Resveratrol and Myopathy

{kind=link}
{kind=link}
Abstract
:1. Introduction
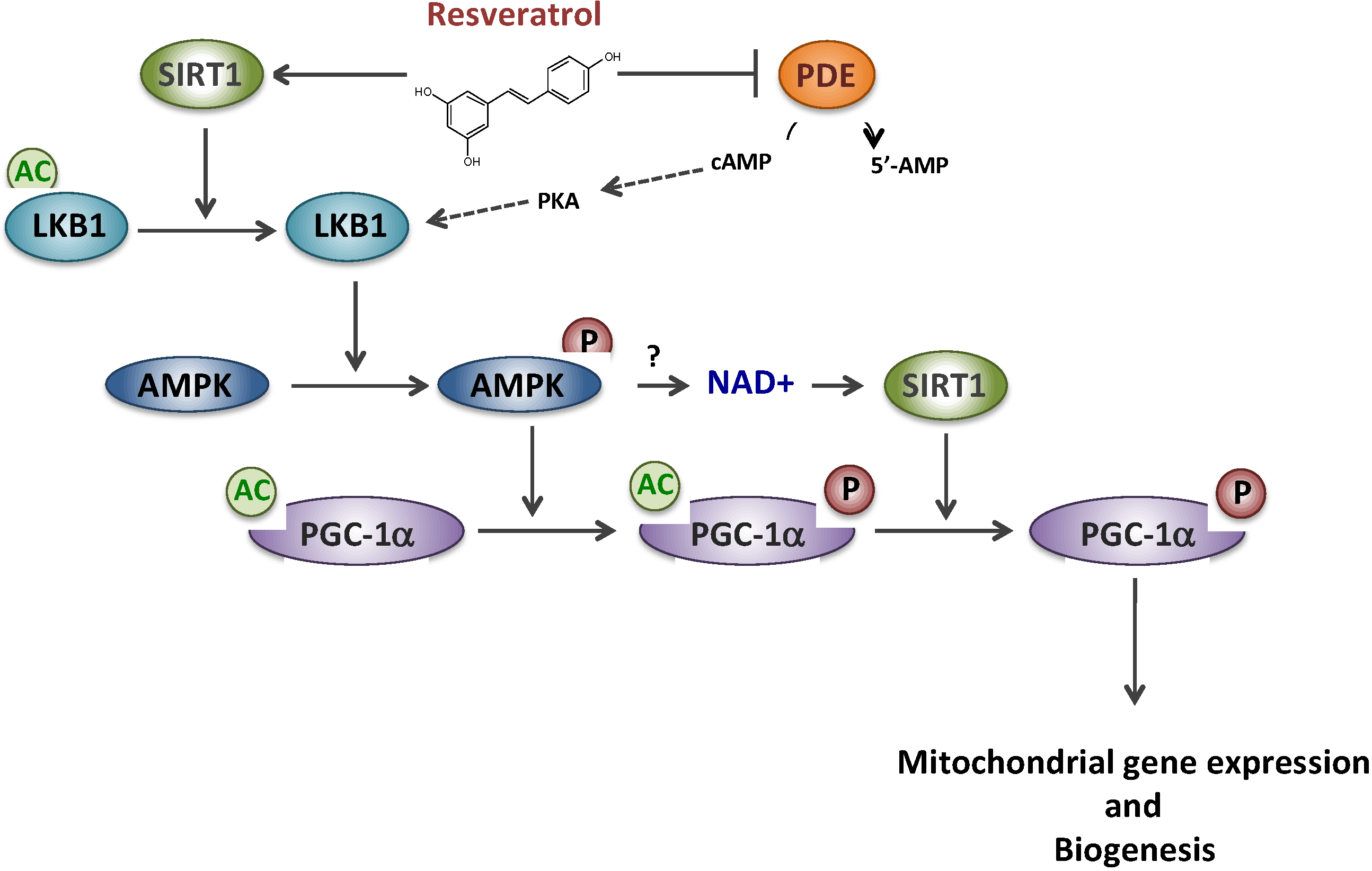
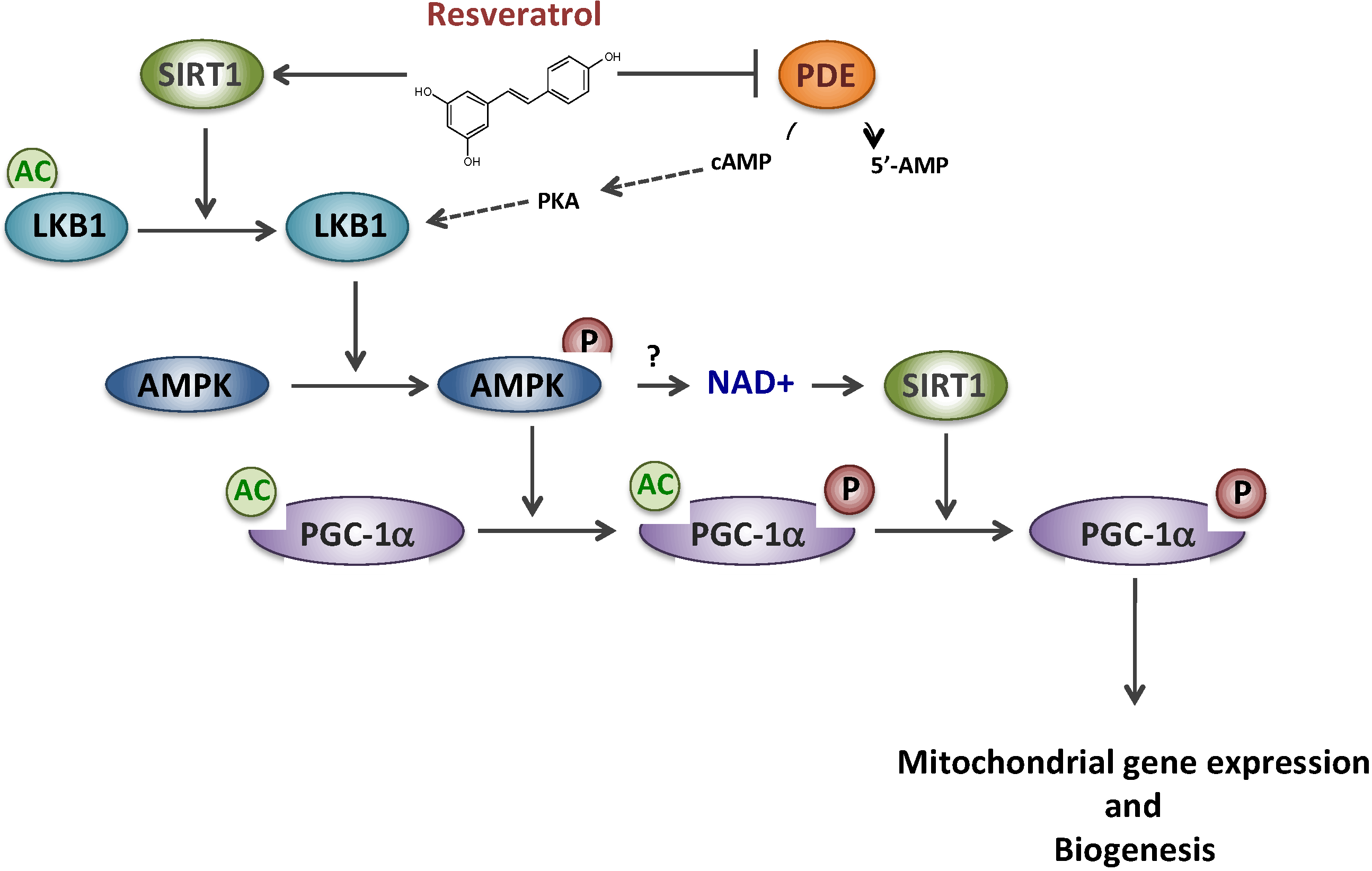
2. Resveratrol and Metabolic Myopathies
2.1. Resveratrol and Mitochondrial Fatty Acid β-Oxidation Disorders
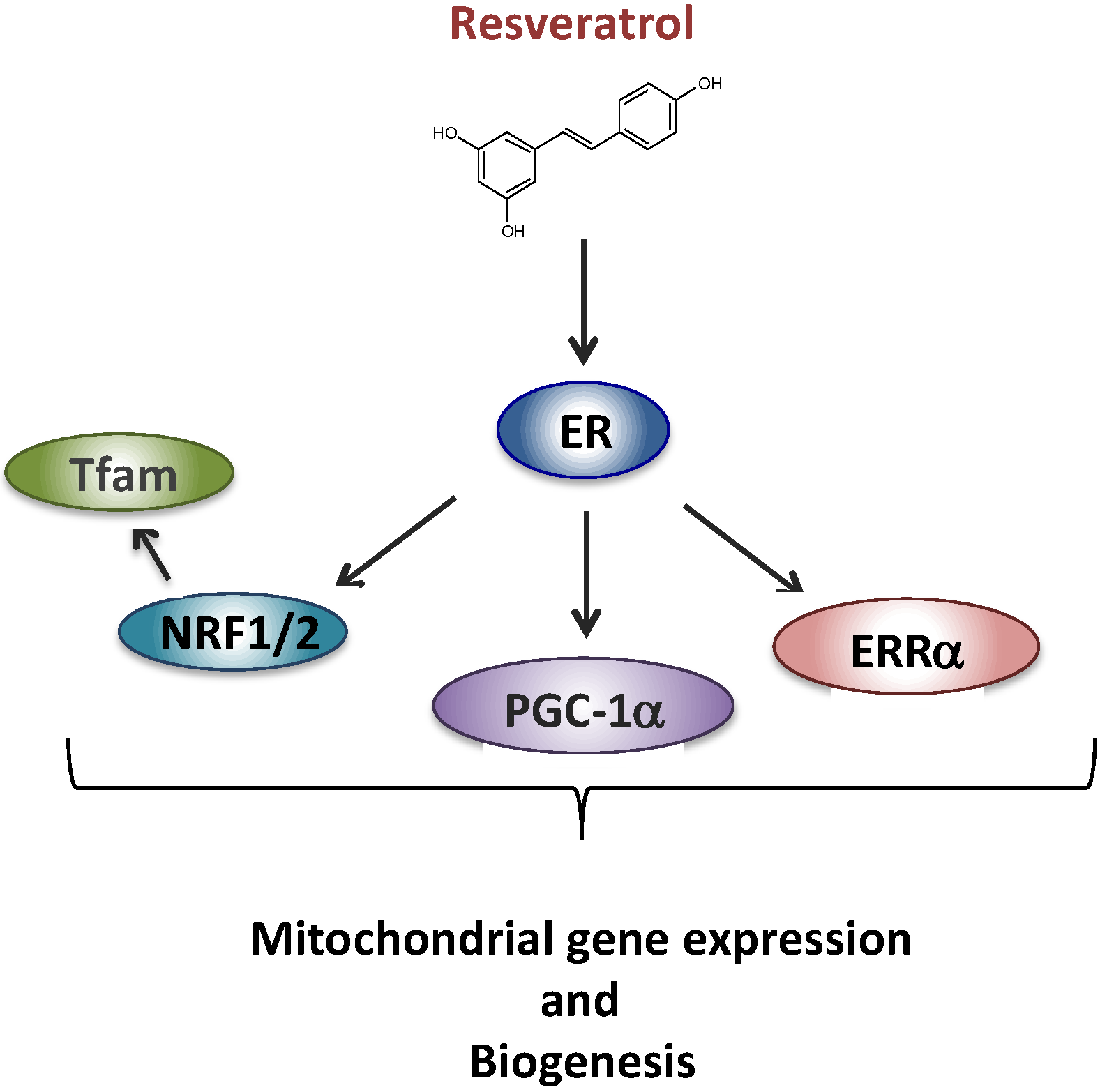
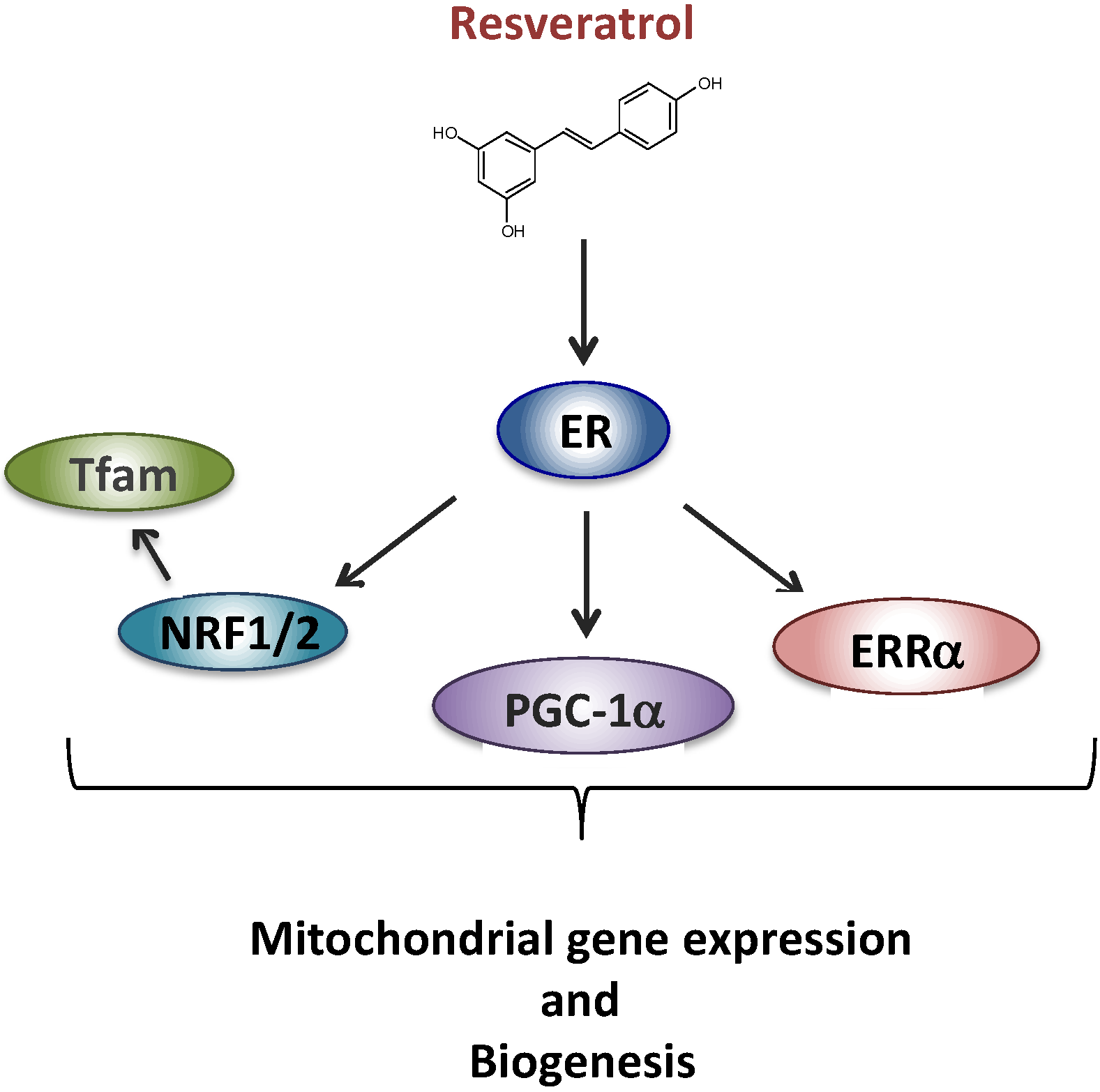
2.2. Resveratrol and Complex I Disorder
3. Resveratrol and Duchenne Myopathy
4. Discussion and Conclusive Remarks
Acknowledgments
Conflicts of Interest
References
- Waterhouse, A.L. Wine phenolics. Ann. N. Y. Acad. Sci. 2002, 957, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M. Of the phenolic substances of white hellbore (veratrum grandiflorum loes. Fil.). J. Fac. Sci. Hokkaido Imp. Univ. 1940, 3, 1–16. [Google Scholar]
- Nonomura, S.; Kanagawa, H.; Makimoto, A. Chemical constituents of polygonaceous plants. I. Studies on the components of ko-j o-kon. (polygonum cuspidatum sieb. Et zucc.). Yakugaku zasshi J. Pharm. Soc. Jpn. 1963, 83, 988–990. [Google Scholar]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Pezzuto, J.M. The pharmacology of resveratrol in animals and humans. Biochim. Biophys. Acta 2015, 1852, 1071–1113. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Tome-Carneiro, J.; Larrosa, M.; Gonzalez-Sarrias, A.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Resveratrol and clinical trials: The crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef] [PubMed]
- Dirks Naylor, A.J. Cellular effects of resveratrol in skeletal muscle. Life Sci. 2009, 84, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, J.; Schrauwen, P. Muscle mitochondria and insulin resistance: A human perspective. Trends Endocrinol. Metab. 2012, 23, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Ann. Rev. Physiol. 2015. [Google Scholar] [CrossRef]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Wieser, T.; Zierz, S. Muscle carnitine palmitoyltransferase ii deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch. Neurol. 2005, 62, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Andresen, B.S.; Corydon, M.J.; Corydon, T.J.; Olsen, R.K.; Bolund, L.; Bross, P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum. Mutat. 2001, 18, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Bastin, J.; Behin, A.; Djouadi, F. Bezafibrate for treatment of an inborn mitochondrial β-oxidation defect. N. Engl. J. Med. 2009, 360, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Bastin, J.; Laforet, P.; Aubey, F.; Mogenet, A.; Romano, S.; Ricquier, D.; Gobin-Limballe, S.; Vassault, A.; Behin, A.; et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin. Pharmacol. Ther. 2010, 88, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Arnold, G.L.; van Hove, J.; Freedenberg, D.; Strauss, A.; Longo, N.; Burton, B.; Garganta, C.; Ficicioglu, C.; Cederbaum, S.; Harding, C.; et al. A delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2009, 96, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Bastin, J.; Gillingham, M.; Morris, A.; Wijburg, F.; Wilcken, B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2010, 33, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bastin, J. Ppars as therapeutic targets for correction of inborn mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2008, 31, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A.G. Drug repositioning for orphan diseases. Brief. Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Bastin, J.; Lopes-Costa, A.; Djouadi, F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum. Mol. Genet. 2011, 20, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Aires, V.; Delmas, D.; le Bachelier, C.; Latruffe, N.; Schlemmer, D.; Benoist, J.F.; Djouadi, F.; Bastin, J. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J. Rare Dis. 2014, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, J.L.; Chung, J.H. Metabolic effects of resveratrol: Addressing the controversies. Cell. Mol. Life Sci. 2015, 72, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human sirt1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef] [PubMed]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates amp kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. Sirt1 regulates hepatocyte lipid metabolism through activating amp-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. Sirt1 is required for ampk activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting camp phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Koopman, W.J.; van den Heuvel, L.P.; Rodenburg, R.J.; Mayatepek, E.; Willems, P.H.; Smeitink, J.A. Mitochondrial complex i deficiency: From organelle dysfunction to clinical disease. Brain J. Neurol. 2009, 132, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochim. Biophys. Acta 2015, 1847, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Lopes Costa, A.; le Bachelier, C.; Mathieu, L.; Rotig, A.; Boneh, A.; de Lonlay, P.; Tarnopolsky, M.A.; Thorburn, D.R.; Bastin, J.; Djouadi, F. Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum. Mol. Genet. 2014, 23, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar] [PubMed]
- Eichner, L.J.; Giguere, V. Estrogen related receptors (errs): A new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011, 11, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial complex i deficient patient’s fibroblasts, reveals aicar as the most beneficial compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; Vandemeulebroecke, K.; Smet, J.; Vanlander, A.; Seneca, S.; Lissens, W.; van Hove, J.L.; Deschepper, E.; Briones, P.; van Coster, R. Effect of resveratrol on cultured skin fibroblasts from patients with oxidative phosphorylation defects. Phytother. Res. PTR 2014, 28, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Polley, K.R.; Jenkins, N.; O’Connor, P.; McCully, K. Influence of exercise training with resveratrol supplementation on skeletal muscle mitochondrial capacity. Appl. Physiol. Nutr. Metab. 2016, 41, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Dyck, J.R. Experimental studies of the molecular pathways regulated by exercise and resveratrol in heart, skeletal muscle and the vasculature. Molecules 2014, 19, 14919–14947. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Timmers, S. Can resveratrol help to maintain metabolic health? Proc. Nutr. Soc. 2014, 73, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatri. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-atp connection: How half a century of research still implicates mitochondrial dysfunction in duchenne muscular dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Ljubicic, V.; Burt, M.; Jasmin, B.J. The therapeutic potential of skeletal muscle plasticity in duchenne muscular dystrophy: Phenotypic modifiers as pharmacologic targets. FASEB J. 2014, 28, 548–568. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1alpha axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef] [PubMed]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and nutriceutical approaches to address energy dysregulation and skeletal muscle wasting in duchenne muscular dystrophy. Nutrients 2015, 7, 9734–9767. [Google Scholar] [CrossRef] [PubMed]
- Moorwood, C.; Lozynska, O.; Suri, N.; Napper, A.D.; Diamond, S.L.; Khurana, T.S. Drug discovery for duchenne muscular dystrophy via utrophin promoter activation screening. PLoS ONE 2011, 6, e26169. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.S.; Delgado Diaz, D.C.; Kostek, M.C. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of duchenne muscular dystrophy. Clin. Nutr. 2013, 32, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Genetic and cellular modifiers of oxidative stress: What can we learn from fatty acid oxidation defects? Mol. Genet. Metab. 2013, 110, S31–S39. [Google Scholar] [CrossRef] [PubMed]
- Roestenberg, P.; Manjeri, G.R.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H.; Koopman, W.J. Pharmacological targeting of mitochondrial complex i deficiency: The cellular level and beyond. Mitochondrion 2012, 12, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bastin, J. Species differences in the effects of bezafibrate as a potential treatment of mitochondrial disorders. Cell Metab. 2011, 14, 715–716. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Hughes, M.C.; Edgett, B.A.; Scribbans, T.D.; Simpson, C.A.; Perry, C.G.; Gurd, B.J. An examination of resveratrol’s mechanisms of action in human tissue: Impact of a single dose in vivo and dose responses in skeletal muscle ex vivo. PLoS ONE 2014, 9, e102406. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Conte, C.; Fontana, L.; Mittendorfer, B.; Imai, S.; Schechtman, K.B.; Gu, C.; Kunz, I.; Rossi Fanelli, F.; Patterson, B.W.; et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012, 16, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254. https://doi.org/10.3390/nu8050254
Bastin J, Djouadi F. Resveratrol and Myopathy. Nutrients. 2016; 8(5):254. https://doi.org/10.3390/nu8050254
Chicago/Turabian StyleBastin, Jean, and Fatima Djouadi. 2016. "Resveratrol and Myopathy" Nutrients 8, no. 5: 254. https://doi.org/10.3390/nu8050254
APA StyleBastin, J., & Djouadi, F. (2016). Resveratrol and Myopathy. Nutrients, 8(5), 254. https://doi.org/10.3390/nu8050254