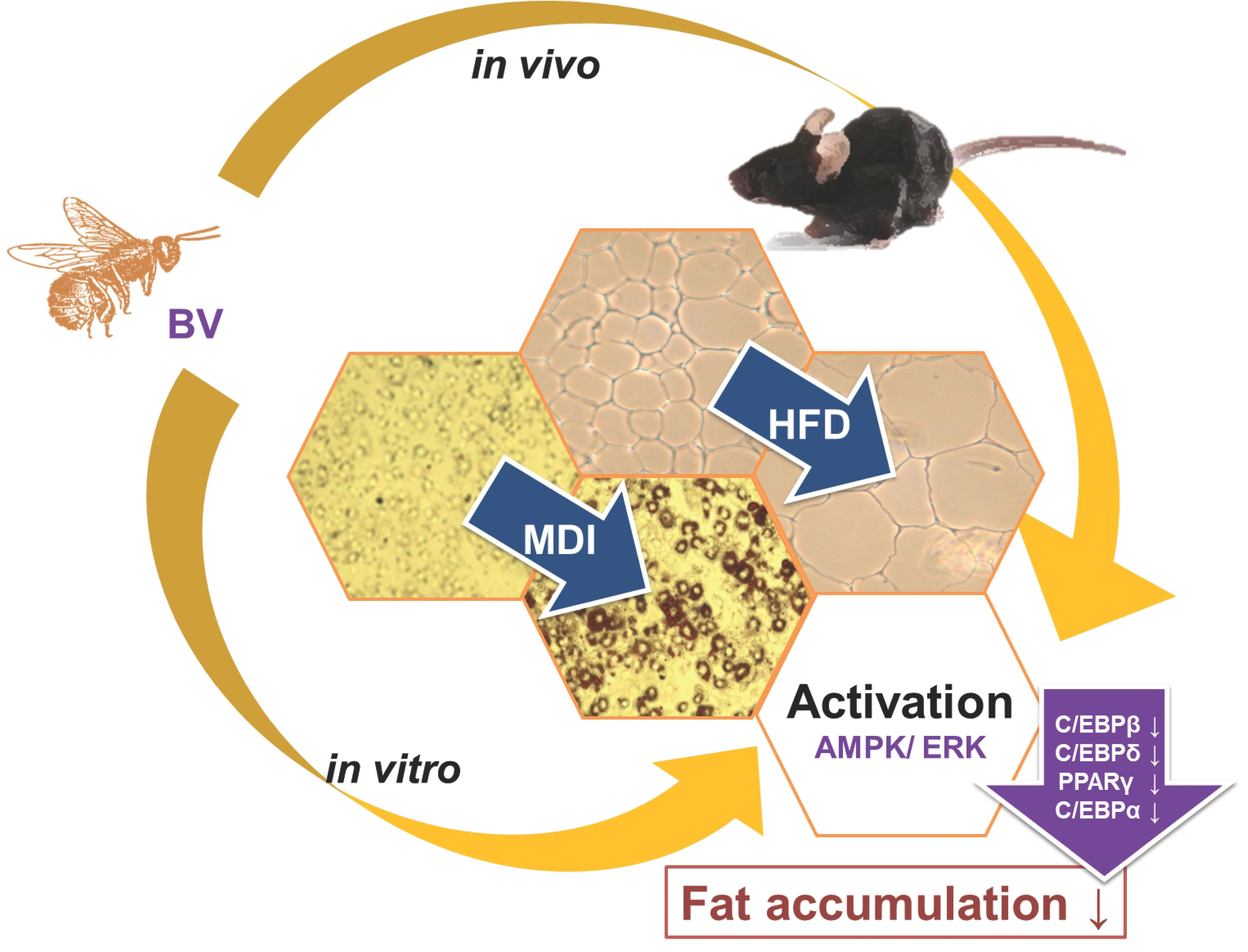
Bee Venom Suppresses the Differentiation of Preadipocytes and High Fat Diet-Induced Obesity by Inhibiting Adipogenesis




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
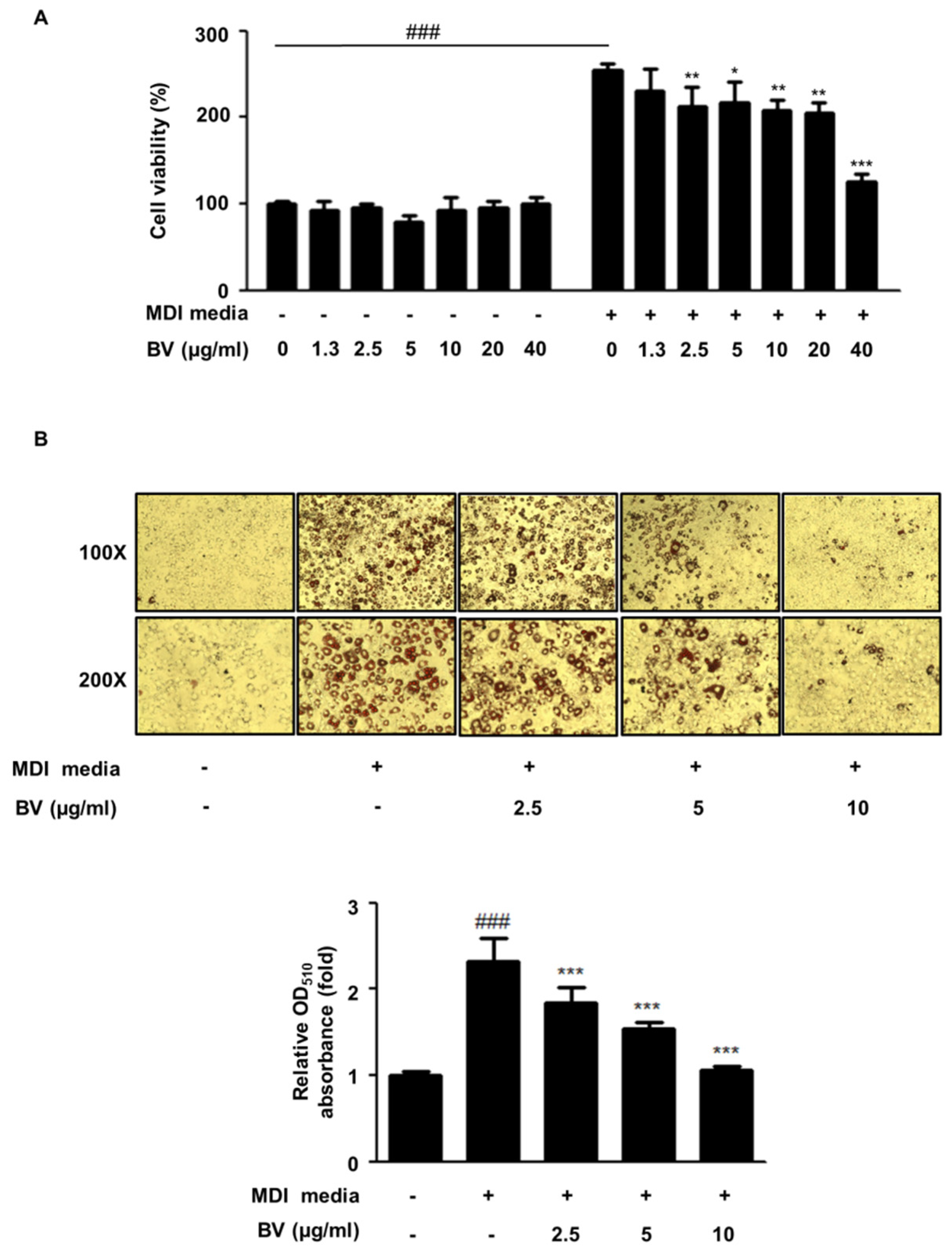
2.1. BV Suppressed Cell Hyperplasia and Lipid Accumulation during Differentiation of 3T3-L1 Adipocytes
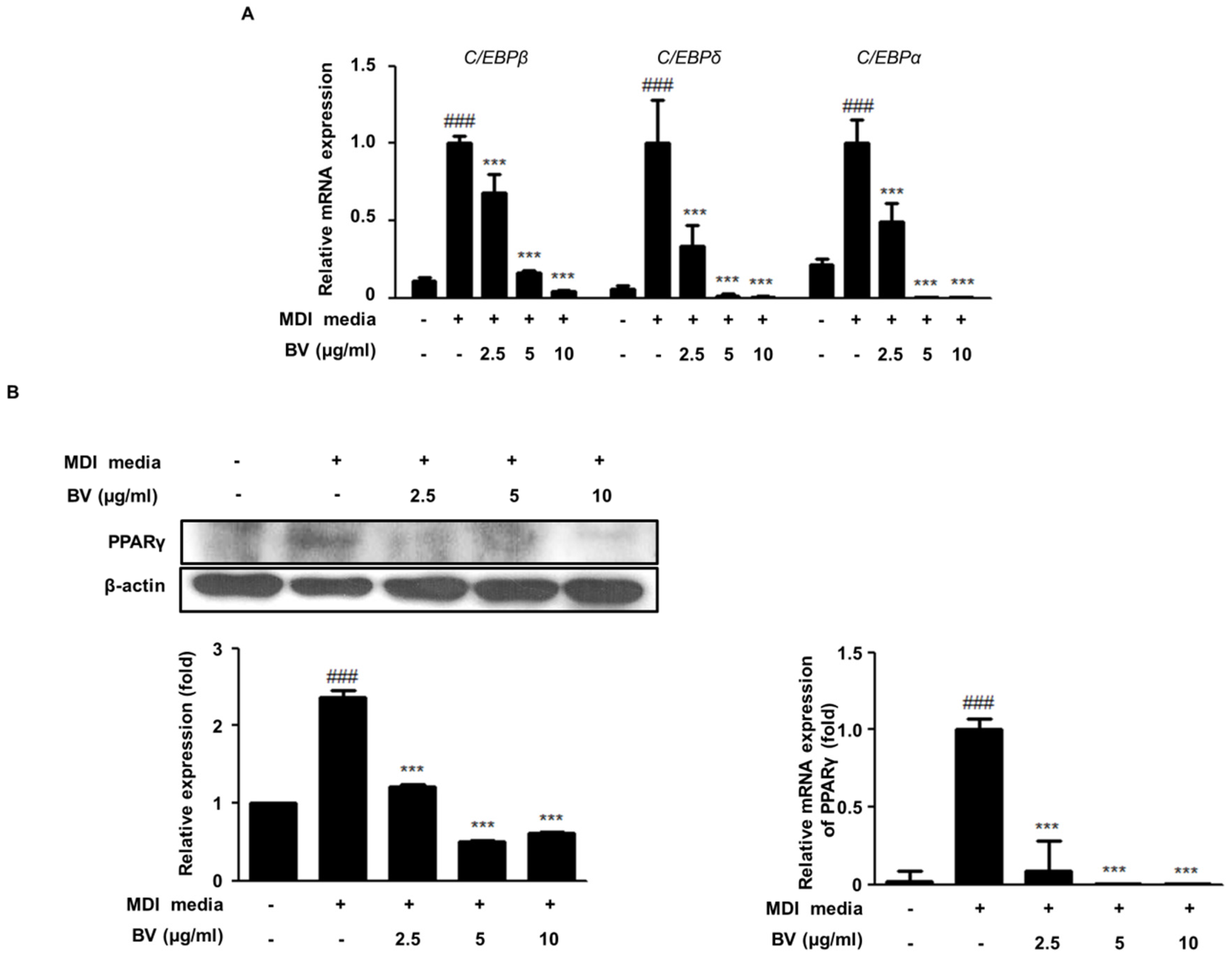
2.2. BV Suppressed the Expression of Adipogenic Markers during Differentiation of 3T3-L1 Adipocytes
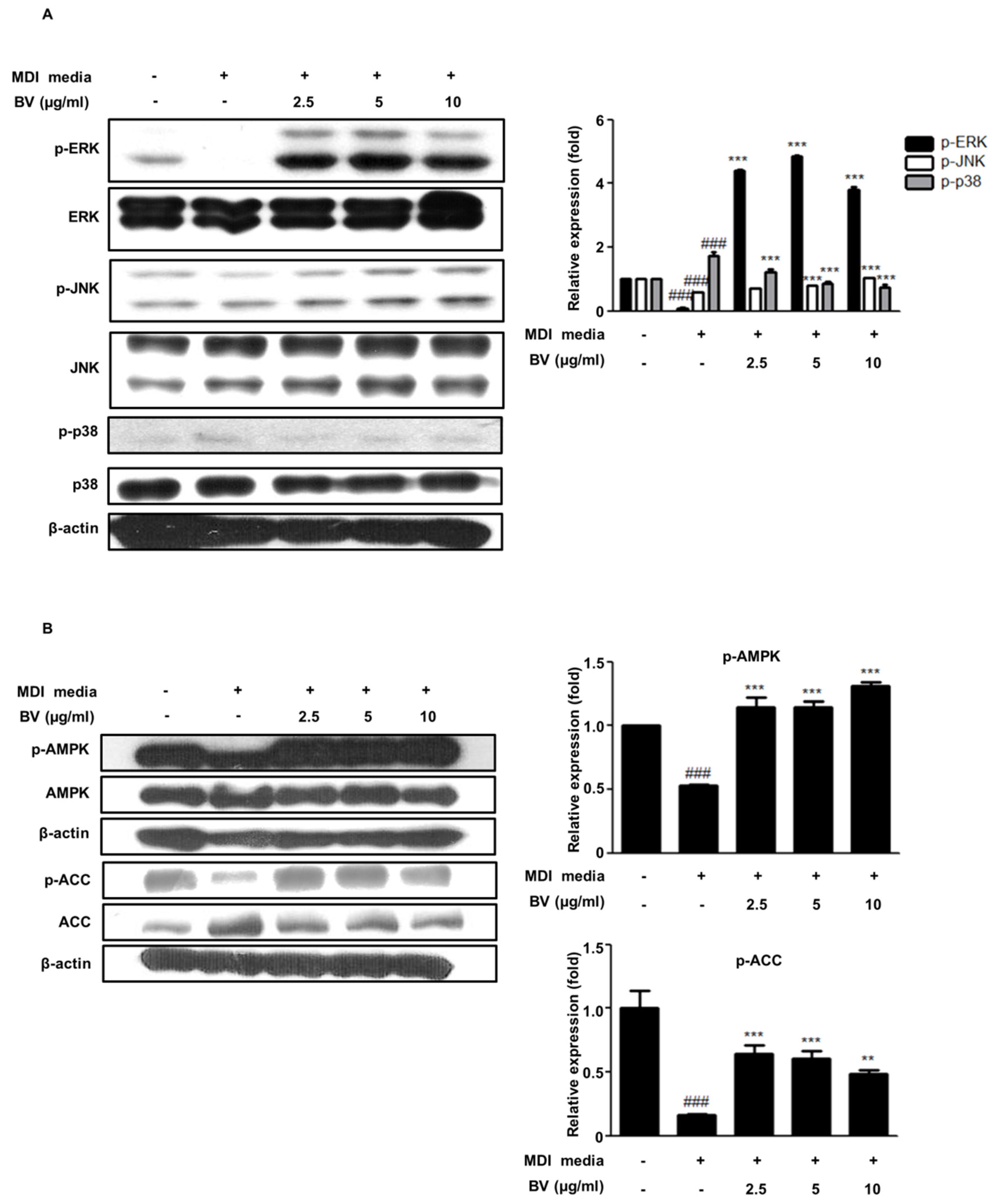
2.3. BV Regulated the MAPK Pathway during Differentiation of 3T3-L1 Preadipocytes
2.4. BV Activated the AMPK Pathway during the Differentiation of 3T3-L1 Preadipocytes
2.5. BV Suppressed Body Weight, Fat, and Lipid Accumulation in HFD-Induced Obese Mice
2.6. BV Suppressed Adipogenic Markers and Activated the AMPK Pathway in HFD-Induced Obese Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Treatment
4.3. MTT Assay
4.4. Oil Red O Staining
4.5. Western Blot Analysis
4.6. Isolation of Total RNA and Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
4.7. Animals
4.8. Histological Analysis
4.9. Analysis of Serum Lipid Profiles
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marti, A.; Martinez-Gonzalez, M.A.; Martinez, J.A. Interaction between genes and lifestyle factors on obesity. Proc. Nutr. Soc. 2008, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Kehinde, O. An established preadipose cell line and its differentiation in culture II. Factors affecting the adipose conversion. Cell 1975, 5, 19–27. [Google Scholar] [CrossRef]
- Fajas, L.; Fruchart, J.C.; Auwerx, J. Transcriptional control of adipogenesis. Curr. Opin. Cell Biol. 1998, 10, 165–173. [Google Scholar] [CrossRef]
- Otto, T.C.; Lane, M.D. Adipose development: From stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. Mitotic clonal expansion: A synchronous process required for adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional regulation of adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Darlington, G.J.; Ross, S.E.; MacDougald, O.A. The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem. 1998, 273, 30057–30060. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Nishizuka, M.; Osada, S.; Imagawa, M. The role of C/EBPdelta in the early stages of adipogenesis. Biochimie 2009, 91, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.C.; Cao, Z.; Classon, M.; McKnight, S.L. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995, 9, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Erbayraktar, Z.; Yilmaz, O.; Artmann, A.T.; Cehreli, R.; Coker, C. Effects of selenium supplementation on antioxidant defense and glucose homeostasis in experimental diabetes mellitus. Biol. Trace Elem. Res. 2007, 118, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sert, A.; Pirgon, O.; Aypar, E.; Yilmaz, H.; Odabas, D. Subclinical hypothyroidism as a risk factor for the development of cardiovascular disease in obese adolescents with nonalcoholic fatty liver disease. Pediatr. Cardiol. 2013, 34, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef]
- Muoio, D.M.; Seefeld, K.; Witters, L.A.; Coleman, R.A. Amp-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: Evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 1999, 338, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Xu, A.; Wu, D. Effects of an amp-activated protein kinase inhibitor, compound C, on adipogenic differentiation of 3T3-L1 cells. Biol. Pharm. Bull. 2008, 31, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.J. “It stings a bit but it cleans well”: Venoms of hymenoptera and their antimicrobial potential. J. Insect Physiol. 2013, 59, 186–204. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C. Honeybee venom: A rich source of pharmacologically active peptides. Endeavour 1988, 12, 60–65. [Google Scholar] [CrossRef]
- Lee, W.R.; Kim, S.J.; Park, J.H.; Kim, K.H.; Chang, Y.C.; Park, Y.Y.; Lee, K.G.; Han, S.M.; Yeo, J.H.; Pak, S.C.; et al. Bee venom reduces atherosclerotic lesion formation via anti-inflammatory mechanism. Am. J. Chin. Med. 2010, 38, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.S.; An, H.J.; Cheon, S.Y.; Kwon, K.R.; Lee, K.H. Bee venom suppresses testosterone-induced benign prostatic hyperplasia by regulating the inflammatory response and apoptosis. Exp. Biol. Med. 2015, 240, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, W.R.; Melzack, R. The bee venom test: A new tonic-pain test. Pain 1996, 66, 271–277. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Kim, K.S.; Park, K.K. Melittin inhibits atherosclerosis in LPS/high-fat treated mice through atheroprotective actions. J. Atheroscler. Thromb. 2011, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Pak, S.C.; Han, S.M.; Park, K.K. The protective effect of apamin on LPS/fat-induced atherosclerotic mice. Evid.-Based Complement. Altern. Med. 2012, 2012, 305454. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Lee, S.H.; Shin, J.Y.; Kim, K.S.; Cho, N.G.; Kwon, K.R.; Rhim, T.J. The effects of bee venom and sweet bee venom to the preadipocyte proliferation and lipolysis of adipocyte, localized fat accumulation. J. Pharmacopunct. 2007, 10, 15. [Google Scholar] [CrossRef]
- Fruhbeck, G.; Gomez-Ambrosi, J.; Muruzabal, F.J.; Burrell, M.A. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E827–E847. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, H.; Ogawa, W.; Nakamura, T.; Mori, T.; Nakamura, K.; Kasuga, M. Role of MAPK phosphatase-1 (mkp-1) in adipocyte differentiation. J. Biol. Chem. 2004, 279, 39951–39957. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. Amp-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Kim, T.S.; Leahy, P.; Freake, H.C. Promoter usage determines tissue specific responsiveness of the rat acetyl-CoA carboxylase gene. Biochem. Biophys. Res. Commun. 1996, 225, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Rayalam, S.; Della-Fera, M.A.; Baile, C.A. Phytochemicals and regulation of the adipocyte life cycle. J. Nutr. Biochem. 2008, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, H.S.; Lee, M.S.; Cho, Y.J.; Kim, Y.S.; Hwang, J.T.; Sung, M.J.; Kim, M.S.; Kwon, D.Y. Vitisin A inhibits adipocyte differentiation through cell cycle arrest in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2008, 372, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Wei, Y.; Chen, N.; Jiang, M.; Wu, J.; Liao, K. DNA synthesis and mitotic clonal expansion is not a required step for 3T3-L1 preadipocyte differentiation into adipocytes. J. Biol. Chem. 2001, 276, 11988–11995. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.Y.; Seo, S.G.; Heo, Y.S.; Yue, S.; Cheng, J.X.; Lee, K.W.; Kim, K.H. Piceatannol, natural polyphenolic stilbene, inhibits adipogenesis via modulation of mitotic clonal expansion and insulin receptor-dependent insulin signaling in early phase of differentiation. J. Biol. Chem. 2012, 287, 11566–11578. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Ann. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.D.; Tang, Q.Q.; Jiang, M.S. Role of the CCAAT enhancer binding proteins (C/EBPS) in adipocyte differentiation. Biochem. Biophys. Res. Commun. 1999, 266, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.F.; Schatzkin, A.; Harris, T.B.; Kipnis, V.; Mouw, T.; Ballard-Barbash, R.; Hollenbeck, A.; Leitzmann, M.F. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N. Engl. J. Med. 2006, 355, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P.; Moorjani, S.; Lupien, P.J.; Tremblay, A.; Nadeau, A.; Bouchard, C. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990, 10, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. Srebp-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Sozio, M.S.; Lu, C.; Zeng, Y.; Liangpunsakul, S.; Crabb, D.W. Activated AMPK inhibits PPAR-α and PPAR-γ transcriptional activity in hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G739–G747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of amp-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.J.; Keyse, S.M. Diverse physiological functions for dual-specificity map kinase phosphatases. J. Cell Sci. 2006, 119, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gibson, T.B.; Robinson, F.; Silvestro, L.; Pearson, G.; Xu, B.; Wright, A.; Vanderbilt, C.; Cobb, M.H. Map kinases. Chem. Rev. 2001, 101, 2449–2476. [Google Scholar] [CrossRef] [PubMed]
- Sale, E.M.; Atkinson, P.G.; Sale, G.J. Requirement of map kinase for differentiation of fibroblasts to adipocytes, for insulin activation of p90 s6 kinase and for insulin or serum stimulation of DNA synthesis. EMBO J. 1995, 14, 674–684. [Google Scholar] [PubMed]
- Kimura, Y.; Taniguchi, M.; Baba, K. Antitumor and antimetastatic activities of 4-hydroxyderricin isolated from Angelica keiskei roots. Planta Med. 2004, 70, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Motoi, T.; Seki, A.; Kikuchi, T.; Fukatsu, M.; Tokuda, H.; Suzuki, N.; Kimura, Y. Cytotoxic activities and anti-tumor-promoting effects of microbial transformation products of prenylated chalcones from Angelica keiskei. Chem. Biodivers. 2012, 9, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through map kinase-mediated phosphorylation of ppargamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sawada, K.; Yamamoto, N.; Ashida, H. 4-hydroxyderricin and xanthoangelol from ashitaba (Angelica keiskei) suppress differentiation of preadiopocytes to adipocytes via AMPK and MAPK pathways. Mol. Nutr. Food Res. 2013, 57, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Lisanti, M.P.; Scherer, P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998, 273, 32111–32120. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheon, S.-Y.; Chung, K.-S.; Roh, S.-S.; Cha, Y.-Y.; An, H.-J. Bee Venom Suppresses the Differentiation of Preadipocytes and High Fat Diet-Induced Obesity by Inhibiting Adipogenesis. Toxins 2018, 10, 9. https://doi.org/10.3390/toxins10010009
Cheon S-Y, Chung K-S, Roh S-S, Cha Y-Y, An H-J. Bee Venom Suppresses the Differentiation of Preadipocytes and High Fat Diet-Induced Obesity by Inhibiting Adipogenesis. Toxins. 2018; 10(1):9. https://doi.org/10.3390/toxins10010009
Chicago/Turabian StyleCheon, Se-Yun, Kyung-Sook Chung, Seong-Soo Roh, Yun-Yeop Cha, and Hyo-Jin An. 2018. "Bee Venom Suppresses the Differentiation of Preadipocytes and High Fat Diet-Induced Obesity by Inhibiting Adipogenesis" Toxins 10, no. 1: 9. https://doi.org/10.3390/toxins10010009
APA StyleCheon, S.-Y., Chung, K.-S., Roh, S.-S., Cha, Y.-Y., & An, H.-J. (2018). Bee Venom Suppresses the Differentiation of Preadipocytes and High Fat Diet-Induced Obesity by Inhibiting Adipogenesis. Toxins, 10(1), 9. https://doi.org/10.3390/toxins10010009