Whole-Genome Sequencing of Chinese Yellow Catfish Provides a Valuable Genetic Resource for High-Throughput Identification of Toxin Genes

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Summary of Sequencing Data and Genome-size Estimation
2.2. Generation of a High-quality Whole-genome Assembly
2.2.1. Primary De Novo Genome Assembly
2.2.2. Genome Scaffolding
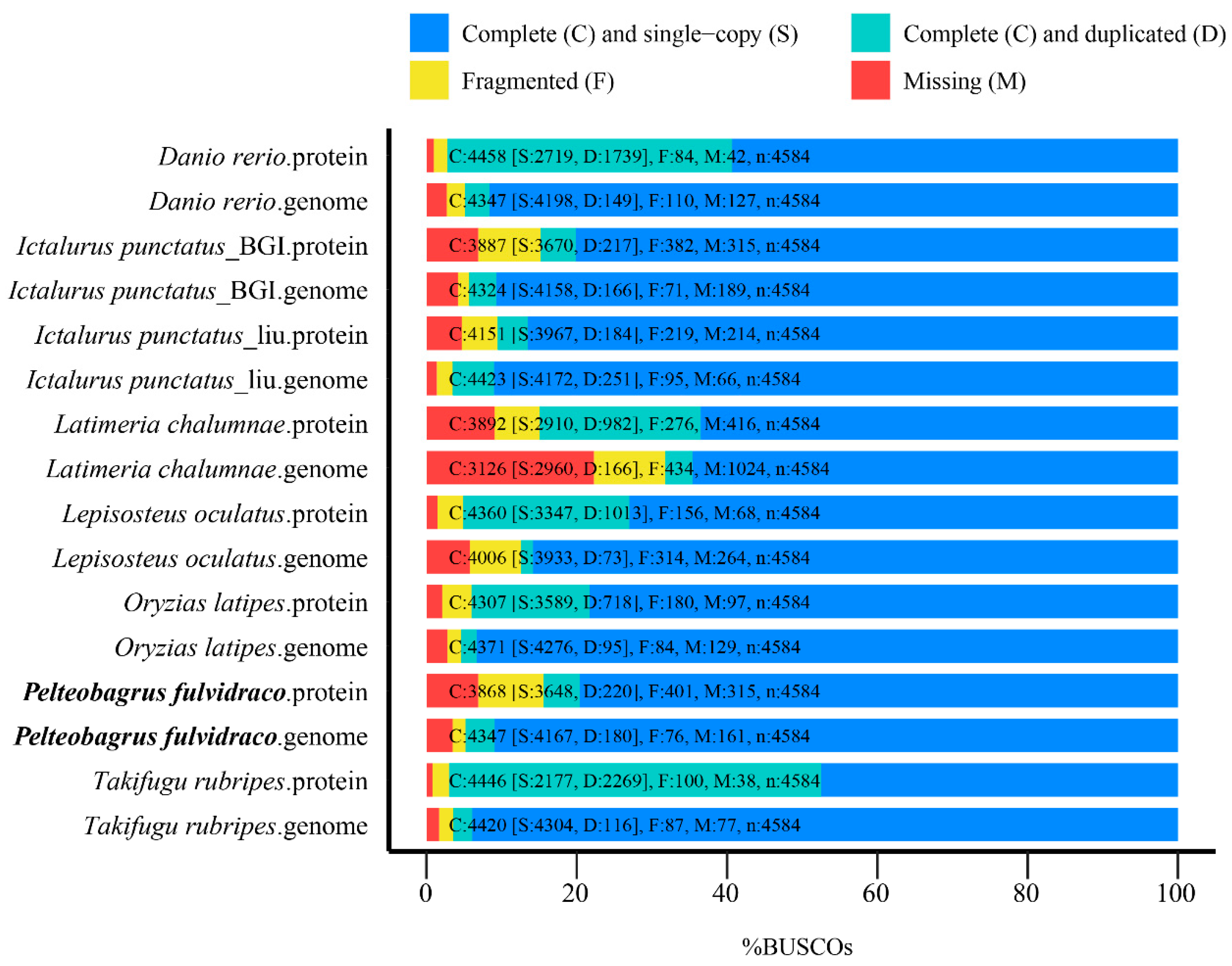
2.2.3. Evaluation of the Achieved Genome Assembly
2.3. Genome Annotation
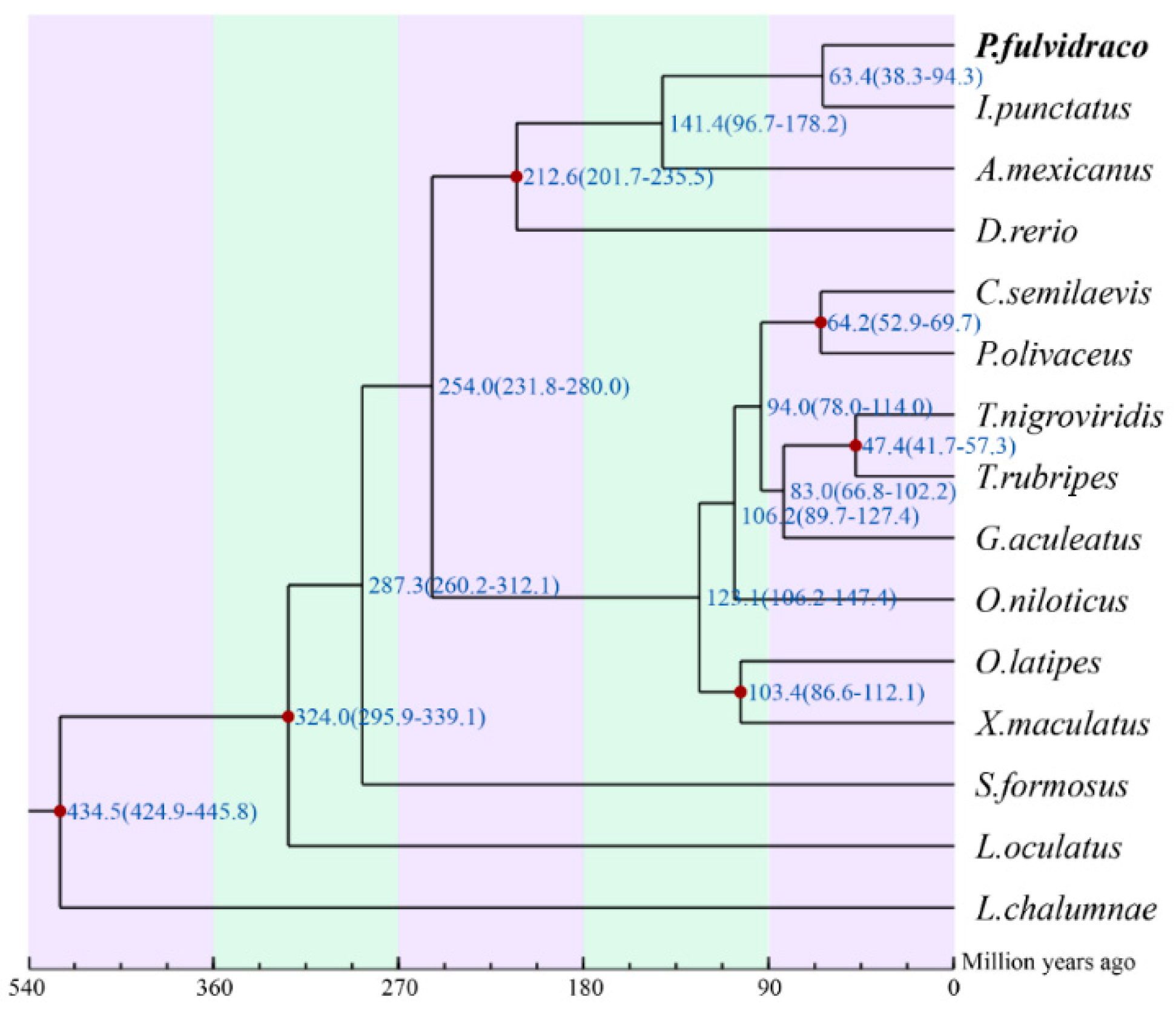
2.4. Phylogenetic Analysis and Divergence-Time Estimation of Chinese Yellow Catfish
2.5. High-Throughput Identification of Toxin Genes
2.5.1. The Short-Length Toxin Genes
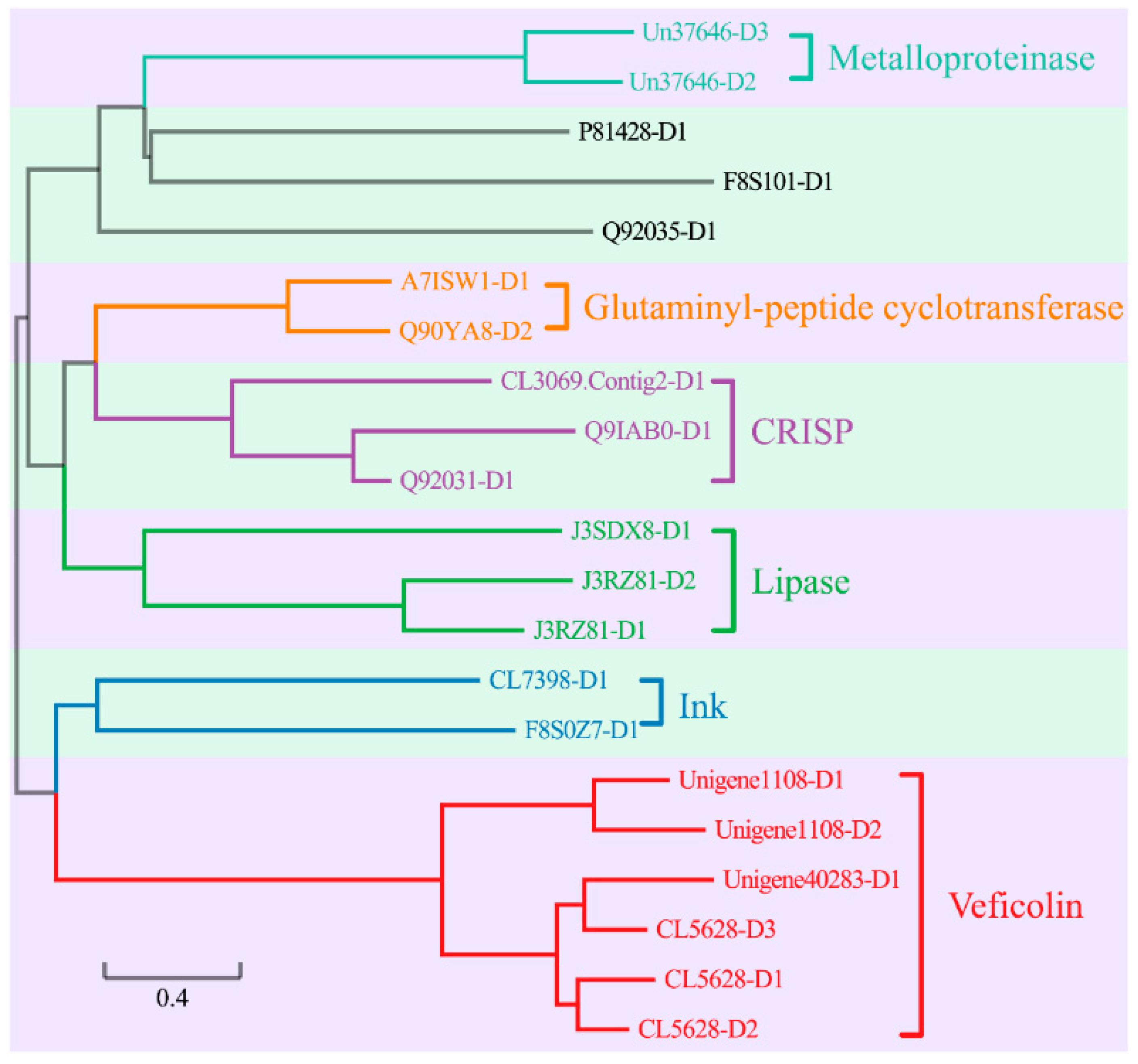
2.5.2. The Medium-Length Toxin Genes
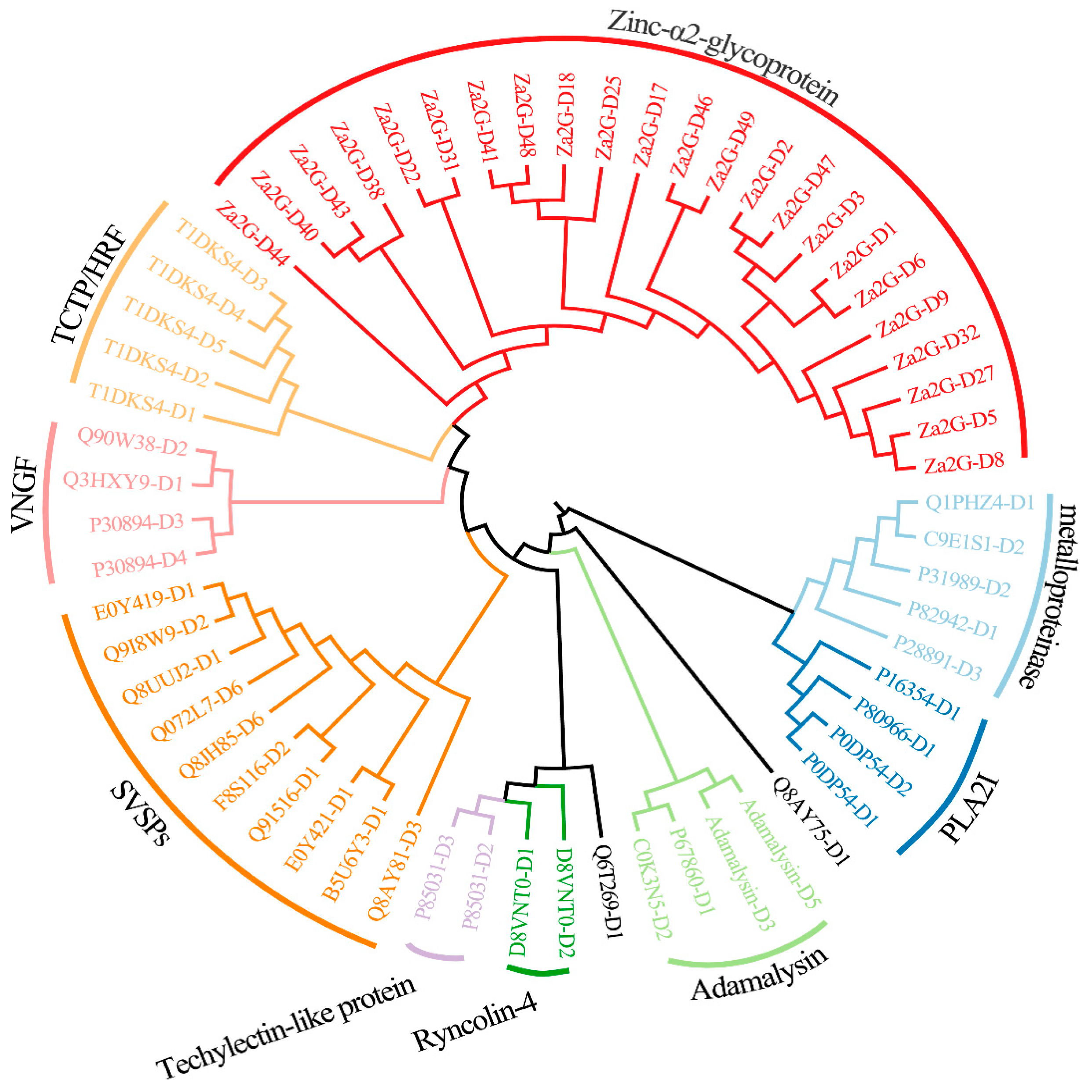
2.5.3. The Long-Length Toxin Genes
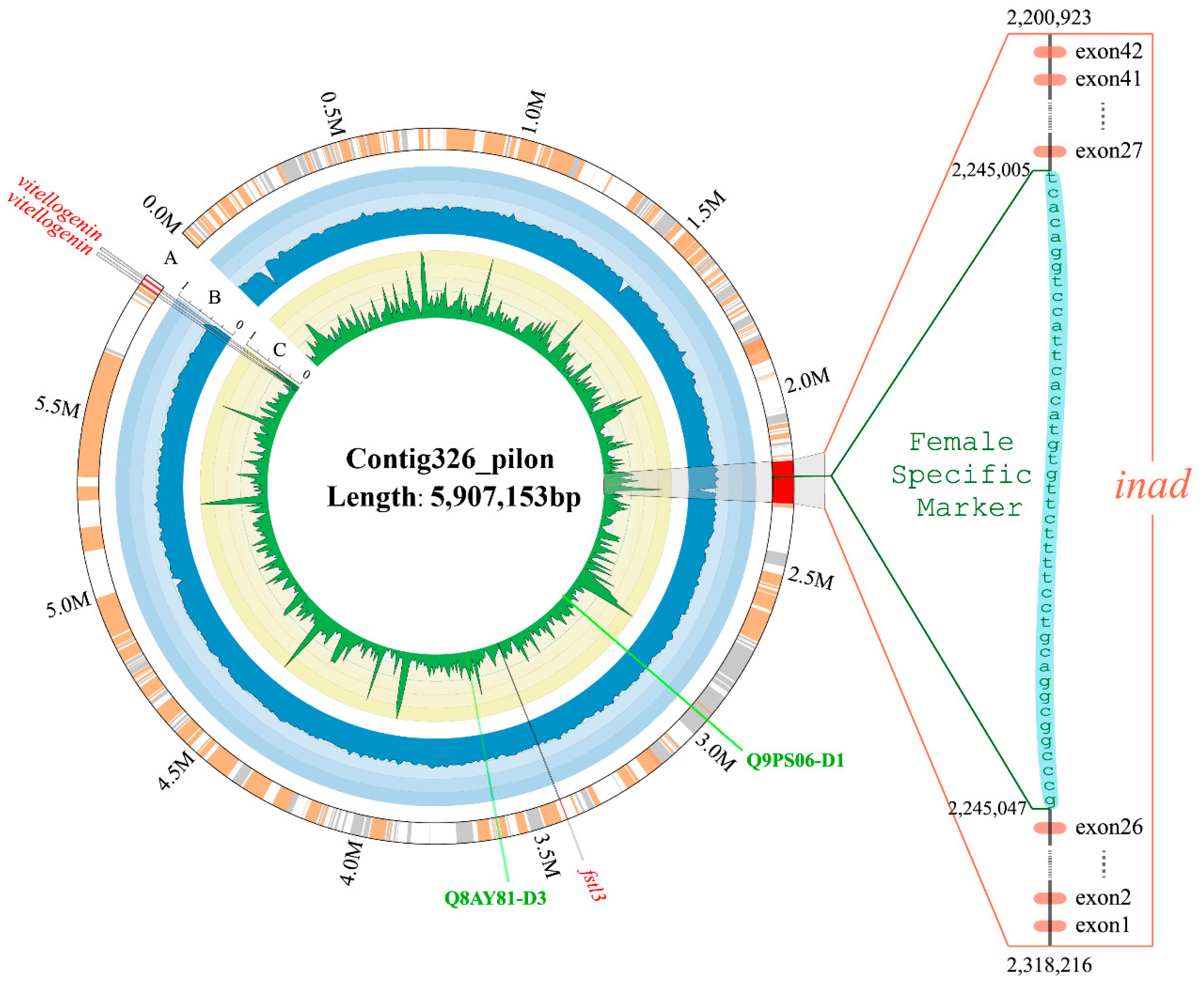
2.6. Identification of Toxin Genes in a Special Sex-Related Region
3. Discussion
3.1. A Good Strategy to Generate the High-Quality Genome Assembly
3.2. High Efficiency to Identify Toxin Sequences
4. Conclusions
5. Materials and Methods
5.1. Sampling and Genome Sequencing
5.2. Estimation of Genome Size
5.3. Genome Annotation
5.3.1. Repeat Annotation
5.3.2. Annotation of Gene Set
5.4. Phylogenetic Analysis
5.5. Prediction of Toxin Genes
5.6. Localization of Potential Toxin Genes in the Sex-Related Region
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, L.O.; Lee, S.-M. Effects of the dietary protein and lipid levels on growth and body composition of bagrid catfish, Pseudobagrus fulvidraco. Aquaculture 2005, 243, 323–329. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, L.; Liu, J.; Li, Z.; Xie, S.; De Silva, S.S. Freshwater aquaculture in PR China: Trends and prospects. Rev. Aquac. 2016, 7, 283–302. [Google Scholar] [CrossRef]
- Xie, B.; Huang, Y.; Baumann, K.; Fry, B.; Shi, Q. From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Mar. Drugs 2017, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Li, X.; Lin, Z.; Ruan, Z.; Wang, M.; Liu, J.; Tong, T.; Li, J.; Huang, Y.; Wen, B.; et al. Prediction of Toxin Genes from Chinese Yellow Catfish Based on Transcriptomic and Proteomic Sequencing. Int. J. Mol. Sci. 2016, 17, 556. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F.; Palumbi, S.R. Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc. Natl. Acad. Sci. USA 1999, 96, 6820–6823. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M. Accelerated evolution of toxin genes: Exonization and intronization in snake venom disintegrin/metalloprotease genes. Toxicon 2018, 148, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Modahl, C.M.; Mrinalini; Frietze, S.; Mackessy, S.P. Adaptive evolution of distinct prey-specific toxin genes in rear-fanged snake venom. Proc. R. Soc. B Biol. Sci. 2018, 285. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Adaptive evolution of animal toxin multigene families. Gene 2000, 261, 43–52. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. SSPACE-LongRead: Scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014, 15, 211. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13 (Suppl. 14), S8. [Google Scholar] [CrossRef] [PubMed]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.F.O.; Waterhouse, M.R.; Ioannidis, P.; Kriventseva, V.E.; Zdobnov, M.E. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef] [PubMed]
- Albertus, D.L.; Seder, C.W.; Chen, G.; Wang, X.; Hartojo, W.; Lin, L.; Silvers, A.; Thomas, D.G.; Giordano, T.J.; Chang, A.C.; et al. AZGP1 autoantibody predicts survival and histone deacetylase inhibitors increase expression in lung adenocarcinoma. J. Thorac. Oncol. 2008, 3, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Zelanis, A.; Huesgen, P.F.; Oliveira, A.K.; Tashima, A.K.; Serrano, S.M.T.; Overall, C.M. Snake venom serine proteinases specificity mapping by proteomic identification of cleavage sites. J. Proteom. 2015, 113, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, M.; Alape-Girón, A.; Barboza-Arguedas, E.; Aguilar-Ulloa, W.; Flores-Díaz, M. Identification of B cell recognized linear epitopes in a snake venom serine proteinase from the central American bushmaster Lachesis stenophrys. Toxicon 2017, 140, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Stocker, W.; Grams, F.; Baumann, U.; Reinemer, P.; Gomis-Ruth, F.X.; McKay, D.B.; Bode, W. The metzincins--topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. 1995, 4, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Moura-da-Silva, A.M.; Butera, D.; Tanjoni, I. Importance of Snake Venom Metalloproteinases in Cell Biology: Effects on Platelets, Inflammatory and Endothelial Cells. Curr. Pharm. Des. 2007, 13, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Rucavado, A.; Escalante, T.; Díaz, C. Hemorrhage induced by snake venom metalloproteinases: Biochemical and biophysical mechanisms involved in microvessel damage. Toxicon 2005, 45, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Senff-Ribeiro, A. Translationally Controlled Tumor Protein (TCTP/HRF) in Animal Venoms. In TCTP/tpt1 Remodeling Signaling from Stem Cell to Disease; Telerman, A., Amson, R., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 193–200. [Google Scholar] [CrossRef]
- Jimenez-Charris, E.; Montealegre-Sanchez, L.; Solano-Redondo, L.; Castro-Herrera, F.; Fierro-Perez, L.; Lomonte, B. Divergent functional profiles of acidic and basic phospholipases A2 in the venom of the snake Porthidium lansbergii lansbergii. Toxicon 2016, 119, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Lei, D.; Jiang, T.; Yang, L.; Zheng, L.; Zhao, J. Nerve growth factor from Chinese cobra venom stimulates chondrogenic differentiation of mesenchymal stem cells. Cell Death Dis. 2017, 8, e2801. [Google Scholar] [CrossRef] [PubMed]
- Liberato, T.; Troncone, L.R.P.; Yamashiro, E.T.; Serrano, S.M.T.; Zelanis, A. High-resolution proteomic profiling of spider venom: Expanding the toxin diversity of Phoneutria nigriventer venom. Amino Acids 2016, 48, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Chung, R.; Dunn, S.R.; Weston, A.J.; Jaimes-Becerra, A.; Marques, A.C.; Morandini, A.C.; Hranueli, D.; Starcevic, A.; Ward, M.; et al. Gene duplications are extensive and contribute significantly to the toxic proteome of nematocysts isolated from Acropora digitifera (Cnidaria: Anthozoa: Scleractinia). BMC Genom. 2015, 16, 774. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Lin, C.-C.; Hsiao, Y.-C.; Wang, P.-J.; Yu, J.-S. Proteomic characterization of six Taiwanese snake venoms: Identification of species-specific proteins and development of a SISCAPA-MRM assay for cobra venom factors. J. Proteom. 2018, 187, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Van Vaerenbergh, M.; Debyser, G.; Smagghe, G.; Devreese, B.; de Graaf, D.C. Unraveling the venom proteome of the bumblebee (Bombus terrestris) by integrating a combinatorial peptide ligand library approach with FT-ICR MS. Toxicon 2015, 102, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Adade, C.M.; Carvalho, A.L.O.; Tomaz, M.A.; Costa, T.F.R.; Godinho, J.L.; Melo, P.A.; Lima, A.P.C.A.; Rodrigues, J.C.F.; Zingali, R.B.; Souto-Padrón, T. Crovirin, a Snake Venom Cysteine-Rich Secretory Protein (CRISP) with Promising Activity against Trypanosomes and Leishmania. PLoS Negl. Trop. Dis. 2014, 8, e3252. [Google Scholar] [CrossRef] [PubMed]
- Koludarov, I.; Jackson, T.N.; Brouw, B.O.D.; Dobson, J.; Dashevsky, D.; Arbuckle, K.; Clemente, C.J.; Stockdale, E.J.; Cochran, C.; Debono, J.; et al. Enter the Dragon: The Dynamic and Multifunctional Evolution of Anguimorpha Lizard Venoms. Toxins 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Butzke, D.; Machuy, N.; Thiede, B.; Hurwitz, R.; Goedert, S.; Rudel, T. Hydrogen peroxide produced by Aplysia ink toxin kills tumor cells independent of apoptosis via peroxiredoxin I sensitive pathways. Cell Death Differ. 2004, 11, 608–617. [Google Scholar] [CrossRef] [PubMed]
- OmPraba, G.; Chapeaurouge, A.; Doley, R.; Devi, K.R.; Padmanaban, P.; Venkatraman, C.; Velmurugan, D.; Lin, Q.; Kini, R.M. Identification of a novel family of snake venom proteins Veficolins from Cerberus rynchops using a venom gland transcriptomics and proteomics approach. J. Proteome Res. 2010, 9, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Flockerzi, V. Molecular characterization of a novel human PDZ domain protein with homology to INAD from Drosophila melanogaster. FEBS Lett. 1997, 413, 243–248. [Google Scholar] [CrossRef]
- Patra, A.; Kalita, B.; Chanda, A.; Mukherjee, A.K. Proteomics and antivenomics of Echis carinatus carinatus venom: Correlation with pharmacological properties and pathophysiology of envenomation. Sci. Rep. 2017, 7, 17119. [Google Scholar] [CrossRef] [PubMed]
- Atoda, H.; Hyuga, M.; Morita, T. The primary structure of coagulation factor IX/factor X-binding protein isolated from the venom of Trimeresurus flavoviridis. Homology with asialoglycoprotein receptors, proteoglycan core protein, tetranectin, and lymphocyte Fc epsilon receptor for immunoglobulin E. J. Biol. Chem. 1991, 266, 14903–14911. [Google Scholar] [PubMed]
- Ullah, A.; Masood, R.; Ali, I.; Ullah, K.; Ali, H.; Akbar, H.; Betzel, C. Thrombin-like enzymes from snake venom: Structural characterization and mechanism of action. Int. J. Biol. Macromol. 2018, 114, 788–811. [Google Scholar] [CrossRef] [PubMed]
- Martinez Barrio, A.; Lamichhaney, S.; Fan, G.; Rafati, N.; Pettersson, M.; Zhang, H.; Dainat, J.; Ekman, D.; Höppner, M.; Jern, P.; et al. The genetic basis for ecological adaptation of the Atlantic herring revealed by genome sequencing. eLife 2016, 5, e12081. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, L.; Bian, C.; Xu, P.; Qiu, Y.; You, X.; Zhang, S.; Huang, Y.; Li, J.; Wang, M.; et al. High-quality genome assembly of channel catfish, Ictalurus punctatus. GigaScience 2016, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Bian, C.; Zan, Q.; Xu, X.; Liu, X.; Chen, J.; Wang, J.; Qiu, Y.; Li, W.; Zhang, X.; et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 2014, 5, 5594. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.-N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, D.; Kan, M.; Lv, Z.; Zhu, A.; Su, Y.; Zhou, D.; Zhang, J.; Zhang, Z.; Xu, M.; et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nat. Commun. 2014, 5, 5227. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef] [PubMed]
- Vij, S.; Kuhl, H.; Kuznetsova, I.S.; Komissarov, A.; Yurchenko, A.A.; Van Heusden, P.; Singh, S.; Thevasagayam, N.M.; Prakki, S.R.S.; Purushothaman, K.; et al. Chromosomal-Level Assembly of the Asian Seabass Genome Using Long Sequence Reads and Multi-layered Scaffolding. PLoS Genet. 2016, 12, e1005954. [Google Scholar] [CrossRef]
- Xu, S.; Xiao, S.; Zhu, S.; Zeng, X.; Luo, J.; Liu, J.; Gao, T.; Chen, N. A draft genome assembly of the Chinese sillago (Sillago sinica), the first reference genome for Sillaginidae fishes. GigaScience 2018, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-Q.; Xing, T.; Rong, T. The muscular nutritional components and flesh quality of farmed Pelteobagrus fulvidraco yearlings. Acta Hydrobiol. Sin. 2015, 39, 193–196. [Google Scholar]
- Tong, J.; Sun, X. Genetic and genomic analyses for economically important traits and their applications in molecular breeding of cultured fish. Sci. China Life Sci. 2015, 58, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Zhu, Z. Molecular basis and genetic improvement of economically important traits in aquaculture animals. Chin. Sci. Bull. 2012, 57, 1751–1760. [Google Scholar] [CrossRef]
- Peng, C.; Yao, G.; Gao, B.-M.; Fan, C.-X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. GigaScience 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Lluisma, A.O.; Milash, B.A.; Moore, B.; Olivera, B.M.; Bandyopadhyay, P.K. Novel venom peptides from the cone snail Conus pulicarius discovered through next-generation sequencing of its venom duct transcriptome. Mar. Genom. 2012, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.S. Fishes of the World, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Boil. 2013, 35, 62–67. [Google Scholar]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2004; pp. 4.10.11–14.10.14. [Google Scholar]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Pevsner, J. Basic Local Alignment Search Tool (BLAST); John Wiley & Sons, Inc.: Hoboken, NY, USA, 2005; pp. 87–125. [Google Scholar]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Mackey, A.J.; Reese, J.T.; Milshina, N.V.; Roos, D.S.; Weinstock, G.M. Creating a honey bee consensus gene set. Genome Biol. 2007, 8, R13. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Li, H.; Coghlan, A.; Ruan, J.; Coin, L.J.; Hériché, J.-K.; Osmotherly, L.; Li, R.; Liu, T.; Zhang, Z.; Bolund, L.; et al. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006, 34, D572–D580. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006, 23, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Wu, J.; Liu, W.; Xiong, S.; Ma, W.; Zhang, J.; Wang, W.; Gui, J.F.; Mei, J. Sex-Biased miRNAs in Gonad and Their Potential Roles for Testis Development in Yellow Catfish. PLoS ONE 2014, 9, e107946. [Google Scholar] [CrossRef] [PubMed]
- Dan, C.; Mei, J.; Wang, D.; Gui, J.F. Genetic differentiation and efficient sex-specific marker development of a pair of Y- and X-linked markers in yellow catfish. Int. J. Biol. Sci. 2013, 9, 1043–1049. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Software | Contig N50 (bp) | Maximum Contig (bp) | Minimum Contig (bp) | Scaffold N50 (bp) | Maximum Scaffold (bp) | Minimum Scaffold (bp) | Total Size (bp) |
|---|---|---|---|---|---|---|---|---|
| Contig assembling | Platanus | 1054 | 49,678 | 109 | - | - | - | 1,010,987,672 |
| DBG2OLC | 707,335 | 6,076,047 | 268 | - | - | - | 706,928,086 | |
| Polishing round 1 | Pilon | 705,180 | 6,050,085 | 270 | - | - | - | 702,622,905 |
| Scaffolding | SSPACELongRead | 982,636 | 6,050,085 | 270 | 1,109,190 | 7,365,535 | 270 | 706,306,982 |
| SSPACE_Standard | 705,180 | 6,050,085 | 270 | 3,655,204 | 19,552,289 | 270 | 712,893,760 | |
| Gap filling | Gapcloser | 813,785 | 11,966,130 | 270 | 3,655,204 | 19,552,617 | 270 | 712,834,712 |
| GapFiller | 859,168 | 11,966,116 | 270 | 3,655,204 | 19,552,752 | 270 | 712,901,309 | |
| PBjelly | 962,661 | 14,953,314 | 270 | 3,655,300 | 19,560,773 | 270 | 714,800,876 | |
| Polishing round 2 | Pilon | 970,098 | 15,455,883 | 277 | 3,653,474 | 19,544,699 | 277 | 713,824,612 |
| Dataset | Number of EST Clusters | Total Length (bp) | Coverage Rate by the Assembly (%) | with >90% Sequence in One Scaffold | with >50% Sequence in One Scaffold | ||
|---|---|---|---|---|---|---|---|
| Number | Percentage (%) | Number | Percentage (%) | ||||
| >0 bp | 78,225 | 57,694,186 | 98.1907917 | 73,167 | 93.53404 | 77,222 | 98.7178 |
| >200 bp | 60,258 | 54,613,314 | 98.2312921 | 56,311 | 93.44983 | 59,575 | 98.86654 |
| >500 bp | 30,229 | 45,487,954 | 98.32383756 | 28,117 | 93.01333 | 29,963 | 99.12005 |
| >1000 bp | 17,675 | 36,547,853 | 98.41627906 | 16,434 | 92.97878 | 17,543 | 99.25318 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Li, J.; Qin, Q.; Liu, W.; Bian, C.; Yi, Y.; Wang, M.; Zhong, L.; You, X.; Tang, S.; et al. Whole-Genome Sequencing of Chinese Yellow Catfish Provides a Valuable Genetic Resource for High-Throughput Identification of Toxin Genes. Toxins 2018, 10, 488. https://doi.org/10.3390/toxins10120488
Zhang S, Li J, Qin Q, Liu W, Bian C, Yi Y, Wang M, Zhong L, You X, Tang S, et al. Whole-Genome Sequencing of Chinese Yellow Catfish Provides a Valuable Genetic Resource for High-Throughput Identification of Toxin Genes. Toxins. 2018; 10(12):488. https://doi.org/10.3390/toxins10120488
Chicago/Turabian StyleZhang, Shiyong, Jia Li, Qin Qin, Wei Liu, Chao Bian, Yunhai Yi, Minghua Wang, Liqiang Zhong, Xinxin You, Shengkai Tang, and et al. 2018. "Whole-Genome Sequencing of Chinese Yellow Catfish Provides a Valuable Genetic Resource for High-Throughput Identification of Toxin Genes" Toxins 10, no. 12: 488. https://doi.org/10.3390/toxins10120488
APA StyleZhang, S., Li, J., Qin, Q., Liu, W., Bian, C., Yi, Y., Wang, M., Zhong, L., You, X., Tang, S., Liu, Y., Huang, Y., Gu, R., Xu, J., Bian, W., Shi, Q., & Chen, X. (2018). Whole-Genome Sequencing of Chinese Yellow Catfish Provides a Valuable Genetic Resource for High-Throughput Identification of Toxin Genes. Toxins, 10(12), 488. https://doi.org/10.3390/toxins10120488