Short-Term Intake of a Fructose-, Fat- and Cholesterol-Rich Diet Causes Hepatic Steatosis in Mice: Effect of Antibiotic Treatment


Abstract
:1. Introduction
2. Materials and Methods
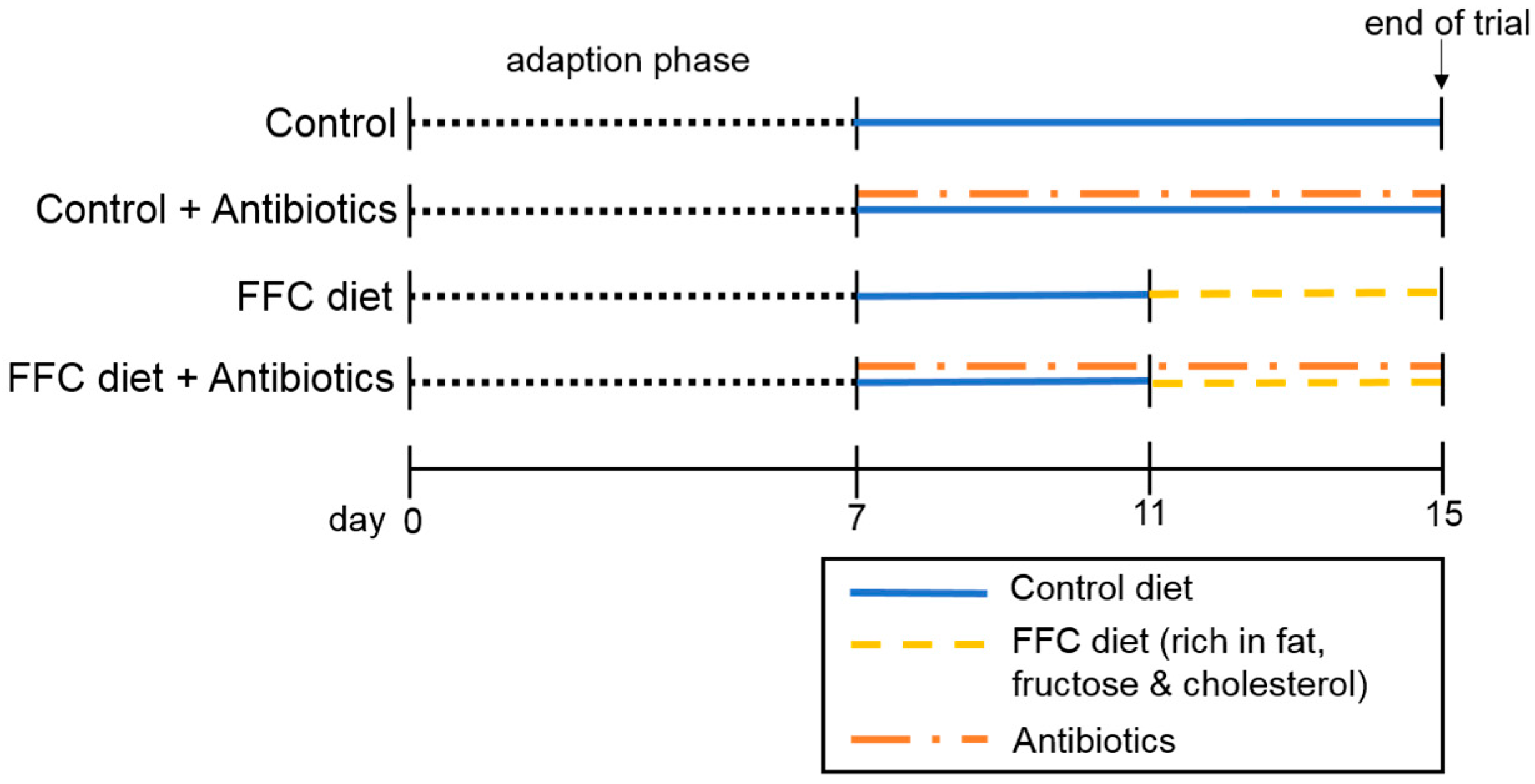
2.1. Animals and Treatment
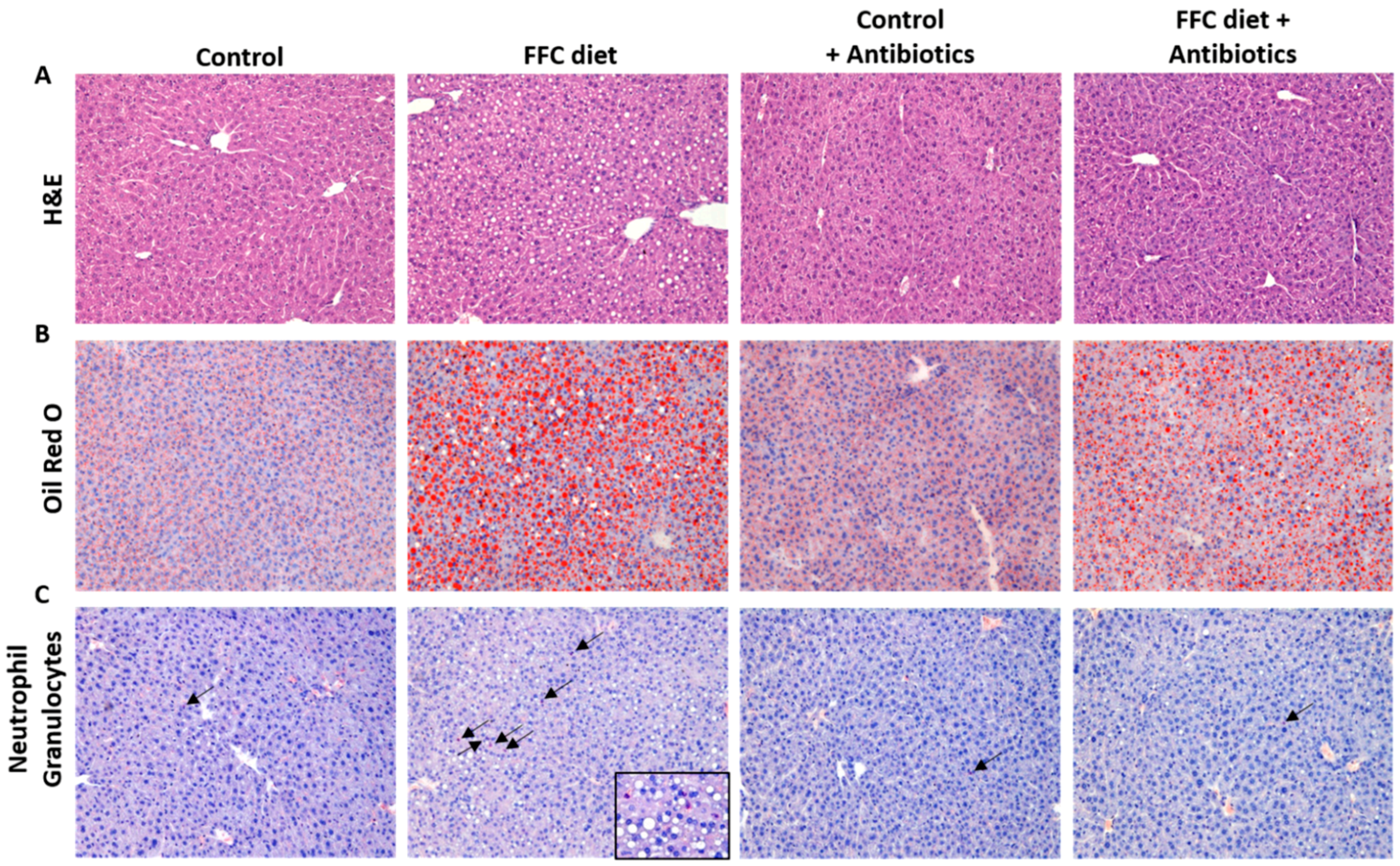
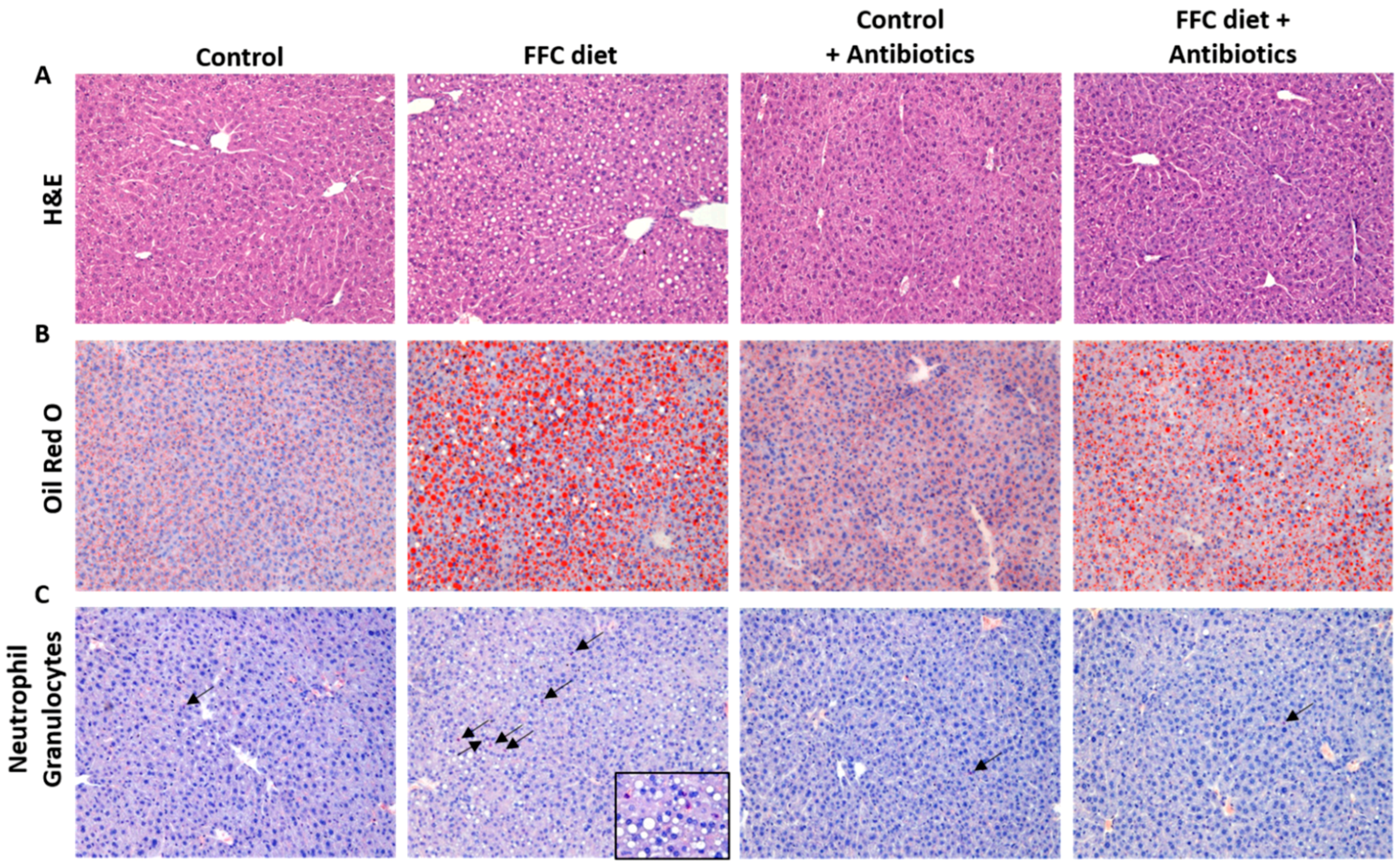
2.2. Histological Evaluation of Liver Sections and Hepatic Lipid Accumulation
2.3. Blood Parameters of Liver Damage, ELISA and Endotoxin Measurement
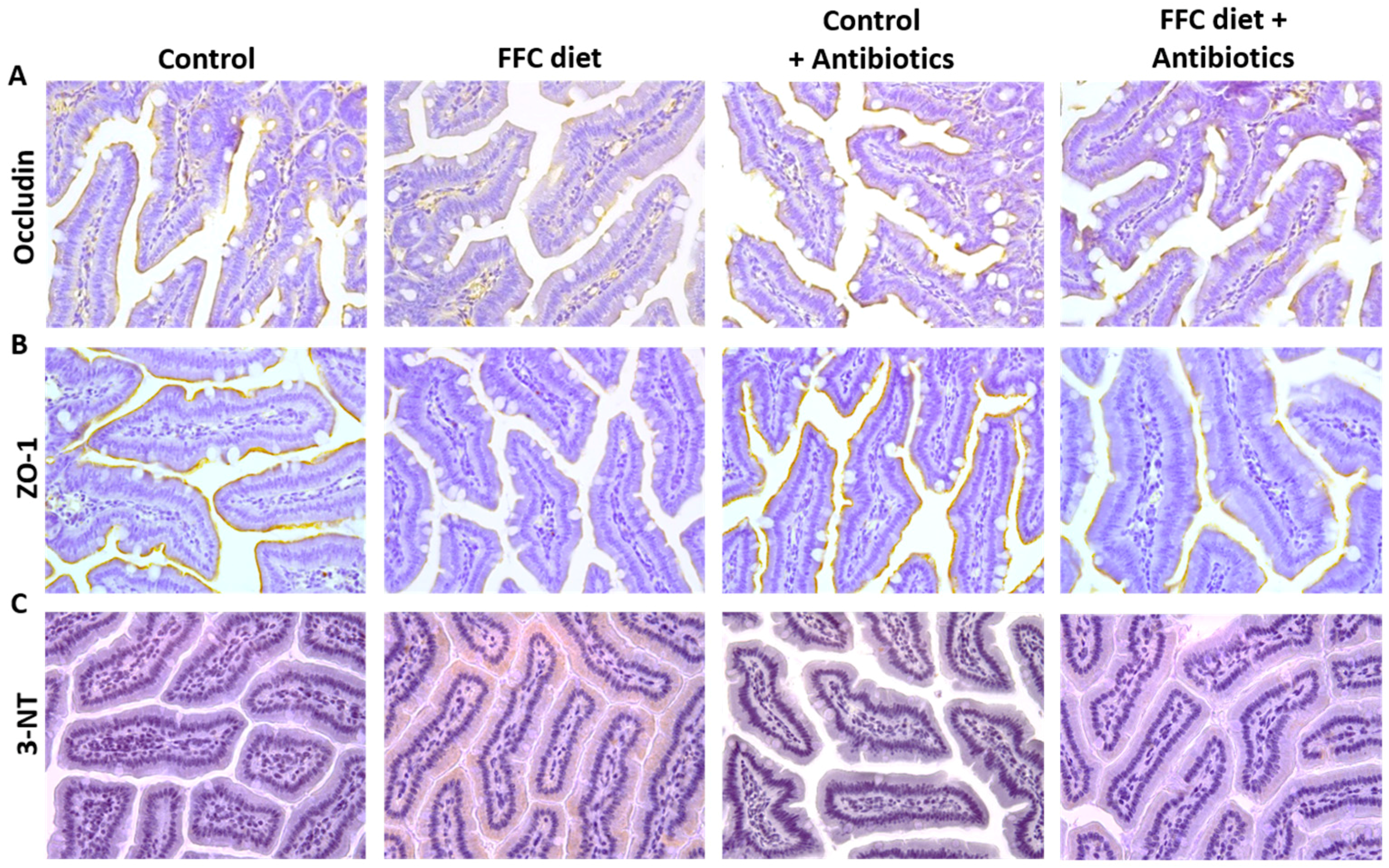
2.4. Immunohistochemical Staining for 4-HNE Protein Adducts and iNOS Protein in the Liver As Well As 3-Nitrotyrosine Protein Adducts, MMP-13, Occludin and ZO-1 Protein in the Small Intestine
2.5. RNA Isolation and Real-Time RT-PCR
2.6. Western Blot
2.7. Statistical Analysis
3. Results
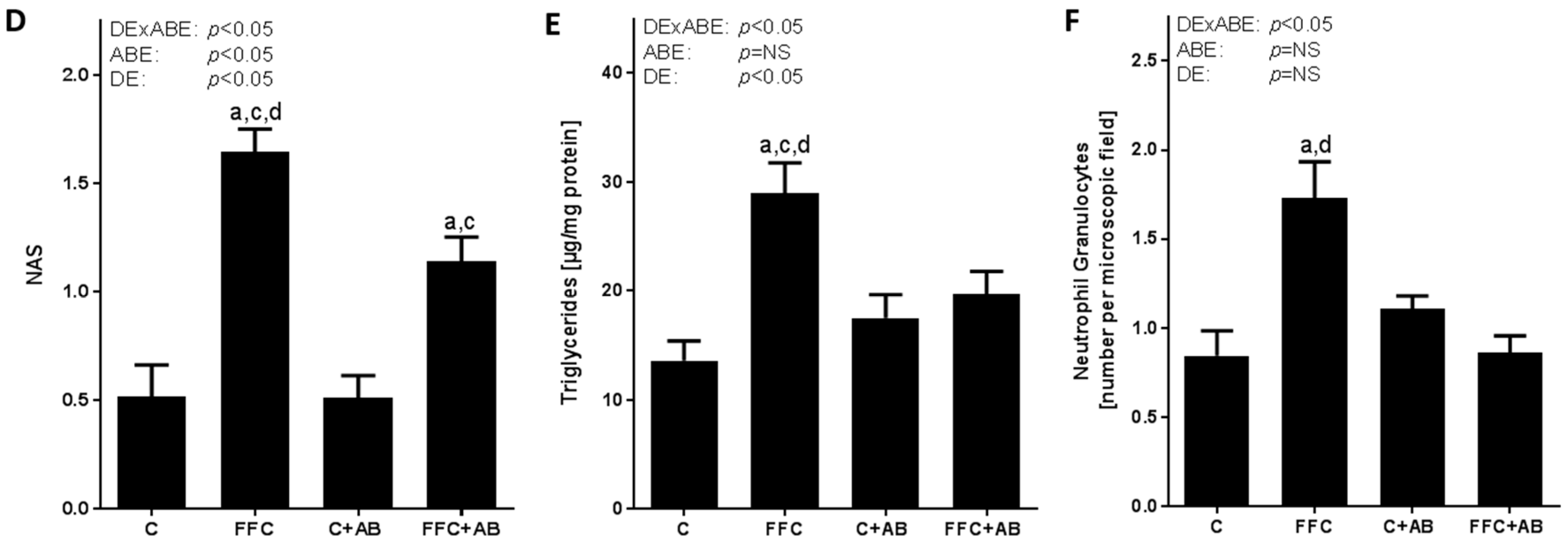
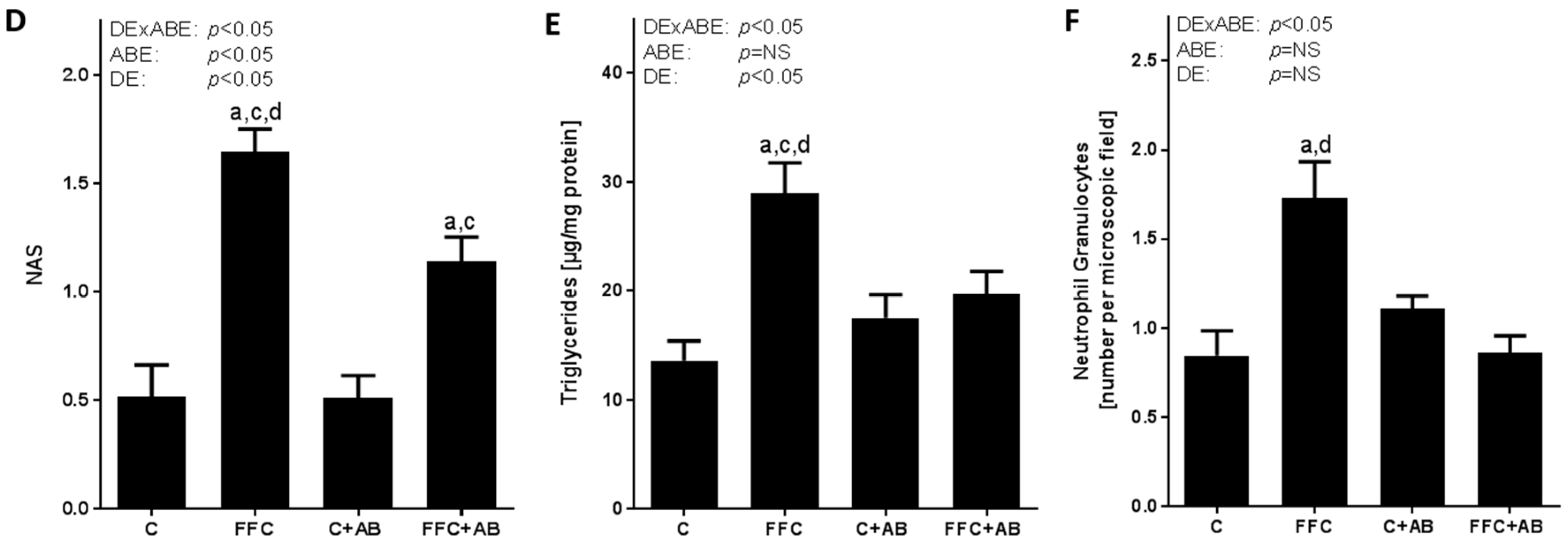
3.1. Body Weight, Liver Steatosis and Inflammatory Markers in Liver
3.2. Markers of Lipogenesis in Liver
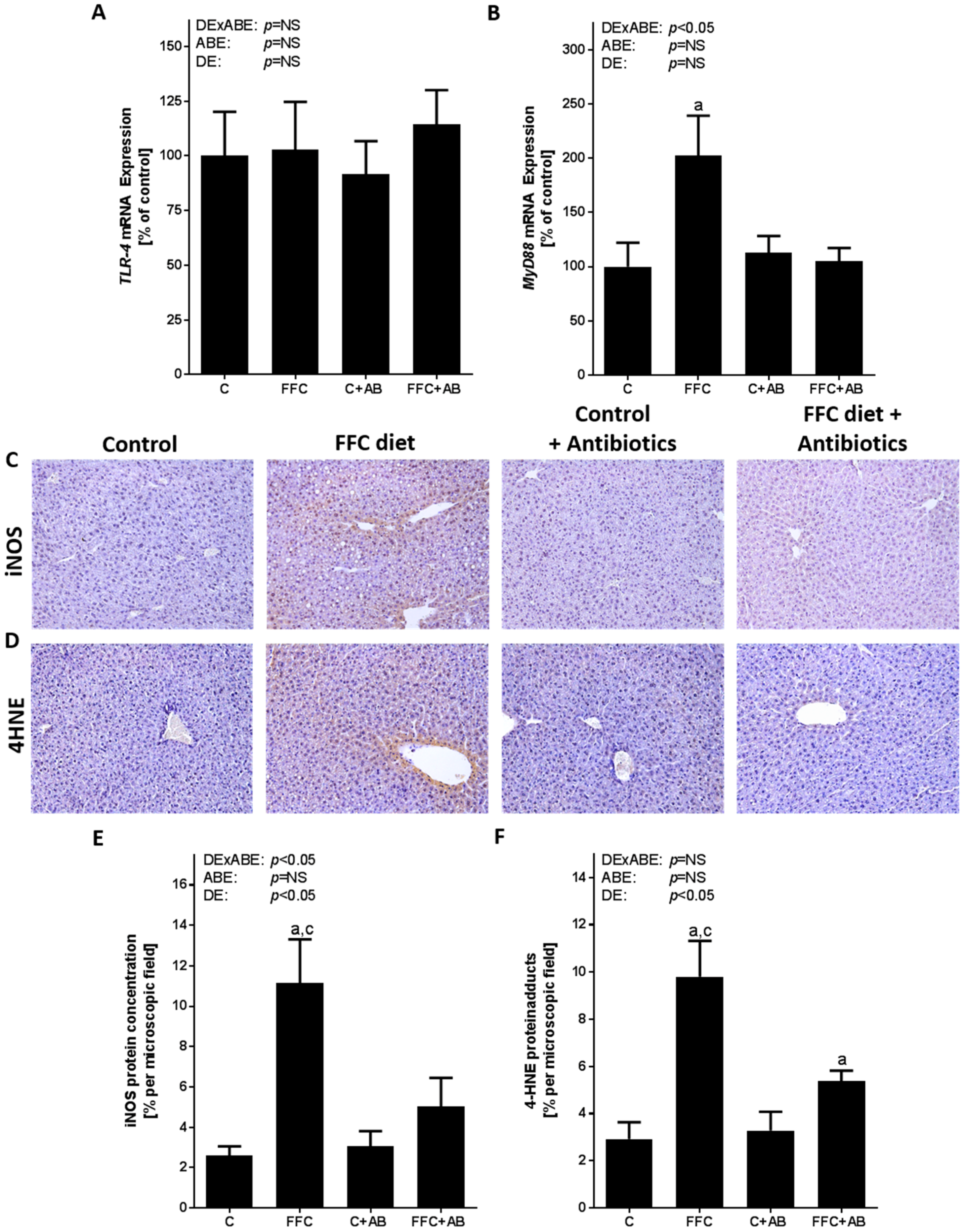
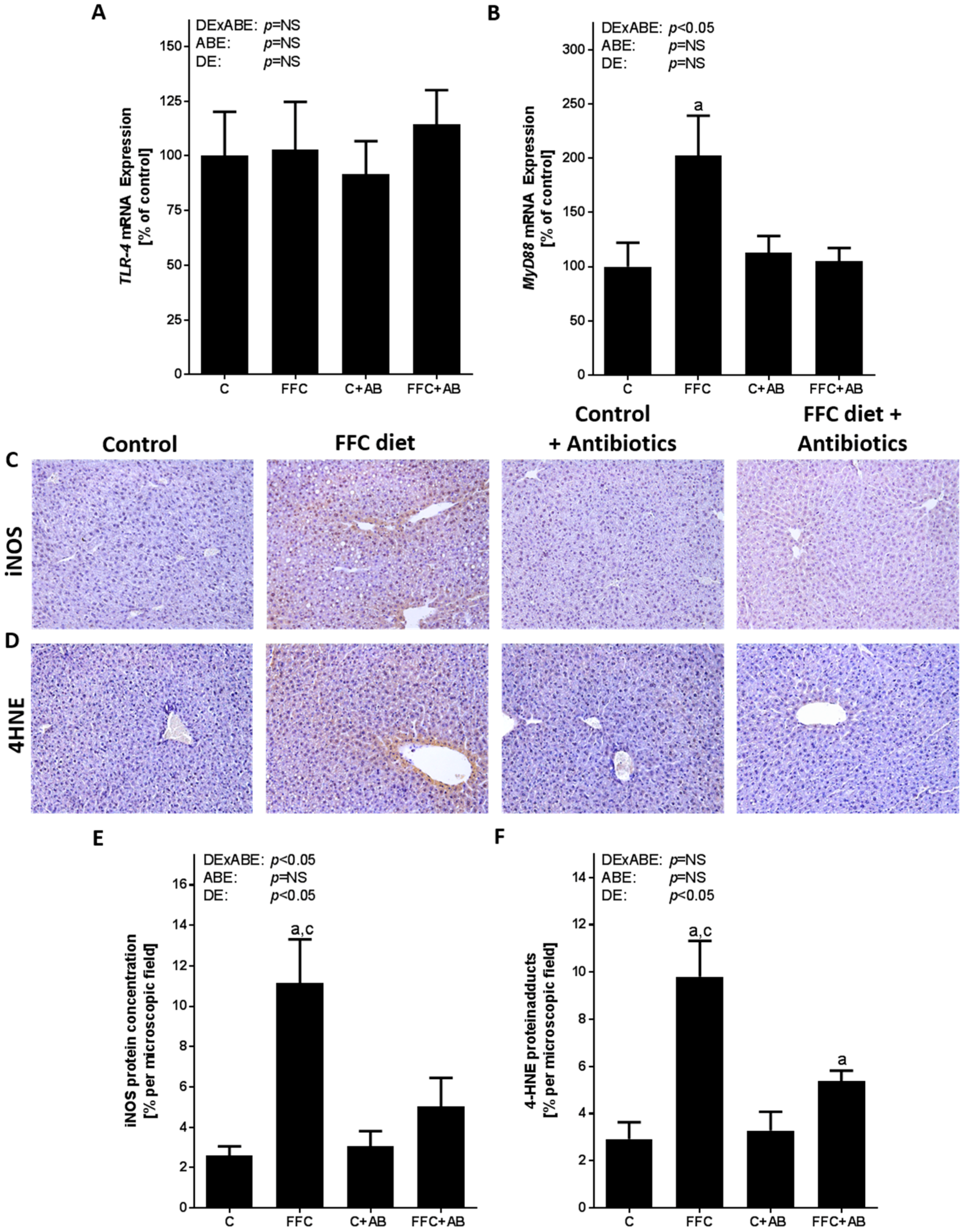
3.3. Markers of TLR-4 Signaling As Well As Lipidperoxidation in Liver Tissue
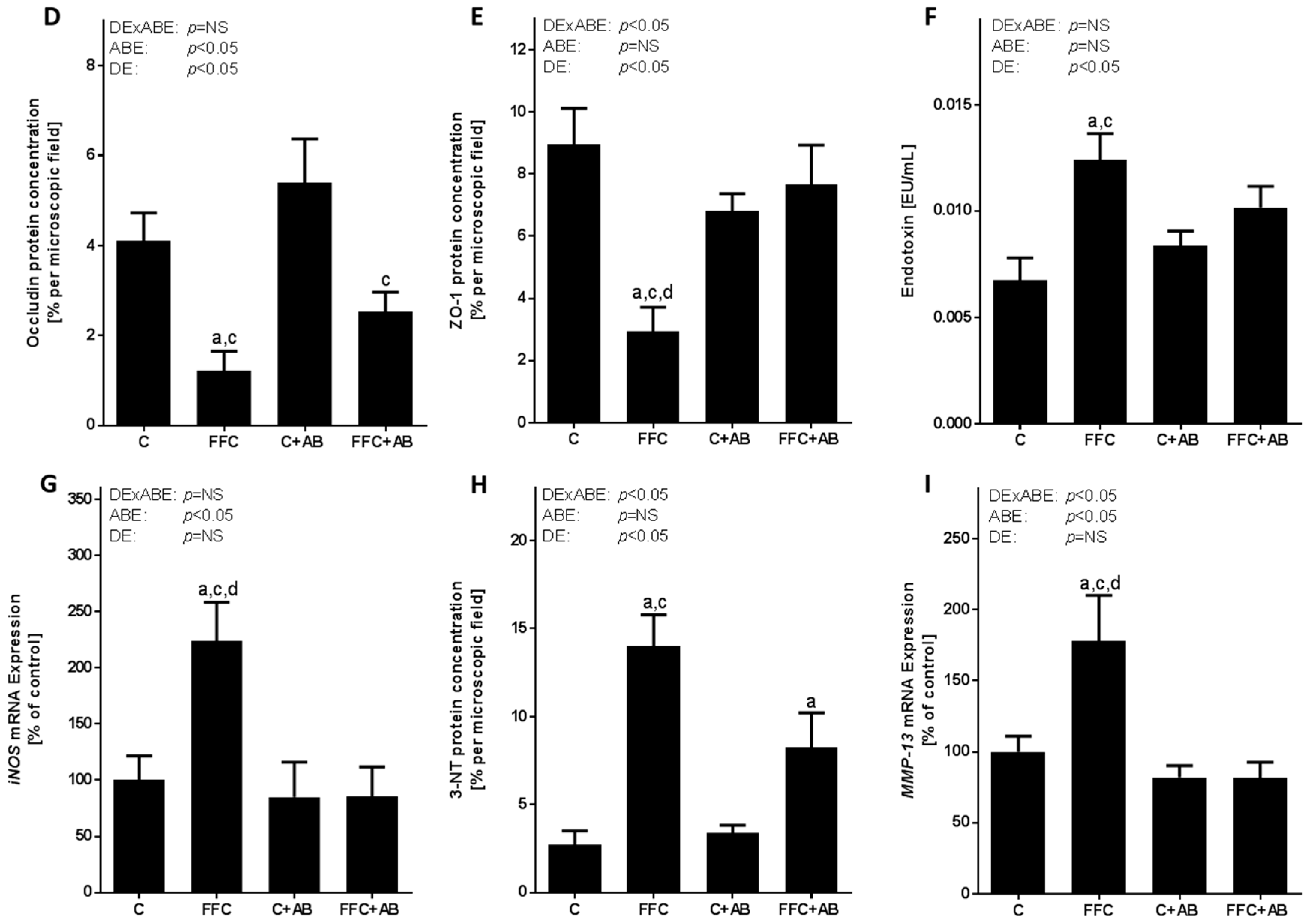
3.4. Markers of Intestinal Barrier Function and Inflammation As Well As Lipidperoxidation in the Small Intestine and Portal Vein
4. Discussion
4.1. Short-Term Isocalcoric Intake of a Fat-, Fructose- and Cholesterol-Rich Diet Leads to Alterations of the Markers of Intestinal Permeability and the Onset of NAFLD
4.2. Antibiotic Treatment Not Only Diminishes the Loss of Tight Junction Proteins and the Induction of iNOS in the Small Intestine and the Increase of Portal Endotoxin Levels, But Also the Accumulation of Triglycerides in the Liver
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.C.; Roudot-Thoraval, F. The burden of liver disease in Europe: A review of available epidemiological data. J. Hepatol. 2013, 58, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD single topic conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Z.; Hollis-Hansen, K.; Wan, X.Y.; Fei, S.J.; Pang, X.L.; Meng, F.D.; Yu, C.H.; Li, Y.M. Clinical guidelines of non-alcoholic fatty liver disease: A systematic review. World J. Gastroenterol. 2016, 22, 8226–8233. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Troeger, J.S.; Dapito, D.H.; Mencin, A.A.; Schwabe, R.F. Toll-like receptor 4 and hepatic fibrogenesis. Semin. Liver Dis. 2010, 30, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Mahana, D.; Trent, C.M.; Kurtz, Z.D.; Bokulich, N.A.; Battaglia, T.; Chung, J.; Muller, C.L.; Li, H.; Bonneau, R.A.; Blaser, M.J. Antibiotic perturbation of the murine gut microbiome enhances the adiposity, insulin resistance, and liver disease associated with high-fat diet. Genome Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: Role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1–9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Garttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Weber, S.; Volynets, V.; Spruss, A.; Bischoff, S.C.; Bergheim, I. Cinnamon extract protects against acute alcohol-induced liver steatosis in mice. J. Nutr. 2009, 139, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Uebel, K.; Bischoff, S.C.; Bergheim, I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid. Redox Signal. 2011, 14, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Gao, J.; Rao, Z.; Shen, Q. Clinicopathological significance and prognostic value of MMP-13 expression in colorectal cancer. Scand. J. Clin. Lab. Investig. 2012, 72, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Spruss, A.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. Fructose-induced steatosis in mice: Role of plasminogen activator inhibitor-1, microsomal triglyceride transfer protein and NKT cells. Lab. Investig. 2011, 91, 885–895. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, A.; Caletti, M.T.; Sasdelli, A.S.; Brodosi, L.; Marchesini, G. Pathophysiology of nonalcoholic fatty liver disease: Lifestyle-gut-gene interaction. Dig. Dis. 2016, 34, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Jin, C.J.; Engstler, A.J.; De Bandt, J.P.; Bergheim, I. Oral citrulline supplementation protects female mice from the development of non-alcoholic fatty liver disease (NAFLD). Eur. J. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Li, S.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS ONE 2014, 9, e115148. [Google Scholar] [CrossRef] [PubMed]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ohashi, N.; Li, W.; Eckman, C.; Nguyen, J.H. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 2009, 50, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Lischper, M.; Beuck, S.; Thanabalasundaram, G.; Pieper, C.; Galla, H.J. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010, 1326, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Hauwermeiren, F.; Lodens, S.; De Rycke, R.; Van Wonterghem, E.; Staes, A.; Gevaert, K.; Lopez-Otin, C.; Libert, C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 2013, 5, 1000–1016. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Forsyth, C.B.; Farhadi, A.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Nitric oxide-mediated intestinal injury is required for alcohol-induced gut leakiness and liver damage. Alcohol Clin. Exp. Res. 2009, 33, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, M.; Fromm, A.; Krug, S.M.; Amasheh, S.; Andres, S.; Zeitz, M.; Fromm, M.; Schulzke, J.D. TNFalpha-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, Akt and NFkappaB signaling. J. Cell Sci. 2010, 123, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Chen, D.; Yu, C.; Lv, B.; Peng, J.; Wang, J.; Lin, Y. Citrus nobiletin ameliorates experimental colitis by reducing inflammation and restoring impaired intestinal barrier function. Mol. Nutr. Food Res. 2015, 59, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Kapus, A.; Marsden, P.A.; Li, Y.H.; Oreopoulos, G.; Marshall, J.C.; Frantz, S.; Kelly, R.A.; Medzhitov, R.; Rotstein, O.D. Regulation of toll-like receptor 4 expression in the lung following hemorrhagic shock and lipopolysaccharide. J. Immunol. 2002, 168, 5252–5259. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Spruss, A.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. Role of tumor necrosis factor alpha (TNFalpha) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem. 2011, 22, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, N.; Molendi-Coste, O.; Horsmans, Y.; van Rooijen, N.; Cani, P.D.; Leclercq, I.A. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G107–G116. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, R.W.; Hammer, S.; Lamb, H.J.; Frolich, M.; Diamant, M.; Rijzewijk, L.J.; de Roos, A.; Romijn, J.A.; Smit, J.W. Effects of short-term high-fat, high-energy diet on hepatic and myocardial triglyceride content in healthy men. J. Clin. Endocrinol. Metab. 2008, 93, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, M.S.; Wueest, S.; Item, F.; Schoenle, E.J.; Konrad, D. Adipose tissue inflammation contributes to short-term high-fat diet-induced hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E388–E395. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016, 151, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Kilic, U.; Gok, O.; Uysal, O.; Senturk, H. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.M.; Guadagnini, D.; Tsukumo, D.M.; Schenka, A.A.; Latuf-Filho, P.; Vassallo, J.; Dias, J.C.; Kubota, L.T.; Carvalheira, J.B.; Saad, M.J. Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia 2012, 55, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K.; et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fink, M.P.; Yang, R.; Delude, R.L. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock 2004, 21, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.; Bowling, J.; Bell, B.; Wang, J.; Ford, H.R. Roles of nitric oxide and intestinal microbiota in the pathogenesis of necrotizing enterocolitis. J. Pediatr. Surg. 2016, 51, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.W.; Hsu, C.M.; Wang, J.S.; Chen, J.S.; Chen, S.C. Specific inhibition of iNOS decreases the intestinal mucosal peroxynitrite level and improves the barrier function after thermal injury. Burns 1998, 24, 699–705. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, L.; Forsyth, C.B.; Shaikh, M.; Song, S.; Keshavarzian, A. The role of miR-212 and iNOS in alcohol-induced intestinal barrier dysfunction and steatohepatitis. Alcohol Clin. Exp. Res. 2015, 39, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Ann, J.Y.; Eo, H.; Lim, Y. Mulberry leaves (Morus alba L.) ameliorate obesity-induced hepatic lipogenesis, fibrosis, and oxidative stress in high-fat diet-fed mice. Genes Nutr. 2015, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Chassaing, B.; Zhang, L.; San Yeoh, B.; Xiao, X.; Kumar, M.; Baker, M.T.; Cai, J.; Walker, R.; Borkowski, K.; et al. Microbiota-dependent hepatic lipogenesis mediated by stearoyl CoA desaturase 1 (SCD1) promotes metabolic syndrome in TLR5-deficient mice. Cell Metab. 2015, 22, 983–996. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) | Accession Number |
|---|---|---|---|
| 18S | GTA ACC CGT TGA ACC CCA TT | CCA TCC AAT CGG TAG TAG CG | NR_003278 |
| ACC | CTT CCT CCT GAT CAG CAA CTC T | CGT GAG TTT TCC CAA AAT AAG C | NM_133904 |
| FASN | TCT GGG CCA ACC TCA TTG GT | GAA GCT GGG GGT CCA TTG TG | NM_007988 |
| Il-1β | TGG CTG TGG AGA AGC TGT GG | GTC CGA CAG CAC GAG GCT TT | NM_008361 |
| Il-6 | CCA CGG CCT TCC CTA CTT CA | TGC AAG TGC ATC ATC GTT GTT C | NM_001314054 |
| iNOS | CCC CTG GAA GTT TCT CTT CAA AGT C | GAT TCT GGA ACA TTC TGT GCT GTC C | NM_010927 |
| MMP-13 | AGA AGT GTG ACC CAG CCC TA | GCG CAA GAA GAA TCT GTC TTT | NM_008607 |
| MMP-9 | TGG TCT TCC CCA AAG ACC TG | GCG GTA CAA GTA TGC CTC TG | NM_013599 |
| MyD88 | CAA AAG TGG GGT GCC TTT GC | AAA TCC ACA GTG CCC CCA GA | NM_010851 |
| SCD1 | CCG ATA AAA GGG GGC TGA GG | TGC TGA GAT CGA GCG TGG AC | NM_009127 |
| SREBP-1c | ACC GGC TAC TGC TGG ACT GC | AGA GCA AGA GGG TGC CAT CG | NM_001313979 |
| TLR-4 | AGC CAT TGC TGC CAA CAT CA | GCT GCC TCA GCA GGG ACT TC | NM_021297 |
| Parameter | Groups | p-Value | |||||
|---|---|---|---|---|---|---|---|
| C | FFC | C + AB | FFC + AB | DE × ABE | ABE | DE | |
| Caloric intake [kcal/mouse/day] | 9.9 ± 0.0 | 10.3 ± 0.2 | 9.7 ± 0.0 | 9.9 ± 0.2 | NS | NS | NS |
| Body weight [g] | 19.1 ± 0.3 | 19.9 ± 0.4 | 19.5 ± 0.3 | 19.6 ± 0.3 | NS | NS | NS |
| Weight gain [g] | 0.8 ± 0.2 | 0.8 ± 0.1 | 0.9 ± 0.2 | 1.3 ± 0.2 | NS | NS | NS |
| Liver weight [g] | 0.9 ± 0.02 | 1.0 ± 0.02 a,c | 0.9 ± 0.01 | 1.0 ± 0.02 a,c | NS | NS | <0.05 |
| Liver:body weight ratio [%] | 4.6 ± 0.07 | 4.9 ± 0.08 a,c | 4.5 ± 0.05 | 5.0 ± 0.08 a,c | NS | NS | <0.05 |
| Plasma ALT [U/I] | 19.6 ± 2.8 | 14.0 ± 0.7 | 14.9 ± 1.0 | 15.5 ± 1.4 | NS | NS | NS |
| Parameter | Groups | p-Value | |||||
|---|---|---|---|---|---|---|---|
| C | FFC | C + AB | FFC + AB | DE × ABE | ABE | DE | |
| Il-6 mRNA | 100 ± 14 | 198 ± 45 | 133 ± 28 | 109 ± 14 | NS | NS | NS |
| Il-1β mRNA | 100 ± 12 | 127 ± 19 c | 61.3 ± 12 | 96.4 ± 16 | NS | NS | <0.05 |
| PAI-1 protein | 100 ± 6 | 123 ± 10 c,d | 75.1 ± 4 | 65.1 ± 13 a | NS | <0.05 | NS |
| SREBP-1c mRNA | 100 ± 20 | 182 ± 10 c | 88.0 ± 15 | 193 ± 25 c | NS | NS | <0.05 |
| ACC mRNA | 100 ± 26 | 298 ± 59 a | 270 ± 50 a | 243 ± 40 | <0.05 | NS | NS |
| FASN mRNA | 100 ± 24 | 146 ± 12 | 228 ± 46 | 258 ± 95 | NS | NS | NS |
| SCD1 mRNA | 100 ± 21 | 429 ± 111 a,c | 109 ± 23 | 286 ± 65 a | NS | NS | <0.05 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, A.; Jin, C.J.; Nolte, K.; Sellmann, C.; Engstler, A.J.; Bergheim, I. Short-Term Intake of a Fructose-, Fat- and Cholesterol-Rich Diet Causes Hepatic Steatosis in Mice: Effect of Antibiotic Treatment. Nutrients 2017, 9, 1013. https://doi.org/10.3390/nu9091013
Brandt A, Jin CJ, Nolte K, Sellmann C, Engstler AJ, Bergheim I. Short-Term Intake of a Fructose-, Fat- and Cholesterol-Rich Diet Causes Hepatic Steatosis in Mice: Effect of Antibiotic Treatment. Nutrients. 2017; 9(9):1013. https://doi.org/10.3390/nu9091013
Chicago/Turabian StyleBrandt, Annette, Cheng Jun Jin, Katja Nolte, Cathrin Sellmann, Anna Janina Engstler, and Ina Bergheim. 2017. "Short-Term Intake of a Fructose-, Fat- and Cholesterol-Rich Diet Causes Hepatic Steatosis in Mice: Effect of Antibiotic Treatment" Nutrients 9, no. 9: 1013. https://doi.org/10.3390/nu9091013
APA StyleBrandt, A., Jin, C. J., Nolte, K., Sellmann, C., Engstler, A. J., & Bergheim, I. (2017). Short-Term Intake of a Fructose-, Fat- and Cholesterol-Rich Diet Causes Hepatic Steatosis in Mice: Effect of Antibiotic Treatment. Nutrients, 9(9), 1013. https://doi.org/10.3390/nu9091013