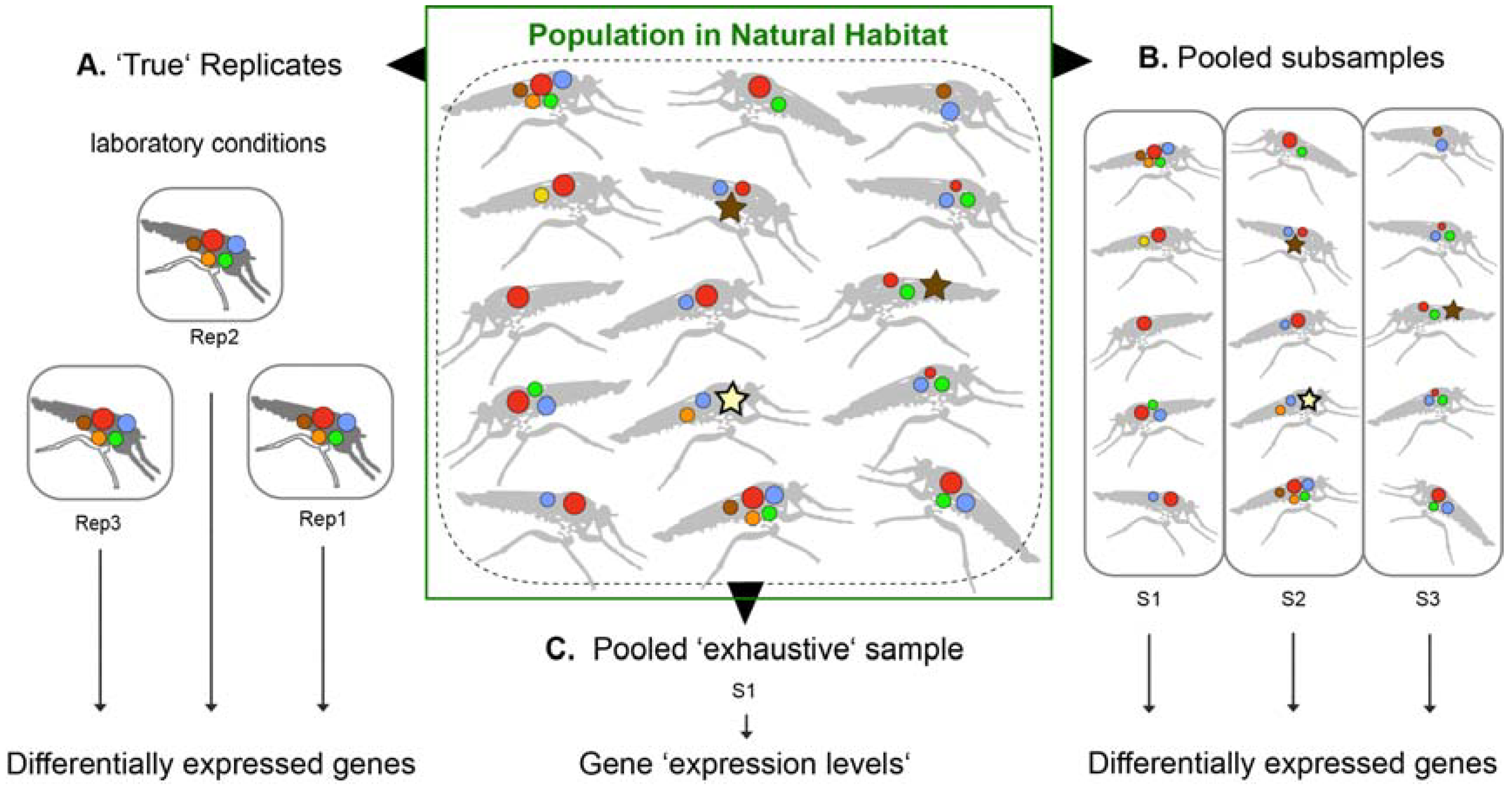
3.1. Implications from Pooled Samples of Small, Neglected Organisms
3.2. Advantages of Dissecting the Whole Venom Delivery System and Its Downside
3.3. Practical Thoughts for the Dissection of Glands, Sample Preservation and Sequencing
4. Transcriptome Analysis and Its Complexity
4.1. Raw Read Filtering (Read Pre-Processing)
- (1)
- Including longer reads increases the assembly performance because orthologous genes are better identified. This effect saturates when read lengths reach a certain threshold. However, this threshold seems sample and taxon dependent, so a general recommendation is not possible (~150 bp for tested human and mouse, and ~75 bp for yeast) [81]. One suggestion is to filter the data multiple times with different read lengths to assess its impact on finally excluded data. Depending on the used sequencing platform and general sequencing length, the impact could be severe. The goal should be to include reads with the longest possible length.
- (2)
- Similarly, the choice of phred quality values is a trade-off between not excluding too many raw reads but retaining as many as possible good quality reads [77]. The phred-value impacts stronger on resulting quality of RNASeq data than on DNA based genome sequencing and is shown to possibly affect later gene expression results [82]. Illumina data should be filtered with a phred value of 30 or more, a phred value of 30 allows for an error rate of 99.9% (one erroneous base per 1000 bases can still be a lot depending on the sequence depth).
4.2. De Novo Transcriptome Read Assembly
4.3. Read Mapping
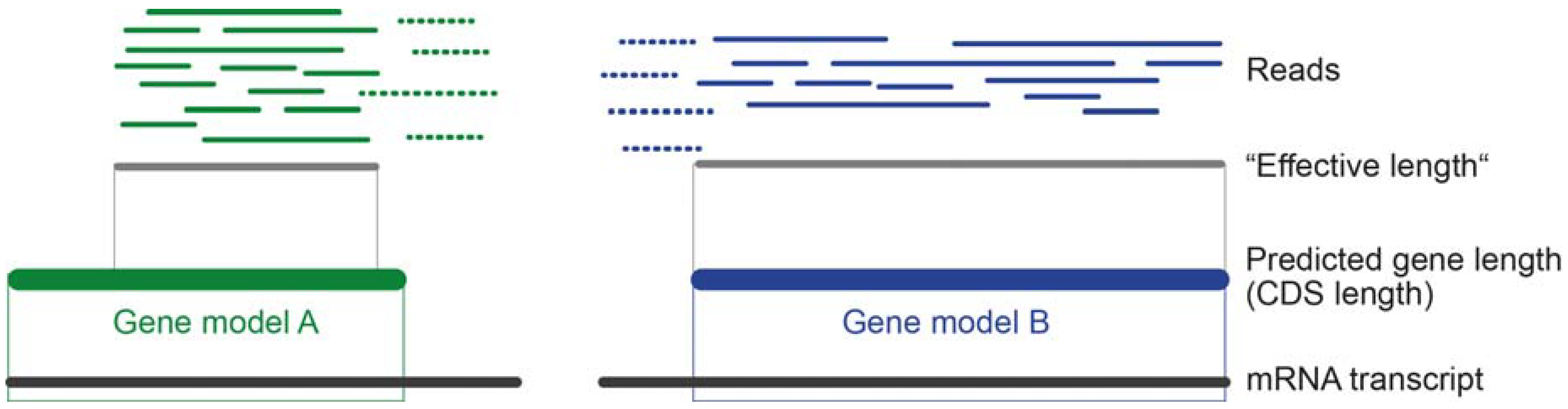
4.4. Transcript Quantification and Gene Expression Level Estimation
5. Identification of Putative Toxins
5.1. First Thresholds to Prevent False Positive Transcripts
5.2. Different Strategies to Identify Putative Toxins
6. Key Advantages of RNASeq
6.1. Small but Mighty –RNASeq Covers Smaller Peptides That are Missed by Proteomics
6.2. With Great Power Comes Great Responsibility—Transcriptome-Only Data
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Bazaa, A.; Marrakchi, N.; El Ayeb, M.; Sanz, L.; Calvete, J.J. Snake venomics: Comparative analysis of the venom proteomes of the Tunisian snakes Cerastes cerastes, Cerastes vipera and Macrovipera lebetina. Proteomics 2005, 5, 4223–4235. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Campbell, L.I.; Jenner, R.A. Quo vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2016, 135, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Drukewitz, S.H.; Fuhrmann, N.; Undheim, E.A.B.; Blanke, A.; Giribaldi, J.; Mary, R.; Laconde, G.; Dutertre, S.; von Reumont, B.M. A dipteran’s novel sucker punch: Evolution of arthropod atypical venom with a neurotoxic component in robber flies (Asilidae, Diptera). Toxins 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Mayhew, M.L.; Jin, J.; Herzig, V.; Undheim, E.A.B.; Sombke, A.; Fry, B.G.; Meritt, D.J.; King, G.F. The assassin bug Pristhesancus plagipennis produces two distinct venoms in separate gland lumens. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Undheim, E.A.B.; Rupasinghe, D.B.; Ikonomopoulou, M.P.; King, G.F. Spider venomics: Implications for drug discovery. Future Med. Chem. 2014, 6, 1699–1714. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Visser, J.C.; Baumann, K.; Dobson, J.; Han, H.; Kuruppu, S.; Morgan, M.; Romilio, A.; Weisbecker, V.; Mardon, K.; et al. The evolution of fangs, venom, and mimicry systems in blenny fishes. Curr. Biol. 2017, 27, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Calvete, J.J. Strategies in “snake venomics” aiming at an integrative view of compositional, functional, and immunological characteristics of venoms. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Sanz, L.; Angulo, Y.; Lomonte, B.; Gutiérrez, J.M. Venoms, venomics, antivenomics. FEBS Lett. 2009, 583, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhao, H.-Y.; Yin, Y.; Shen, S.-S.; Shan, L.-L.; Chen, C.-X.; Zhang, Y.-X.; Gao, J.-F.; Ji, X. Combined venomics, antivenomics and venom gland transcriptome analysis of the monocoled cobra (Naja kaouthia) from China. J. Proteom. 2017, 159, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Menez, A.; Stöcklin, R.; Mebs, D. “Venomics” or: The venomous systems genome project. Toxicon 2006, 47, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.B.; Fry, B.G.; King, G.F. Centipede venom: Recent discoveries and current state of knowledge. Toxins 2015, 7, 679–704. [Google Scholar] [CrossRef] [PubMed]
- Martinson, E.O.; Mrinalini; Kelkar, Y.D.; Chang, C.-H.; Werren, J.H. The evolution of venom by co-option of single-copy genes. Curr. Biol. 2017, 27, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Gendreau, K.L.; Haney, R.A.; Schwager, E.E.; Wierschin, T.; Stanke, M.; Richards, S.; Garb, J.E. House spider genome uncovers evolutionary shifts in the diversity and expression of black widow venom proteins associated with extreme toxicity. BMC Genom. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gorson, J.; Holford, M. Small packages, big returns: Uncovering the venom diversity of small invertebrate conoidean snails. Integr. Comp. Biol. 2016, 56, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bleidorn, C. Phylogenomics; Springer International Publishing AG: Cham, Switzerland, 2017. [Google Scholar]
- Earl, D.; Bradnam, K.; St John, J.; Darling, A.; Lin, D.; Fass, J.; Yu, H.O.K.; Buffalo, V.; Zerbino, D.R.; Diekhans, M.; et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21, 2224–2241. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Whelan, N.V.; Kocot, K.M.; Halanych, K.M. Employing phylogenomics to tesolve the relationships among cnidarians, ctenophores, sponges, placozoans, and bilaterians. Integr. Comp. Biol. 2015, 55, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- ENCODE. Available online: https://www.encodeproject.org/ (accessed on 10 July 2018).
- i5K. Available online: https://www.hgsc.bcm.edu/arthropods/i5k (accessed on 10 July 2018).
- Koepfli, K.-P.; Paten, B.; Genome 10K Community of Scientists; O’Brien, S.J. The Genome 10K Project: A way forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef] [PubMed]
- GIGA. Available online: http://giga-cos.org/ (accessed on 10 July 2018).
- 1KITE. Available online: http://www.1kite.org/ (accessed on 10 July 2018).
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Bleidorn, C. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 2016, 14, 1–8. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High throughput sequencing: An overview of sequencing chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Jenner, R.A.; Wills, M.A.; Dell’Ampio, E.; Pass, G.; Ebersberger, I.; Meyer, B.; Koenemann, S.; Iliffe, T.M.; Stamatakis, A.; et al. Pancrustacean phylogeny in the light of new phylogenomic data: Support for Remipedia as the possible sister group of Hexapoda. Mol. Biol. Evol. 2012, 29, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The first venomous crustacean revealed by transcriptomics and functional morphology: Remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 2014, 31, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Undheim, E.A.B.; Jauss, R.-T.; Jenner, R.A. Venomics of remipede crustaceans reveals novel peptide diversity and illuminates the venom’s biological role. Toxins 2017, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Garb, J.E. Extraction of venom and venom gland microdissections from spiders for proteomic and transcriptomic analyses. J. Vis. Exp. 2014, 93, e51618. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.D.; Scortecci, K.C.; Kobashi, L.S.; Agnez-Lima, L.F.; Medeiros, S.R.B.; Silva-Junior, A.A.; De Junqueira-de-Azevedo, I.L.M.; De Fernandes-Pedrosa, M.F. Profiling the resting venom gland of the scorpion Tityus stigmurus through a transcriptomic survey. BMC Genom. 2012, 13, 362. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Verdes, A.; Simpson, D.; Holford, M. Are Fireworms venomous? Evidence for the convergent evolution of toxin homologs in three species of fireworms (Annelida, Amphinomidae). Genome Biol. Evol. 2018, 10, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ontano, A.Z.; Harvey, M.S.; Sharma, P.P. Transcriptomic analysis of pseudoscorpion venom reveals a unique cocktail dominated by enzymes and protease inhibitors. Toxins 2018, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, J.; White, K.P. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics 2014, 30, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, D.; Rohde, B.H.; King, G.F.; Tal, T.; Sher, D.; Zlotkin, E. The tale of a resting gland: Transcriptome of a replete venom gland from the scorpion Hottentotta judaicus. Toxicon 2011, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Kelln, W.J.; Hayes, W.K. Venom regeneration in the centipede Scolopendra polymorpha: Evidence for asynchronous venom component synthesis. Zoology 2014, 117, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Chippaux, J.P.; Williams, V.; White, J. Snake-venom variability—Methods of study, results and interpretation. Toxicon 1991, 29, 1279–1303. [Google Scholar] [CrossRef]
- Calvete, J.J.; Escolano, J.; Sanz, L. Snake venomics of Bitis species reveals large intragenus venom toxin composition variation: Application to taxonomy of congeneric taxa. J. Proteome Res. 2007, 6, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Neale, V.; Sotillo, J.; Seymour, J.E.; Wilson, D. The venom of the spine-bellied sea snake (Hydrophis curtus): Proteome, toxin diversity and intraspecific variation. Int. J. Mol. Sci. 2017, 18, 2695. [Google Scholar] [CrossRef] [PubMed]
- Nunez, V.; Cid, P.; Sanz, L.; De La Torre, P.; Angulo, Y.; Lomonte, B.; Maria Gutierrez, J.; Calvete, J.J. Snake venomics and antivenomics of Bothrops atrox venoms from Colombia and the Amazon regions of Brazil, Peru and Ecuador suggest the occurrence of geographic variation of venom phenotype by a trend towards paedomorphism. J. Proteom. 2009, 73, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Lomonte, B.; Leon, G.; Alape-Girón, A.; Flores-Diaz, M.; Sanz, L.; Angulo, Y.; Calvete, J.J. Snake venomics and antivenomics: Proteomic tools in the design and control of antivenoms for the treatment of snakebite envenoming. J. Proteom. 2009, 72, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Biass, D.; Stoecklin, R.; Favreau, P. Dramatic intraspecimen variations within the injected venom of Conus consors: An unsuspected contribution to venom diversity. Toxicon 2010, 55, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Undheim, E.A.B. True lies: Using proteomics to assess the accuracy of transcriptome-based venomics in centipedes uncovers false positives and reveals startling intraspecific variation in Scolopendra subspinipes. Toxins 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Touchard, A.; Dejean, A.; Escoubas, P.; Orivel, J. Intraspecific variations in the venom peptidome of the ant Odontomachus haematodus (Formicidae: Ponerinae) from French Guiana. J. Hymenopt. Res. 2015, 47, 87–101. [Google Scholar] [CrossRef]
- Ruiming, Z.; Yibao, M.; Yawen, H.; Zhiyong, D.; Yingliang, W.; Zhijian, C.; Wenxin, L. Comparative venom gland transcriptome analysis of the scorpion Lychas mucronatus reveals intraspecific toxic gene diversity and new venomous components. BMC Genom. 2010, 11, 452. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.A.; Omran, M.A.A.; Abdel-Nabi, I.M.; Ueda, H.; McVean, A. Intraspecific variation in the Egyptian scorpion Scorpio maurus palmatus venom collected from different biotopes. Toxicon 2009, 53, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.C.; Furtado, M.F.; Travaglia-Cardoso, S.R.; Camargo, A.C.M.; Serrano, S.M.T. Sex-based individual variation of snake venom proteome among eighteen Bothrops jararaca siblings. Toxicon 2006, 47, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, D.C.; Prezoto, B.C.; Konno, K.; Melo, R.L.; Furtado, M.F.; Camargo, A.C.M.; Serrano, S.M.T. Mass spectrometric analysis of the individual variability of Bothrops jararaca venom fraction. Evidence for sex-based variation among the bradykinin-potentiating peptides. Rapid Commun. Mass Spectrom. 2007, 21, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Binford, G.J. An analysis of geographic and intersexual chemical variation in venoms of the spider Tegenaria agrestis (Agelenidae). Toxicon 2001, 39, 955–968. [Google Scholar] [CrossRef]
- Binford, G.J.; Gillespie, R.G.; Maddison, W.P. Sexual dimorphism in venom chemistry in Tetragnatha spiders is not easily explained by adult niche differences. Toxicon 2016, 114, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.S.; Tae, H.; Yang, Y.; Mockaitis, K.; Van Hemert, J.L.; Proulx, S.R.; Choi, J.-H.; Bronikowski, A.M. A garter snake transcriptome: Pyrosequencing, de novo assembly, and sex-specific differences. BMC Genom. 2010, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ferreira, M.; Sosa-Rosales, I.; Bruni, F.M.; Ramos, A.D.; Vieira Portaro, F.C.; Conceicao, K.; Lima, C. Analysis of the intersexual variation in Thalassophryne maculosa fish venoms. Toxicon 2016, 115, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Jones, A.D.; Goldston, J.S.; Rowe, M.P.; Rowe, A.H. Sex Differences in Defensive Behavior and Venom of The Striped Bark Scorpion Centruroides vittatus (Scorpiones: Buthidae). Integr. Comp. Biol. 2016, 56, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Hodgson, W.C. Intersexual variations in the pharmacological properties of Coremiocnemis tropix (Araneae, Theraphosidae) spider venom. Toxicon 2009, 53, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Rosenthal, M.; Undheim, E.E.A.; King, G.F. Harvesting venom toxins from assassin bugs and other heteropteran insects. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Besson, T.; Debayle, D.; Diochot, S.; Salinas, M.; Lingueglia, E. Low cost venom extractor based on Arduino. Toxicon 2016, 118, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.M.; Farrell, P.M. A portable device for the electrical extraction of scorpion venom. Toxicon 2011, 57, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, C. Comments on the natural expression and artificial extraction of venom gland components from spiders. Toxin Rev. 2005, 24, 257–271. [Google Scholar] [CrossRef]
- Oukkache, N.; Chgoury, F.; Lalaoui, M.; Cano, A.A.; Ghalim, N. Comparison between two methods of scorpion venom milking in Morocco. J. Venom. Anim. Toxins Incl. Trop. Dis. 2013, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Sahayaraj, K.; Muthukumar, S.; Prem Anandh, G. Evaluation of milking and electric shock methods for venom collection from hunter reduviids. Entomon 2006, 31, 65–68. [Google Scholar]
- Cooper, A.M.; Fox, G.A.; Nelsen, D.R.; Hayes, W.K. Variation in venom yield and protein concentration of the centipedes Scolopendra polymorpha and Scolopendra subspinipes. Toxicon 2014, 82, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.B.; Hamilton, B.R.; Kurniawan, N.D.; Bowlay, G.; Cribb, B.W.; Merritt, D.J.; Fry, B.G.; King, G.F.; Venter, D.J. Production and packaging of a biological arsenal: Evolution of centipede venoms under morphological constraint. Proc. Natl. Acad. Sci. USA 2015, 112, 4026–4031. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Lum, J.; Cordero, F.; Beccuti, M.; Poidinger, M.; Donatelli, S.; Calogero, R.A.; Zolezzi, F. Alternative splicing detection workflow needs a careful combination of sample prep and bioinformatics analysis. BMC Bioinform. 2015, 16, S2. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Khaleel, S.S.; Huang, H.; Wu, C.H. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 2014, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Wang, Z.; Li, G. The impacts of read length and transcriptome complexity for de novo Assembly: A simulation study. PLoS ONE 2014, 9, e94825. [Google Scholar] [CrossRef] [PubMed]
- De Sa, P.H.C.G.; Veras, A.A.O.; Carneiro, A.R.; Pinheiro, K.C.; Pinto, A.C.; Soares, S.C.; Schneider, M.P.C.; Azevedo, V.; Silva, A.; Ramos, R.T.J. The impact of quality filter for RNA-Seq. Gene 2015, 563, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.; Schroeder, C.; Bauer, P. SeqPurge: Highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinform. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Didion, J.P.; Martin, M.; Collins, F.S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017, 5, e3720. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.B.; Zadlock, F.J.; Zhang, Z.; Murphy, W.R.; Bentivegna, C.S. Comparison of De Novo Transcriptome assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS ONE 2016, 11, e0153104. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Holding, M.L.; Margres, M.J.; Mason, A.J.; Parkinson, C.L.; Rokyta, D.R. Evaluating the performance of de novo assembly methods for venom-gland transcriptomics. Toxins 2018, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Cabau, C.; Escudie, F.; Djari, A.; Guiguen, Y.; Bobe, J.; Klopp, C. Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ 2017, 5, e2988. [Google Scholar] [CrossRef] [PubMed]
- Pevzner, P.A.; Tang, H.; Waterman, M.S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA 2001, 98, 9748–9753. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, S.T.; Emrich, S.J. Assessing de novo transcriptome assembly metrics for consistency and utility. BMC Genom. 2013, 14, 465. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Campbell, L.I.; Richter, S.; Hering, L.; Sykes, D.; Hetmank, J.; Jenner, R.A.; Bleidorn, C. A Polychaete’s powerful punch: Venom gland transcriptomics of Glycera reveals a complex cocktail of toxin homologs. Genome Biol. Evol. 2014, 6, 2406–2423. [Google Scholar] [CrossRef] [PubMed]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, O.; Hara, Y.; Kuraku, S. gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 2017, 33, 3635–3637. [Google Scholar] [CrossRef] [PubMed]
- Pop, M. Genome assembly reborn: Recent computational challenges. Brief. Bioinform. 2009, 10, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.-I.; Nam, J.-W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 2018, 19, 23–40. [Google Scholar] [PubMed]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Zhou, P.; Mudge, J.; Gurtowski, J.; Lee, H.; Ramaraj, T.; Walenz, B.P.; Liu, J.; Stupar, R.M.; Denny, R.; et al. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genom. 2017, 18, 541. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 2014, 30, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Marioni, J.; Brazma, A. RNA-Seq gene profiling—A systematic empirical comparison. PLoS ONE 2014, 9, e107026. [Google Scholar] [CrossRef] [PubMed]
- Thankaswamy-Kosalai, S.; Sen, P.; Nookaew, I. Evaluation and assessment of read-mapping by multiple next-generation sequencing aligners based on genome-wide characteristics. Genomics 2017, 109, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Sarkar, H.; Gupta, N.; Patro, R. RapMap: A rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes. Bioinformatics 2016, 32, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Prachter, L. Models for transcript quantification from RNA-Seq. arXiv, 2011; arXiv:arXiv:1104. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Maza, E. In Papyro Comparison of TMM (edgeR), RLE (DESeq2), and MRN Normalization Methods for a Simple Two-Conditions-Without-Replicates RNA-Seq Experimental Design. Front. Genet. 2016, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Mount, S.M.; Kingsford, C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014, 32, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Pla, D.; Petras, D.; Saviola, A.J.; Modahl, C.M.; Sanz, L.; Pérez, A.; Juarez, E.; Frietze, S.; Dorrestein, P.C.; Mackessy, S.P.; et al. Transcriptomics-guided bottom-up and top-down venomics of neonate and adult specimens of the arboreal rear-fanged Brown Treesnake, Boiga irregularis, from Guam. J. Proteom. 2018, 174, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.D.; Tatonetti, N.P. VenomKB, a new knowledge base for facilitating the validation of putative venom therapies. Sci. Data 2015, 2, 150065. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.D.; Tatonetti, N.P. Using a novel ontology to inform the discovery of therapeutic peptides from animal venoms. AMIA Jt. Summits Transl. Sci. Proc. 2016, 2016, 209–218. [Google Scholar] [PubMed]
- Gacesa, R.; Barlow, D.J.; Long, P.F. Machine learning can differentiate venom toxins from other proteins having non-toxic physiological functions. PeerJ Comput. Sci. 2016, 2, e90. [Google Scholar] [CrossRef]
- Wood, D.L.A.; Miljenović, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. ArachnoServer: A database of protein toxins from spiders. BMC Genom. 2009, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Xu, Y. Protein databases on the internet. Curr. Protoc. Mol. Biol. 2004. [Google Scholar] [CrossRef]
- Luna-Ramírez, K.; Tonk, M.; Rahnamaeian, M.; Vilcinskas, A. bioactivity of natural and engineered antimicrobial peptides from venom of the scorpions Urodacus yaschenkoi and U. manicatus. Toxins 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Podsiadlowski, L.; Muhammed, M.; Vilcinskas, A. Diversity, evolution and medical applications of insect antimicrobial peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150290. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Brock, G.N.; Rouchka, E.C.; Cooper, N.G.F.; Wu, D.; O’Toole, T.E.; Gill, R.S.; Eteleeb, A.M.; O’Brien, L.; Rai, S.N. A comparison of per sample global scaling and per gene normalization methods for differential expression analysis of RNA-seq data. PLoS ONE 2017, 12, e0176185. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, A.D.; Swain, M.T.; Hegarty, M.J.; Logan, D.W.; Mulley, J.F. Restriction and recruitment-gene duplication and the origin and evolution of snake venom toxins. Genome Biol. Evol. 2014, 6, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed]
- Schwager, E.E.; Sharma, P.P.; Clarke, T.; Leite, D.J.; Wierschin, T.; Pechmann, M.; Akiyama-Oda, Y.; Esposito, L.; Bechsgaard, J.; Bilde, T.; et al. The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biol. 2017, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.W.; Papenfuss, A.T.; Whittington, C.M.; Warren, W.C.; Belov, K. A limited role for gene duplications in the evolution of platypus venom. Mol. Biol. Evol. 2012, 29, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Columbus-Shenkar, Y.Y.; Sachkova, M.Y.; Macrander, J.; Fridrich, A.; Modepalli, V.; Reitzel, A.M.; Sunagar, K.; Moran, Y. Dynamics of venom composition across a complex life cycle. eLife 2018, 7, e35014. [Google Scholar] [CrossRef] [PubMed]
- Dowell, N.L.; Giorgianni, M.W.; Kassner, V.A.; Selegue, J.E. The Deep Origin and Recent Loss of Venom Toxin Genes in Rattlesnakes. Curr. Biol. 2016, 26, 2434–2445. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Meaning and Formula (Source) | More Detailed Description and Calculation Steps |
|---|---|---|
| Read count | Read number estimated for a transcript | This reflects the “raw” read number per transcript, which is given as first result by most read mappers |
| CPM | Read number counts per million | This is the read count normalized by the number of sequenced reads (library size). |
| RPKM | Reads per kilobase (kb) per million. Reads are normalized with library size and then read length. | (1) Total reads are divided by 1,000,000 to scale per million. (2) Mapped reads are divided by the scaling factor normalizing for sequencing depth resulting in reads per million. (3) Reads per million are divided by the transcript length (in kb). |
| FPKM | Fragments per kilobase (kb) per million. | Same as RPKM, but paired ends are taken into account, in case a fragment occurs in both reads it is only counted once. |
| TPM | Transcripts per million. Transcripts are normalized with read length first and then by the number of read numbers of the library. | (1) Mapped reads are divided by transcript length (in kb) resulting in reads per kb. (2) All reads per kb values are counted up and divided by 1,000,000 to receive a per million scaling factor. (3) The reads per kb are finally divided by the scaling factor. |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Reumont, B.M. Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide. Toxins 2018, 10, 292. https://doi.org/10.3390/toxins10070292
Von Reumont BM. Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide. Toxins. 2018; 10(7):292. https://doi.org/10.3390/toxins10070292
Chicago/Turabian StyleVon Reumont, Björn Marcus. 2018. "Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide" Toxins 10, no. 7: 292. https://doi.org/10.3390/toxins10070292
APA StyleVon Reumont, B. M. (2018). Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide. Toxins, 10(7), 292. https://doi.org/10.3390/toxins10070292