The Fever Tree: from Malaria to Neurological Diseases


Abstract

{kind=link}
{kind=link}
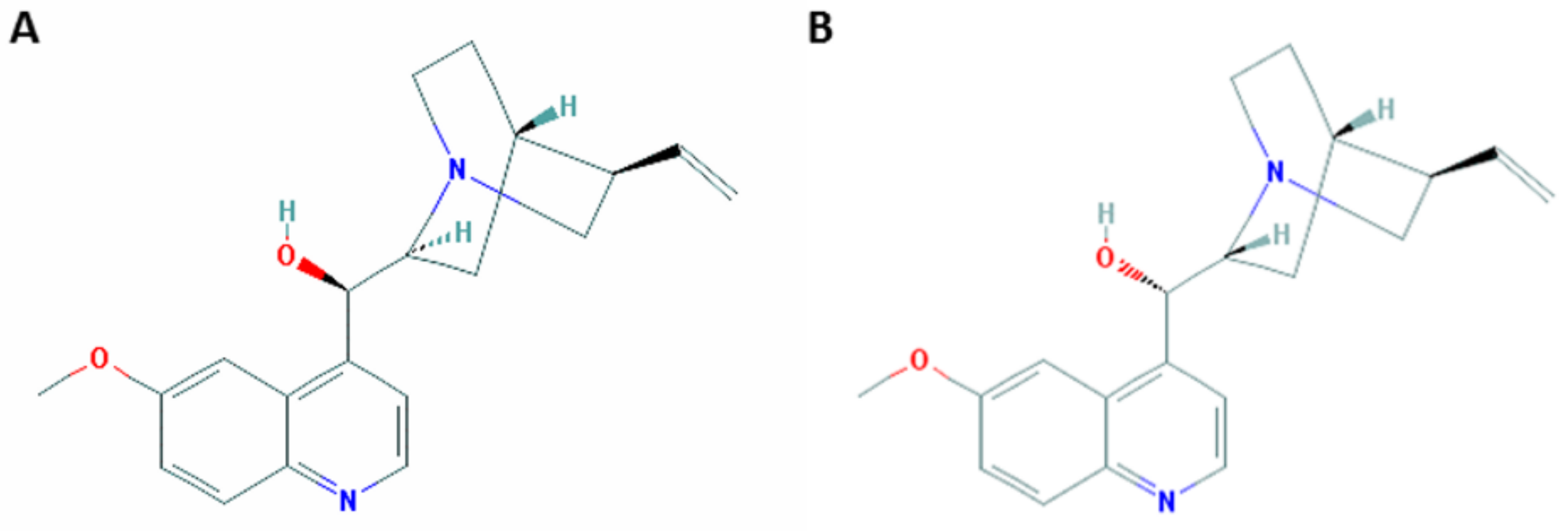{kind=link}
{kind=link}
{kind=link}
1. From Bark to Chemical Synthesis
2. The Rise and Descent of Cinchona Alkaloids as Antimalarial Drugs
3. Introduction of Cinchona Alkaloids as Antiarrhythmic Agents
4. Toxicity of Quinine and Quinidine Still Limits Their Use
5. Current Uses of Quinine and Quinidine
6. Conclusions and Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, C.J.S. The history and lore of cinchona. Br. Med. J. 1928, 2, 1188–1190. [Google Scholar]
- Jaramillo-Arango, J. A Critical Review of the Basic Facts in the History of Cinchona. Bot. J. Linn. Soc. 1949, 53, 272–311. [Google Scholar] [CrossRef]
- Sequeira, J.H. The History of Cinchona. Br. Med. J. 1929, 1, 621–622. [Google Scholar] [CrossRef]
- Thompson, K.A.; Blair, I.A.; Woosley, R.L.; Roden, D.M. Comparative in vitro electrophysiology of quinidine, its major metabolites and dihydroquinidine. J. Pharmacol. Exp. Ther. 1987, 241, 84–90. [Google Scholar] [PubMed]
- Burba, J. Cinchona Bark. University of Minnesota Libraries. 2018. Available online: https://www.lib.umn.edu/bell/tradeproducts/cinchonabark#n14 (accessed on 3 October 2018).
- Harrison, N. In celebration of the Jesuit’s powder: A history of malaria treatment. Lancet 2015, 15, 1143. [Google Scholar] [CrossRef]
- Klein, W.; Pieters, T. The Hidden History of a Famous Drug: Tracing the Medical and Public Acculturation of Peruvian Bark in Early Modern Western Europe (c. 1650–1720). J. Hist Med. Allied Sci. 2016, 71, 400–421. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Azoulay, S. Stories About the Origin of Quinquina and Quinidine. J. Cardiovasc. Electrophysiol. 1994, 5, 635–636. [Google Scholar] [CrossRef] [PubMed]
- De Jussieu, J. Description de L’arbre a Quinquina; Société du Traitement des Quinquinas: Paris, France, 1936. [Google Scholar]
- Rabe, P.; Kindler, K. Über die partielle Synthese des Chinins. Zur Kenntnis der China-Alkaloide XIX. Berichte der deutschen chemischen Gesellschaft 1918, 51, 466–467. [Google Scholar] [CrossRef]
- Sanderson, K. A Tonic for Quinine Chemistry. Nature 2008. [Google Scholar] [CrossRef]
- Woodward, R.B.; Doering, W.E. The total synthesis of quinine. J. Am. Chem Soc. 1944, 66, 849. [Google Scholar] [CrossRef]
- Woodward, R.B.; Doering, W.E. The Total Synthesis of Quinine. J. Am. Chem Soc. 1945, 67, 860–874. [Google Scholar] [CrossRef]
- Ball, P. Quinine steps back in time. Nature 2008, 451, 1065. [Google Scholar] [CrossRef] [PubMed]
- Stork, G.; Niu, D.; Fujimoto, R.A.; Koft, E.R.; Balkovec, J.M.; Tata, J.R.; Dake, G.R. The first stereoselective total synthesis of quinine. J. Am. Chem. Soc. 2001, 123, 3239–3242. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Williams, R.M. Rabe rest in peace: Confirmation of the rabe-kindler conversion of D-quinotoxine into quinine: Experimental affirmation of the Woodward-Doering formal total synthesis of quinine. Angew. Chem. 2008, 47, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Dymock, W.; Warden, C.J.H.; Hooper, D. Pharmacographia Indica: A History of the Principal Drugs of Vegetable Origin Met with in British India; Kegan Paul, Trench, Trübner: London, UK, 1891; Volume II, pp. 180–181. Available online: http://www.jameslindlibrary.org/dymock-w-warden-cjh-hooper-d-1891/ (accessed on 13 November 2018).
- Sanders, J.P.; Dawson, W.T. Quinidine. J. Am. Med. Assoc. 1932, 99, 1773–1777. [Google Scholar] [CrossRef]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Shanks, G.D. How World War 1 changed global attitudes to war and infectious diseases. Lancet 2014, 384, 1699–1707. [Google Scholar] [CrossRef]
- Brabin, B.J. Malaria’s contribution to World War One–the unexpected adversary. Malar. J. 2014, 13, 497. [Google Scholar] [CrossRef] [PubMed]
- Vinetz, J.M. Chemotherapy of Malaria. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Ross, R. On some Peculiar Pigmented Cells Found in Two Mosquitos Fed on Malarial Blood. Br. Med. J. 1897, 2, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Martin, N. Surgeon-Major Ronald Ross, IMS Nobel laureate in medicine (1902) for his work on malaria. J. R. Army Med. Corps 1999, 145, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.F.; Cerami, A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature 1992, 355, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Olafson, K.N.; Nguyen, T.Q.; Rimer, J.D.; Vekilov, P.G. Antimalarials inhibit hematin crystallization by unique drug-surface site interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 7531–7536. [Google Scholar] [CrossRef] [PubMed]
- Wenckebach, K.F. Cinchona derivatives in the treatment of heart disorders. J. Am. Med. Assoc. 1923, 81, 472–474. [Google Scholar] [CrossRef]
- Smulyan, H. The Beat Goes on: The Story of 5 Ageless Cardiac Drugs. Am. J. Med. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Frey, W. Weitere Erfahrungen mit Chimdin bei Abseluten Herzunregelmassigkeit. Wiener Klinische Wochenschrift 1918, 55, 849. [Google Scholar]
- Lewis, T.; Drury, A.N.; Iliescu, C.C.; Wedd, A.M. The manner in which quinidine sulphate acts in auricular fibrillation. Br. Med. J. 1921, 2, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Selzer, A.; Wray, H.W. Quinidine syncope. Paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation 1964, 30, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Hanon, S.; Lam, P.; Schweitzer, P. Quinidine revisited. Am. J. Med. 2009, 122, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Camm, A.J. Quinidine. N. Engl. J. Med. 1998, 338, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Roden, D.M. Antiarrhythmic Drugs. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- White, N.J. The treatment of malaria. N. Engl. J. Med. 1996, 335, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Qualaquin, Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021799s024lbl.pdf. (accessed on 7 October 2018).
- Bar-Oz, B.; Levichek, Z.; Koren, G. Medications that can be fatal for a toddler with one tablet or teaspoonful. Paediatr. Drugs 2004, 6, 123–126. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Morton, J.M.; Liles, N.W.; Nester, C.M. After the Party’s Over. N. Engl. J. Med. 2017, 376, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Liles, N.W.; Page, E.E.; Liles, A.L.; Vesely, S.K.; Raskob, G.E.; George, J.N. Diversity and severity of adverse reactions to quinine: A systematic review. Am. J. Hematol. 2016, 91, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Treatment of Malaria: Guidelines for Clinicians (United States). Available online: https://www.cdc.gov/malaria/diagnosis_treatment/clinicians2.html (accessed on 7 October 2018).
- Rosenthal, P.J. Antiprotozoal Drugs. In Basic & Clinical Pharmacology, 14th ed.; Katzung, B.G., Ed.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- WHO. WHO Model List of Essential Medicines. 19th List (April 2015) (Amended August 2015). Available online: http://www.who.int/selection_medicines/committees/expert/20/EML_2015_FINAL_amended_AUG2015.pdf?ua=1&ua=1.&ua=1 (accessed on 6 October 2018).
- Fardet, L.; Nazareth, I.; Petersen, I. Association between Long-term Quinine Exposure and All-Cause Mortality. JAMA 2017, 317, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Medscape. Quinine and Leg Cramps: Not Worth the Risk. 2012. Available online: https://www.medscape.com/viewarticle/771699 (accessed on 6 October 2018).
- Houstoun, M.; Reichman, M.E.; Graham, D.J.; Nambiar, S.; Shamsuddin, H.; Jones, S.C.; Cao, K.; Wernecke, M.; Lam, C.; Worrall, C.M.; et al. Use of an active surveillance system by the FDA to observe patterns of quinine sulfate use and adverse hematologic outcomes in CMS Medicare data. Pharmacoepidemiol. Drug Saf. 2014, 23, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Quinine-Containing Shampoo from Amazon.com. 2018. Available online: https://www.amazon.com/Klorane-Shampoo-Quinine-Vitamins-Thinning/dp/B001LFJMGG?th=1 (accessed on 5 October 2018).
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Leshem, E.; Rahkovich, M.; Mazo, A.; Suleiman, M.; Blich, M.; Laish-Farkash, A.; Konstantino, Y.; Fogelman, R.; Strasberg, B.; Geist, M.; et al. Arrhythmic Events in Brugada Syndrome: A Nationwide Israeli Survey of the Clinical Characteristics, Treatment; and Long-Term Follow-up (ISRABRU-VF). Isr. Med. Assoc. J. IMAJ 2018, 5, 269–276. [Google Scholar] [PubMed]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T.; Dempsey, J.L.; Mimche, S.M.; Lamb, T.J. Physiological Regulation of Drug Metabolism and Transport: Pregnancy, Microbiome, Inflammation, Infection, and Fasting. Drug Metab. Dispos. 2018, 46, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. Combination drug shows promise for treating agitation in patients with Alzheimer’s disease. BMJ 2015, 351, h5015. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Deeks, E.D. Dextromethorphan/quinidine: A review of its use in adults with pseudobulbar affect. Drugs 2015, 75, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Lyketsos, C.G.; Peskind, E.R.; Porsteinsson, A.P.; Mintzer, J.E.; Scharre, D.W.; De La Gandara, J.E.; Agronin, M.; Davis, C.S.; Nguyen, U.; et al. Effect of Dextromethorphan-Quinidine on Agitation in Patients With Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA 2015, 314, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Smith, K.R.; Bahlo, M.; Nobili, L.; Kahana, E.; Licchetta, L.; Oliver, K.L.; Mazarib, A.; Afawi, Z.; Korczyn, A.; et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 2012, 44, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Barcia, G.; Fleming, M.R.; Deligniere, A.; Gazula, V.R.; Brown, M.R.; Langouet, M.; Chen, H.; Kronengold, J.; Abhyankar, A.; Cilio, R.; et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 2012, 44, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.J.; Li, M.; Gazina, E.V.; Heron, S.E.; Nair, U.; Trager, C.; Reid, C.A.; Venkat, A.; Younkin, D.P.; Dlugos, D.J.; et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 2014, 75, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Bearden, D.; Strong, A.; Ehnot, J.; DiGiovine, M.; Dlugos, D.; Goldberg, E.M. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 2014, 76, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.F.; Nakamura, R.; Saitsu, H.; Matsumoto, N.; Kira, R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann. Neurol. 2016, 79, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Kuki, I.; Kawawaki, H.; Okazaki, S.; Kim, K.; Hattori, Y.; Tsuji, H.; Nukui, M.; Inoue, T.; Yoshida, Y.; et al. Quinidine therapy for West syndrome with KCNTI mutation: A case report. Brain Dev. 2017, 39, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Mikati, M.A.; Jiang, Y.H.; Carboni, M.; Shashi, V.; Petrovski, S.; Spillmann, R.; Milligan, C.J.; Li, M.; Grefe, A.; McConkie, A.; et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol. 2015, 78, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.A.; Carney, P.W.; Roten, A.; Ching, M.; Lightfoot, P.A.; Churilov, L.; Nair, U.; Li, M.; Berkovic, S.F.; Petrou, S.; et al. Precision therapy for epilepsy due to KCNT1 mutations: A randomized trial of oral quinidine. Neurology 2018, 90, e67–e72. [Google Scholar] [CrossRef] [PubMed]
- Leahey, E.B., Jr.; Reiffel, J.A.; Drusin, R.E.; Heissenbuttel, R.H.; Lovejoy, W.P.; Bigger, J., Jr. Interaction between quinidine and digoxin. JAMA 1978, 240, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.; Kim, R.; Stein, C.; Wilkinson, G.; Roden, D. Inhibition of P-glycoprotein-mediated drug transport: A unifying mechanism to explain the interaction between digoxin and quinidine. Circulation 1999, 99, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications Guidance for Industry. Available online: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf (accessed on 12 October 2018).
- Eyal, S.; Hsiao, P.; Unadkat, J.D. Drug interactions at the blood-brain barrier: Fact or fantasy? Pharmacol. Ther. 2009, 123, 80–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Collier, A.C.; Link, J.M.; Domino, K.B.; Mankoff, D.A.; Eary, J.F.; Spiekerman, C.F.; Hsiao, P.; Deo, A.K.; Unadkat, J.D. Modulation of P-glycoprotein at the human blood-brain barrier by quinidine or rifampin treatment: A positron emission tomography imaging study. Drug Metab. Dispos. 2015, 43, 1795–1804. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eyal, S. The Fever Tree: from Malaria to Neurological Diseases. Toxins 2018, 10, 491. https://doi.org/10.3390/toxins10120491
Eyal S. The Fever Tree: from Malaria to Neurological Diseases. Toxins. 2018; 10(12):491. https://doi.org/10.3390/toxins10120491
Chicago/Turabian StyleEyal, Sara. 2018. "The Fever Tree: from Malaria to Neurological Diseases" Toxins 10, no. 12: 491. https://doi.org/10.3390/toxins10120491
APA StyleEyal, S. (2018). The Fever Tree: from Malaria to Neurological Diseases. Toxins, 10(12), 491. https://doi.org/10.3390/toxins10120491