Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and ERS-Mimicking Condition (EMM)
2.2. Animal Study
2.3. Immunoblotting
2.4. Proliferation Assay
2.5. Immunohistochemistry
2.6. Quantitative Real-Time Polymerase Chain Reaction
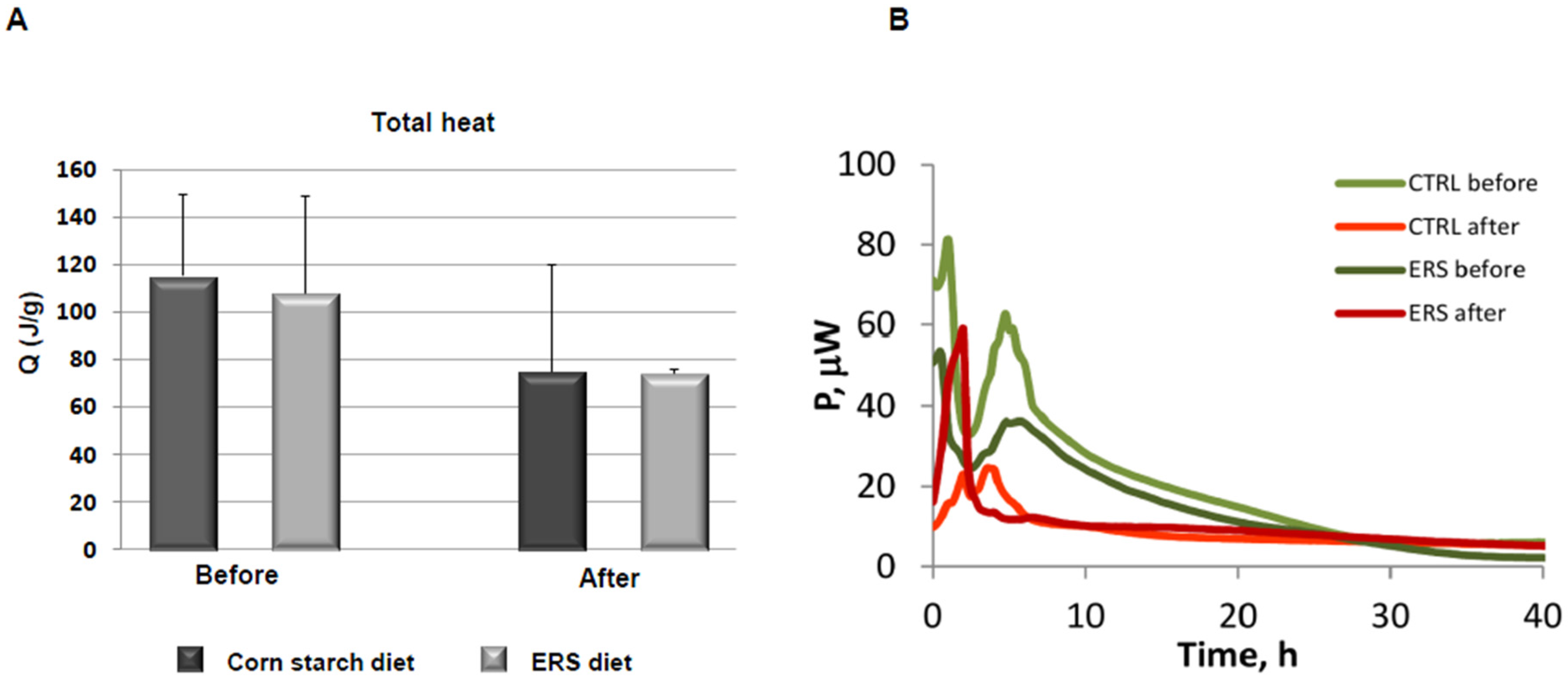
2.7. Isothermal Microcalorimetry
2.8. Determination of Metabolites
2.9. Microbiome Analyses
2.10. Statistical Analysis
3. Results
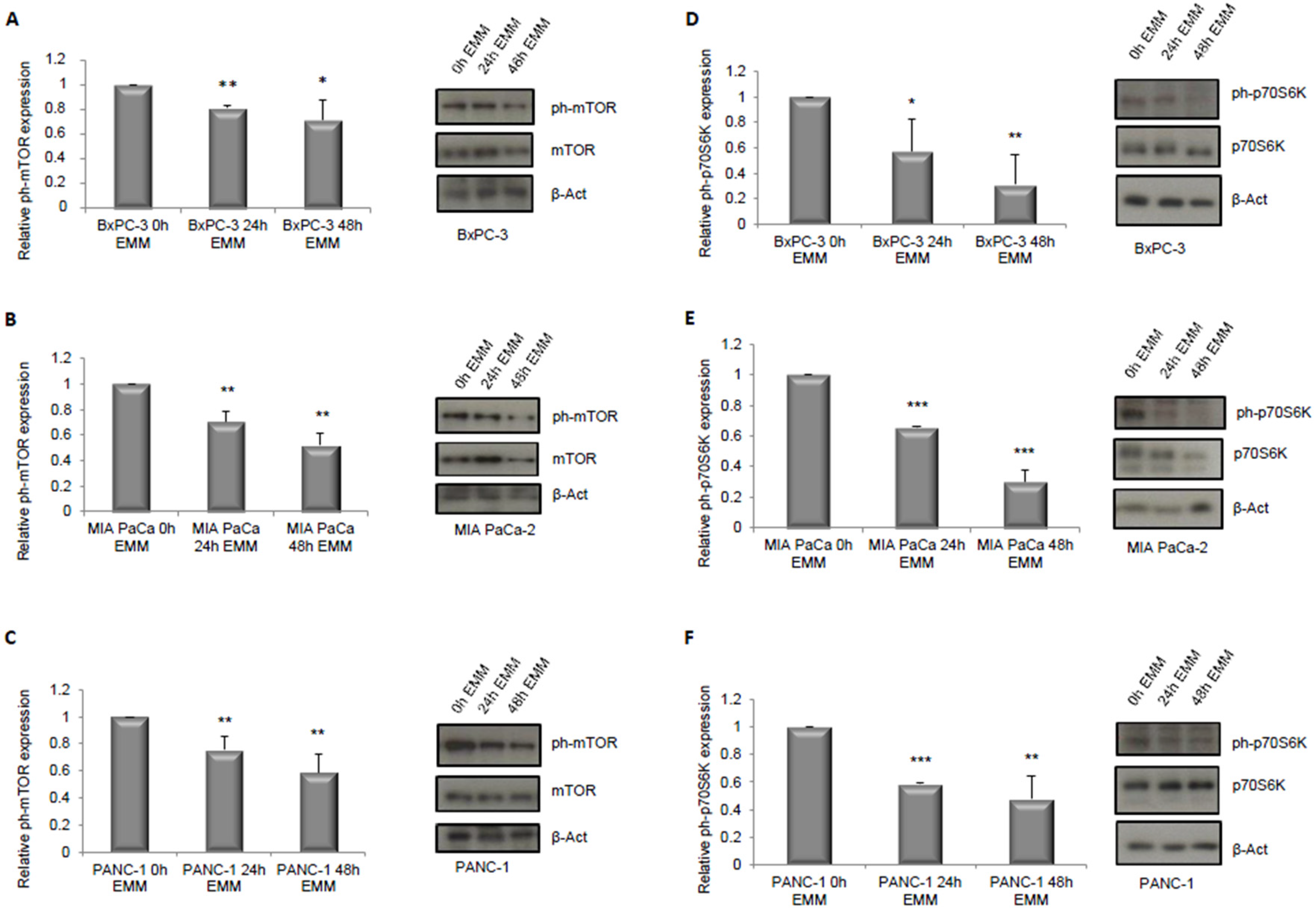
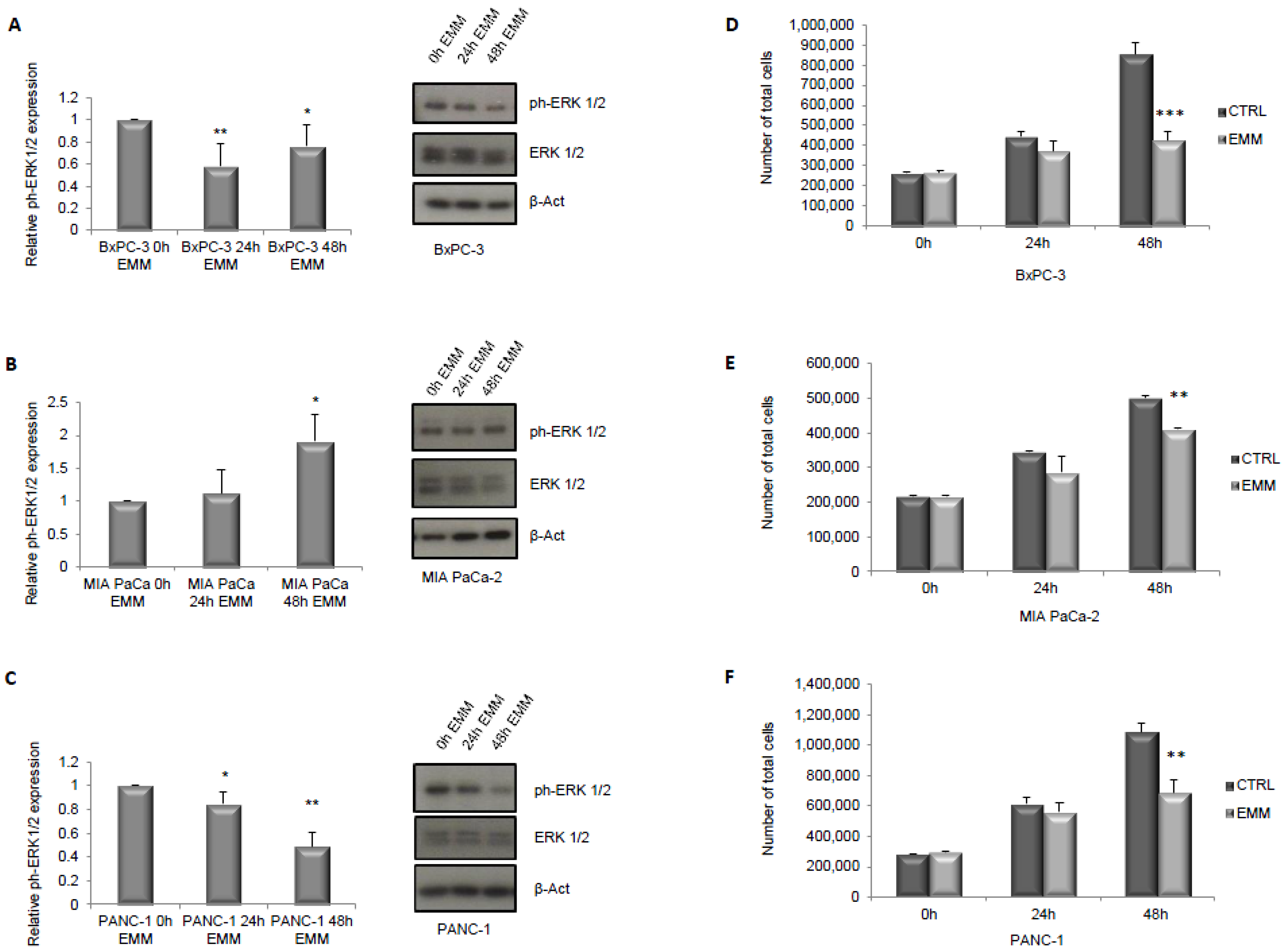
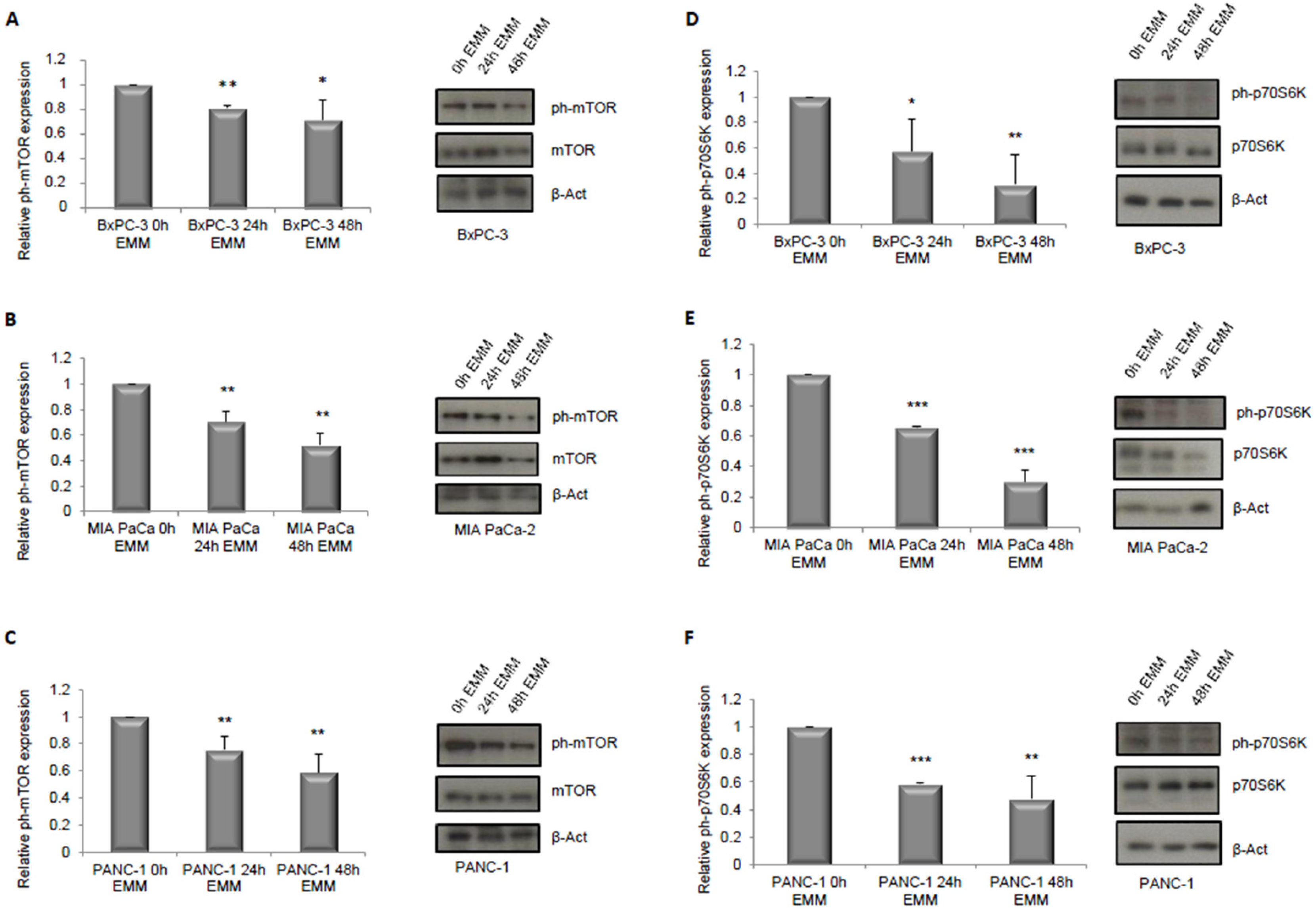
3.1. ERK1/2 and mTOR Pathways Detection in Pancreatic Cancer Cells under ERS-Mimicking Culture Conditions
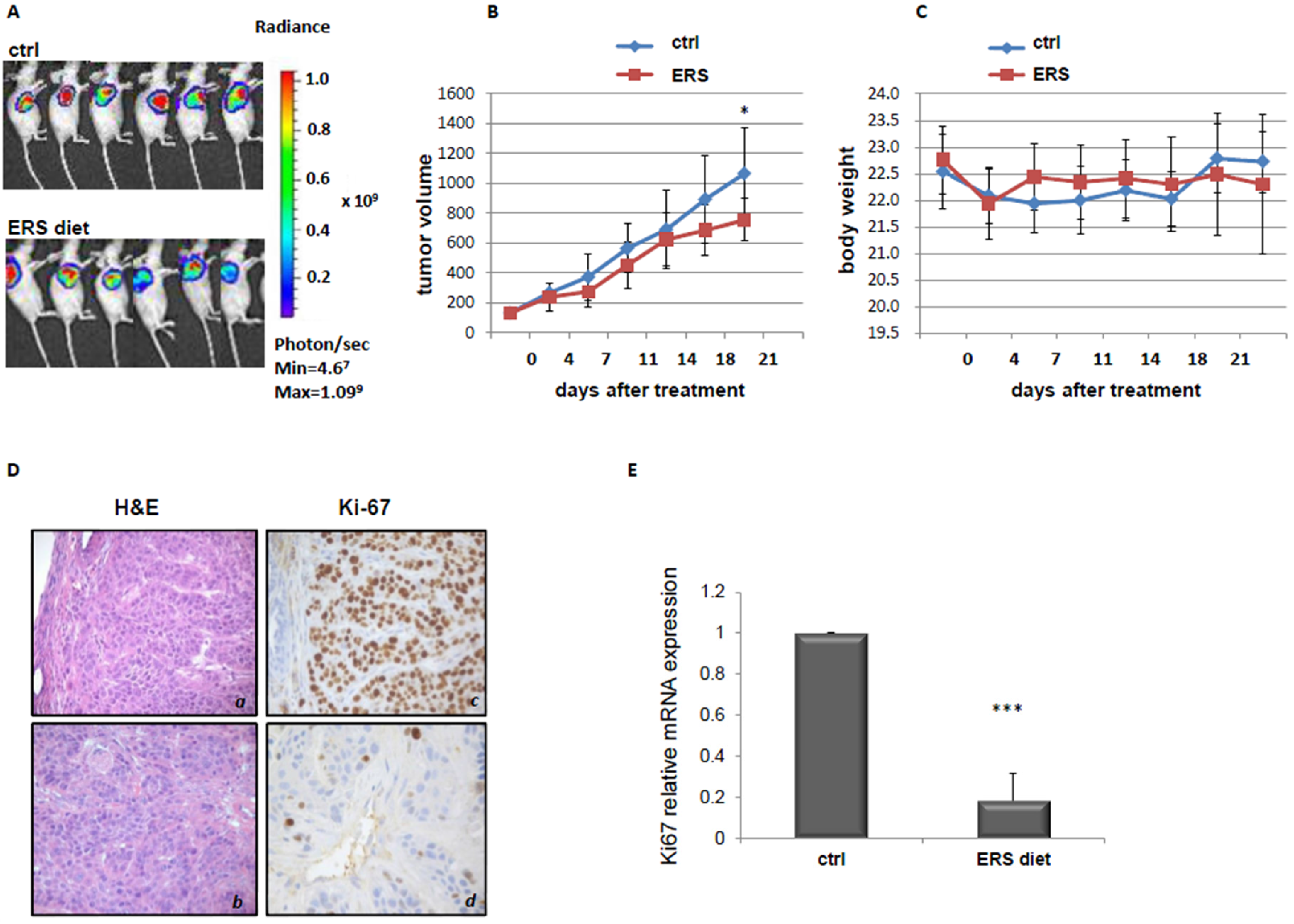
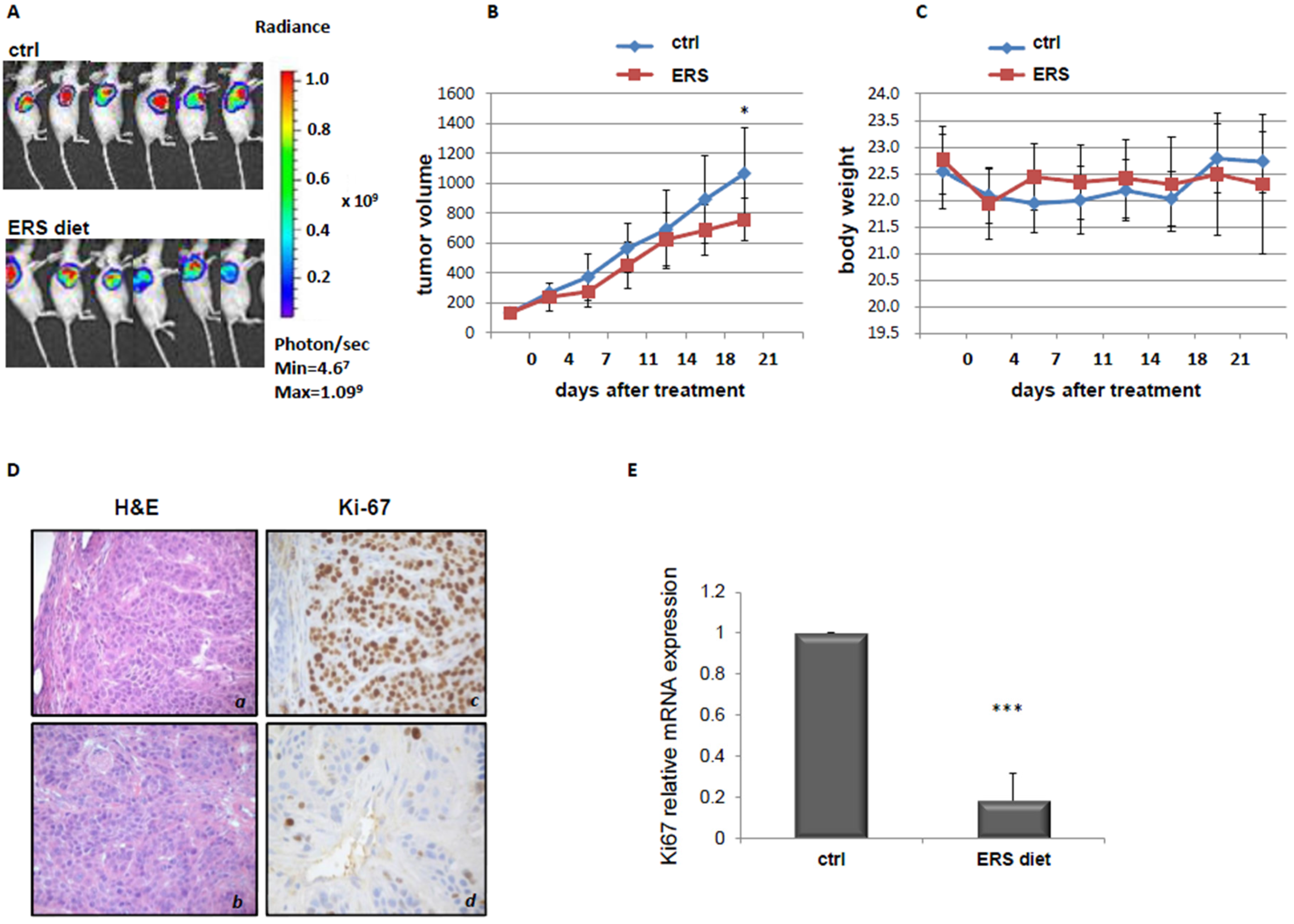
3.2. Effect of ERS Diet on Pancreatic Cancer Xenograft Mice Tumor Growth
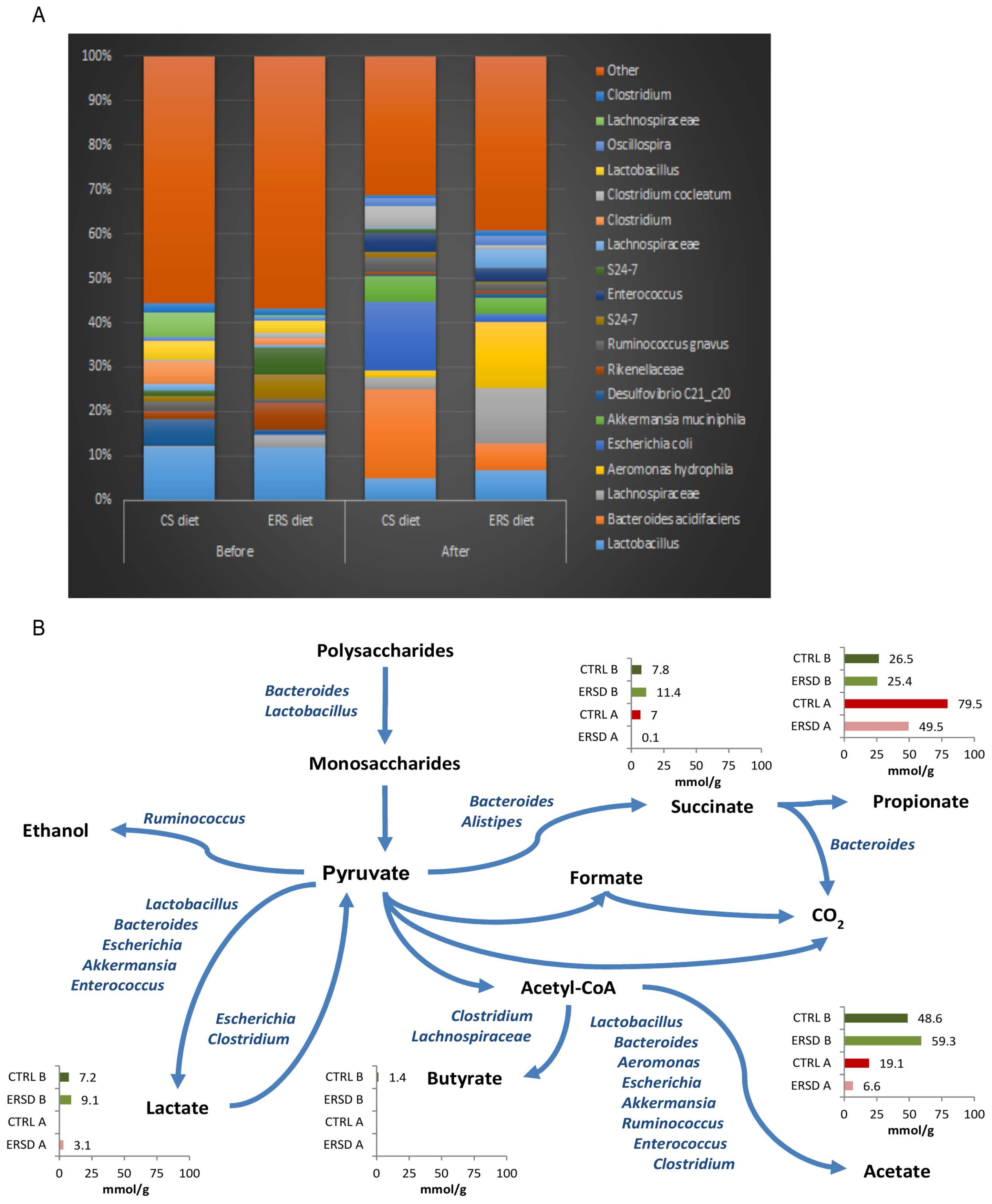
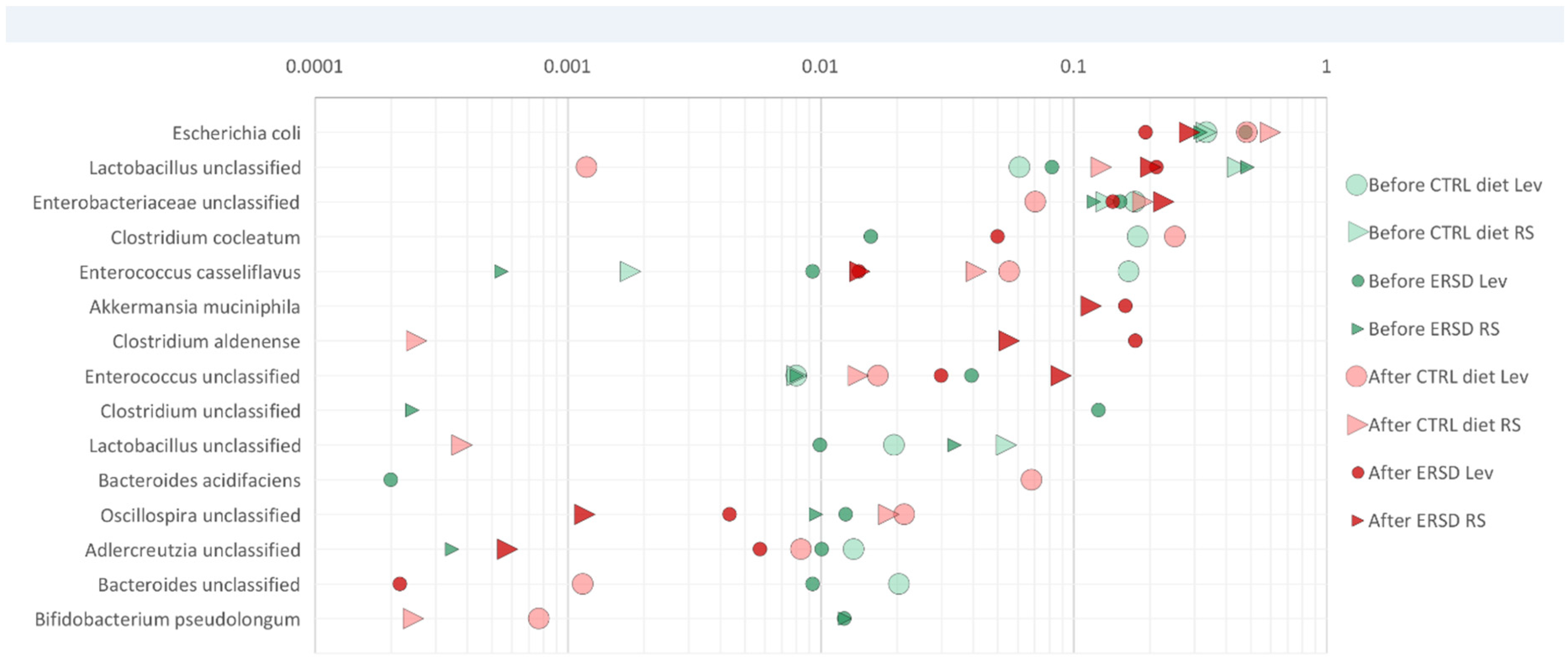
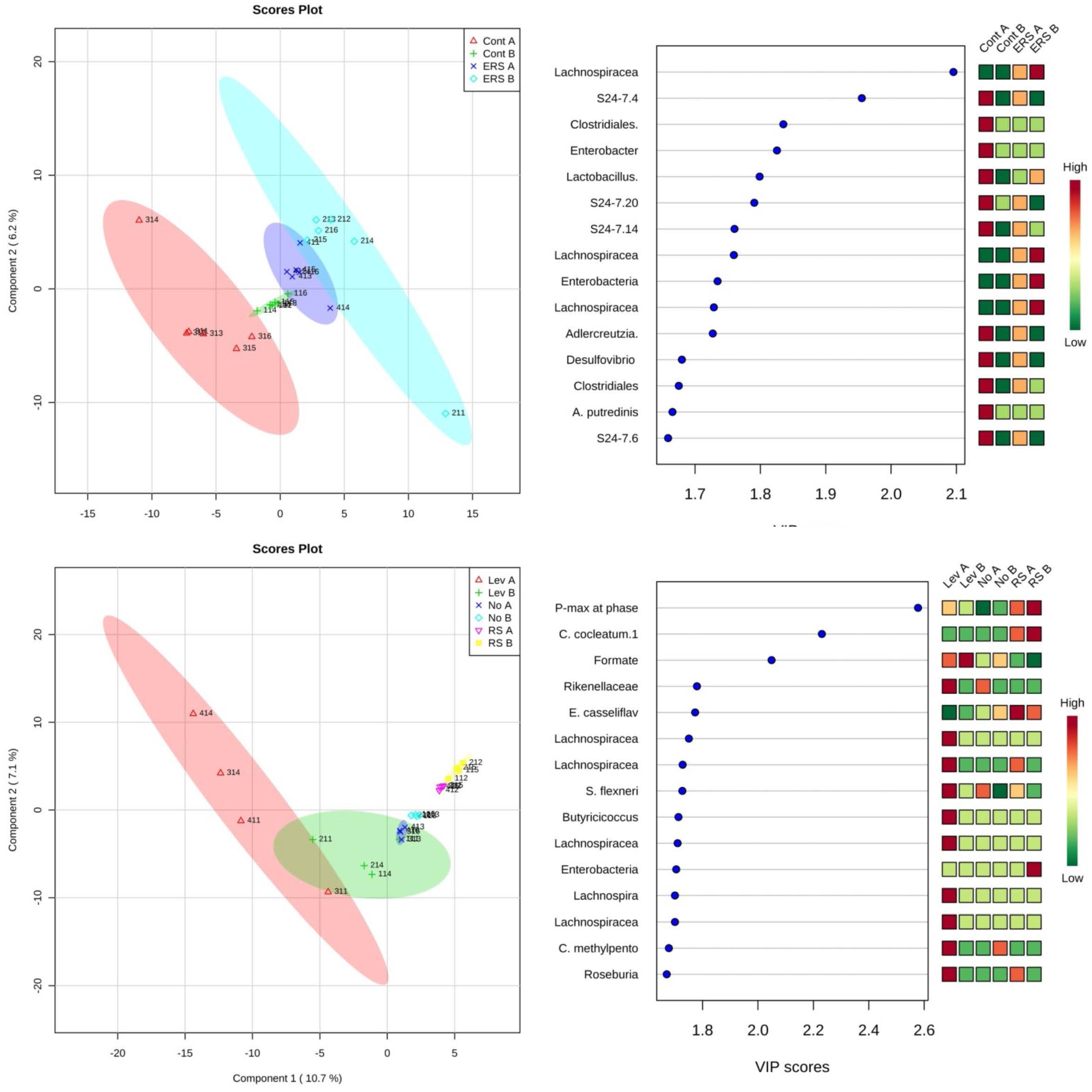
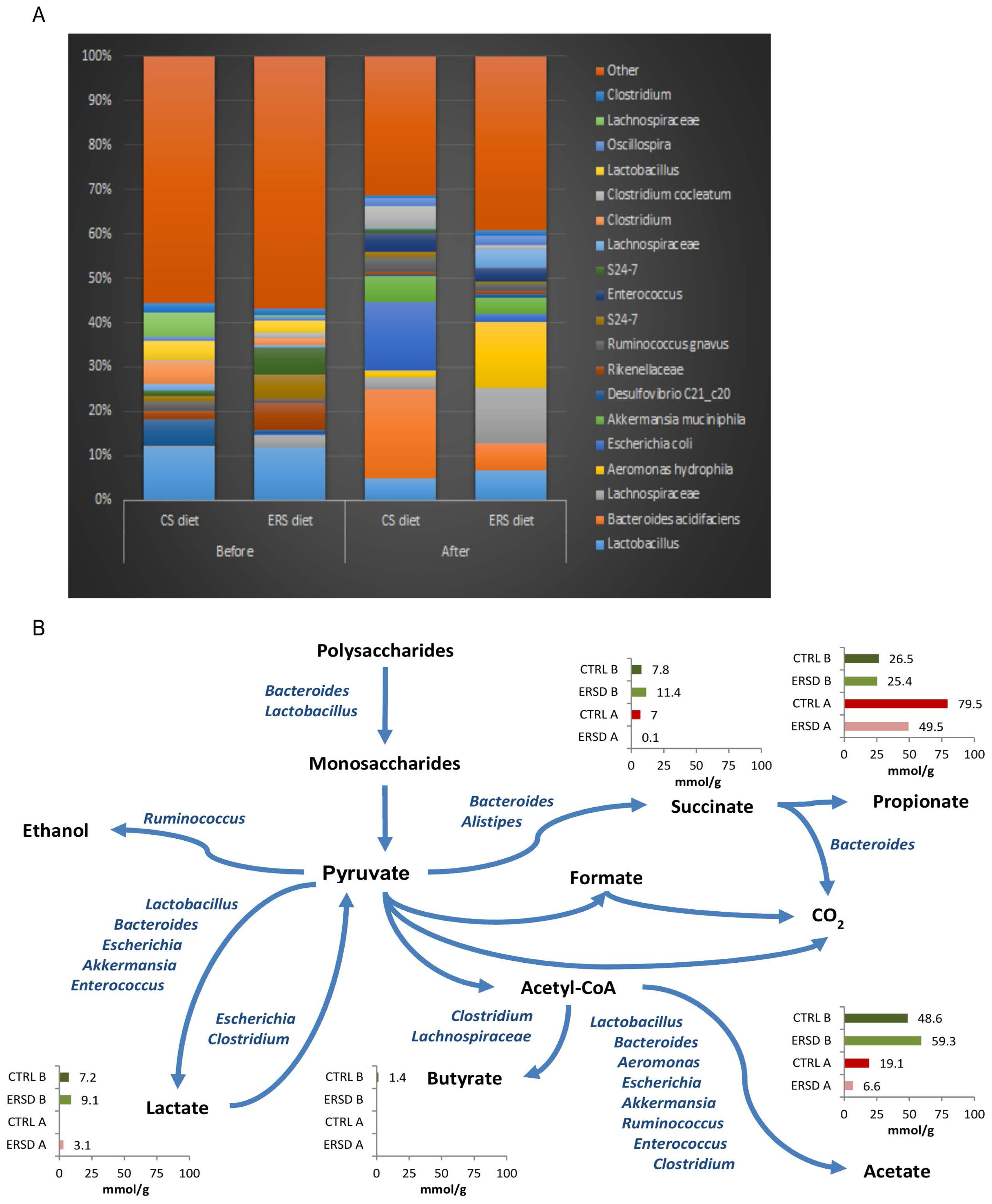
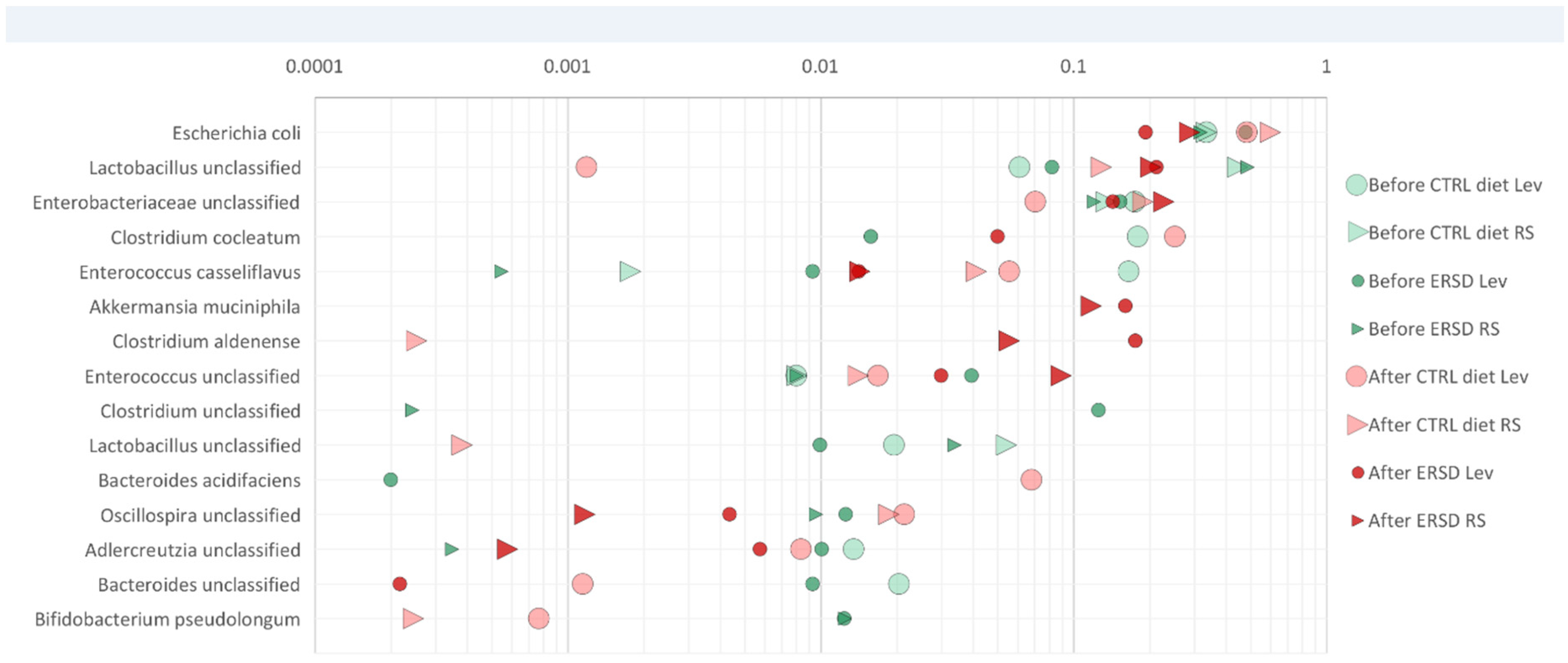
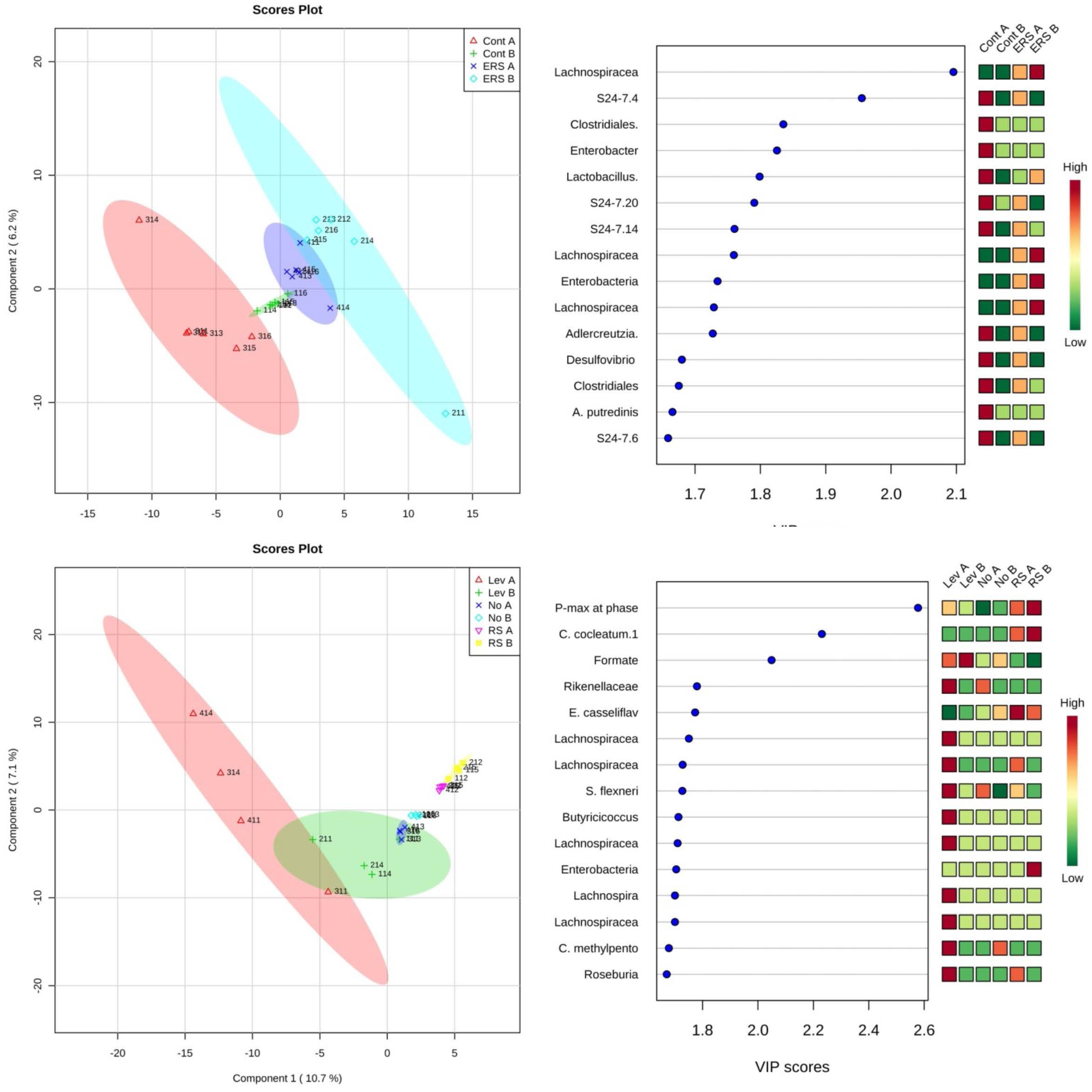
3.3. Characterization of Microbiota and Metabolites of Fecal Samples
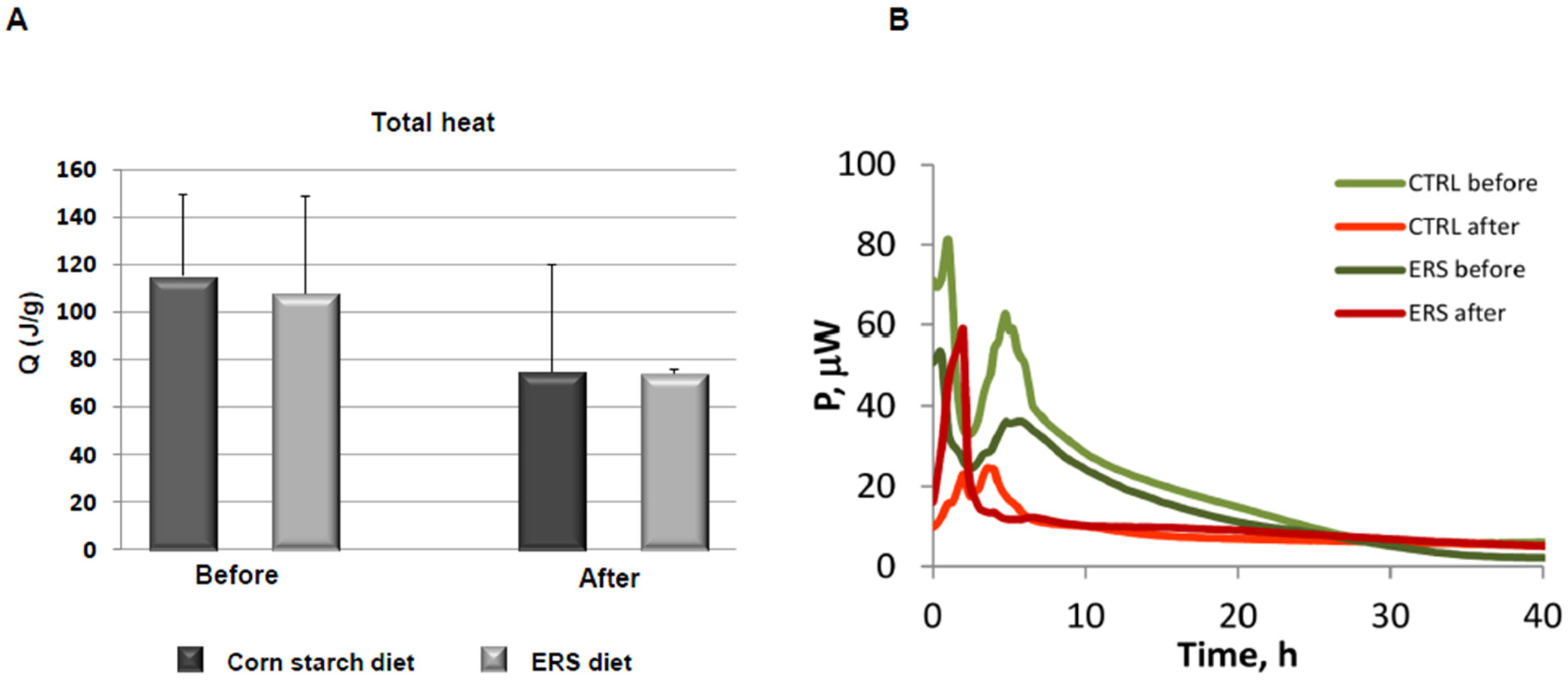
3.4. Growth Experiments with Fecal Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2015, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cid-Arregui, A.; Juarez, V. Perspectives in the treatment of pancreatic adenocarcinoma. World J. Gastroenterol. 2015, 21, 9297–9316. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, J. Promising molecular mechanism responsible for gemcitabine resistance in cancer. Genes Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef]
- Korkeila, E.A. Advanced pancreatic cancer—How to choose an adequate treatment option. World J. Gastroenterol. 2015, 21, 10709–10713. [Google Scholar] [CrossRef] [PubMed]
- D’Aronzo, M.; Vinciguerra, M.; Mazza, T.; Panebianco, C.; Saracino, C.; Pereira, S.P.; Graziano, P.; Pazienza, V. Fasting cycles potentiate the efficacy of gemcitabine treatment in in vitro and in vivo pancreatic cancer models. Oncotarget 2015, 6, 18545–18557. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.E.; Lashinger, L.M.; Hays, D.; Harrison, L.M.; Lewis, K.; Fischer, S.M.; Hursting, S.D. Calorie restriction decreases murine and human pancreatic tumor cell growth, nuclear factor-kappaB activation, and inflammation-related gene expression in an insulin-like growth factor-1-dependent manner. PLoS ONE 2014, 9, e94151. [Google Scholar] [CrossRef] [PubMed]
- Mathews, E.H.; Liebenberg, L. Short-term starvation for cancer control in humans. Exp. Gerontol. 2013, 48, 1293. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Kammerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.W.; Leung, K.; Hsu, A.; Luk, B.; Lai, J.; Shen, S.Y.; Minchinton, A.I.; Waterhouse, D.; Bally, M.B.; Lin, W.; et al. A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res. 2011, 71, 4484–4493. [Google Scholar] [CrossRef] [PubMed]
- Birt, D.F.; Boylston, T.; Hendrich, S.; Jane, J.L.; Hollis, J.; Li, L.; McClelland, J.; Moore, S.; Phillips, G.J.; Rowling, M.; et al. Resistant starch: promise for improving human health. Adv. Nutr. 2013, 4, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Graf, D.; Di Cagno, R.; Fak, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of diet to the composition of the human gut microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Erejuwa, O.O.; Sulaiman, S.A.; Ab Wahab, M.S. Modulation of gut microbiota in the management of metabolic disorders: The prospects and challenges. Int. J. Mol. Sci. 2014, 15, 4158–4188. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vetizou, M.; Daillere, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef]
- Farrell, J.J.; Zhang, L.; Zhou, H.; Chia, D.; Elashoff, D.; Akin, D.; Paster, B.J.; Joshipura, K.; Wong, D.T. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012, 61, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Leal-Lopes, C.; Velloso, F.J.; Campopiano, J.C.; Sogayar, M.C.; Correa, R.G. Roles of Commensal Microbiota in Pancreas Homeostasis and Pancreatic Pathologies. J. Diabetes Res. 2015, 2015, 284680. [Google Scholar] [CrossRef] [PubMed]
- Zambirinis, C.P.; Pushalkar, S.; Saxena, D.; Miller, G. Pancreatic cancer, inflammation, and microbiome. Cancer J. 2014, 20, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [PubMed]
- Benegiamo, G.; Vinciguerra, M.; Mazzoccoli, G.; Piepoli, A.; Andriulli, A.; Pazienza, V. DNA methyltransferases 1 and 3b expression in Huh-7 cells expressing HCV core protein of different genotypes. Dig. Dis. Sci. 2012, 57, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Adamberg, S.; Tomson, K.; Vija, H.; Puurand, M.; Kabanova, N.; Visnapuu, T.; Jogi, E.; Alamae, T.; Adamberg, K. Degradation of Fructans and Production of Propionic Acid by Bacteroides thetaiotaomicron are Enhanced by the Shortage of Amino Acids. Front. Nutr. 2014, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, N.; Kazarjan, A.; Stulova, I.; Vilu, R. Microcalorimetric study of growth of Lactococcuslactis IL1403 at different glucose concentrations in broth. Thermochim. Acta 2009, 496, 87–92. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Apontes, P.; Leontieva, O.V.; Demidenko, Z.N.; Li, F.; Blagosklonny, M.V. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011, 2, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Duysen, E.; Yaktine, A.L.; Au, A.; Wang, W.; Birt, D.F. Dietary energy restriction inhibits ERK but not JNK or p38 activity in the epidermis of SENCAR mice. Carcinogenesis 2001, 22, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fontana, L. Intermittent supplementation with rapamycin as a dietary restriction mimetic. Aging 2011, 3, 1039–1040. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Jiao, X.; Jiang, X.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Matsuda, Y.; Kawahara, K.; Naito, Z.; Ishiwata, T. Keratinocyte growth factor stimulates growth of MIA PaCa-2 cells through extracellular signal-regulated kinase phosphorylation. Oncol. Lett. 2012, 3, 307–310. [Google Scholar] [PubMed]
- Chang, Q.; Chen, E.; Hedley, D. Effects of combined inhibition of MEK and mTOR on downstream signaling and tumor growth in pancreatic cancer xenograft models. Cancer Biol. Ther. 2009, 8, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Soares, H.P.; Ni, Y.; Kisfalvi, K.; Sinnett-Smith, J.; Rozengurt, E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS ONE 2013, 8, e57289. [Google Scholar] [CrossRef] [PubMed]
- Boureau, H.; Decre, D.; Carlier, J.P.; Guichet, C.; Bourlioux, P. Identification of a Clostridium cocleatum strain involved in an anti-Clostridium difficile barrier effect and determination of its mucin-degrading enzymes. Res. Microbiol. 1993, 144, 405–410. [Google Scholar] [CrossRef]
- Hursting, S.D.; Lavigne, J.A.; Berrigan, D.; Perkins, S.N.; Barrett, J.C. Calorie restriction, aging, and cancer prevention: Mechanisms of action and applicability to humans. Annu. Rev. Med. 2003, 54, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra127. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.A.; Brown, I.L. Resistant starch: A promising dietary agent for the prevention/treatment of inflammatory bowel disease and bowel cancer. Curr. Opin. Gastroenterol. 2013, 29, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Malcomson, F.C.; Willis, N.D.; Mathers, J.C. Is resistant starch protective against colorectal cancer via modulation of the WNT signalling pathway? Proc. Nutr. Soc. 2015, 74, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Dronamraju, S.S.; Coxhead, J.M.; Kelly, S.B.; Burn, J.; Mathers, J.C. Cell kinetics and gene expression changes in colorectal cancer patients given resistant starch: a randomised controlled trial. Gut 2009, 58, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Tajaddini, A.; Pourzand, A.; Sanaat, Z.; Pirouzpanah, S. Dietary resistant starch contained foods and breast cancer risk: A case-control study in northwest of Iran. Asian. Pac. J. Cancer Prev. 2015, 16, 4185–4192. [Google Scholar] [CrossRef] [PubMed]
- Harnden, K.K.; Blackwell, K.L. Increased Fiber Intake Decreases Premenopausal Breast Cancer Risk. Pediatrics 2016, 137, e20154376. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Berry, D.; Rauch, I.; Rennisch, I.; Ramesmayer, J.; Hainzl, E.; Heider, S.; Decker, T.; Kenner, L.; Muller, M.; et al. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J. 2014, 8, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Png, C.W.; Linden, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zingarelli, B.; Peck, O.M.; Teti, G.; Tempel, G.E.; Halushka, P.V.; Spicher, K.; Boulay, G.; Birnbaumer, L.; Cook, J.A. Lipopolysaccharide- and gram-positive bacteria-induced cellular inflammatory responses: Role of heterotrimeric Galpha(i) proteins. Am. J. Physiol. Cell Physiol. 2005, 289, C293–C301. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.C.; Chiang, S.R.; Yan, J.J.; Chuang, Y.C. Comparative pathogenicity of bacteraemic isolates of Aeromonas hydrophila and Klebsiella pneumoniae. Clin. Microbiol. Infect. 2005, 11, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Abuelsaad, A.S.; Allam, G.; Al-Solumani, A.A. Hesperidin inhibits inflammatory response induced by Aeromonas hydrophila infection and alters CD4+/CD8+ T cell ratio. Mediat. Inflamm. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, S.; Kong, B.; Michalski, C.; Erkan, M.; Friess, H. The role of inflammation in pancreatic cancer. Adv. Exp. Med. Biol. 2014, 816, 129–151. [Google Scholar] [PubMed]
- Levrat, M.A.; Remesy, C.; Demigne, C. High propionic acid fermentations and mineral accumulation in the cecum of rats adapted to different levels of inulin. J. Nutr. 1991, 121, 1730–1737. [Google Scholar] [PubMed]
- Archer, S.Y.; Meng, S.; Shei, A.; Hodin, R.A. p21WAF1 is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6791–6796. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Chopin, V.; Toillon, R.A.; Jouy, N.; Le Bourhis, X. Sodium butyrate induces P53-independent, Fas-mediated apoptosis in MCF-7 human breast cancer cells. Br. J. Pharmacol. 2002, 135, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Scheppach, W.; Weiler, F. The butyrate story: Old wine in new bottles? Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Velcich, A.; Palumbo, L.; Jarry, A.; Laboisse, C.; Racevskis, J.; Augenlicht, L. Patterns of expression of lineage-specific markers during the in vitro-induced differentiation of HT29 colon carcinoma cells. Cell Growth Differ. 1995, 6, 749–757. [Google Scholar] [PubMed]
- Gaschott, T.; Maassen, C.U.; Stein, J. Tributyrin, a butyrate precursor, impairs growth and induces apoptosis and differentiation in pancreatic cancer cells. Anticancer Res. 2001, 21, 2815–2819. [Google Scholar] [PubMed]
- Natoni, F.; Diolordi, L.; Santoni, C.; Gilardini Montani, M.S. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and the extrinsic apoptotic pathways. Biochim. Biophys. Acta 2005, 1745, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Rychahou, P.; O’Connor, K.L.; Evers, B.M. Butyrate inhibits pancreatic cancer invasion. J. Gastrointest. Surg. 2003, 7, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Lockyer, S.; Nugent, A.P. Health effects of resistant starch. Nutr. Bull. 2017, 42, 10–41. [Google Scholar] [CrossRef]
- Nofrarías, M.; Martínez-Puig, D.; Pujols, J.; Majó, N.; Pérez, J.F. Long-term intake of resistant starch improves colonic mucosal integrity and reduces gut apoptosis and blood immune cells. Nutrition 2007, 23, 861–870. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panebianco, C.; Adamberg, K.; Adamberg, S.; Saracino, C.; Jaagura, M.; Kolk, K.; Di Chio, A.G.; Graziano, P.; Vilu, R.; Pazienza, V. Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models. Nutrients 2017, 9, 331. https://doi.org/10.3390/nu9040331
Panebianco C, Adamberg K, Adamberg S, Saracino C, Jaagura M, Kolk K, Di Chio AG, Graziano P, Vilu R, Pazienza V. Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models. Nutrients. 2017; 9(4):331. https://doi.org/10.3390/nu9040331
Chicago/Turabian StylePanebianco, Concetta, Kaarel Adamberg, Signe Adamberg, Chiara Saracino, Madis Jaagura, Kaia Kolk, Anna Grazia Di Chio, Paolo Graziano, Raivo Vilu, and Valerio Pazienza. 2017. "Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models" Nutrients 9, no. 4: 331. https://doi.org/10.3390/nu9040331
APA StylePanebianco, C., Adamberg, K., Adamberg, S., Saracino, C., Jaagura, M., Kolk, K., Di Chio, A. G., Graziano, P., Vilu, R., & Pazienza, V. (2017). Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models. Nutrients, 9(4), 331. https://doi.org/10.3390/nu9040331