Validation of Correction Algorithms for Near-IR Analysis of Human Milk in an Independent Sample Set—Effect of Pasteurization

 , and
, and 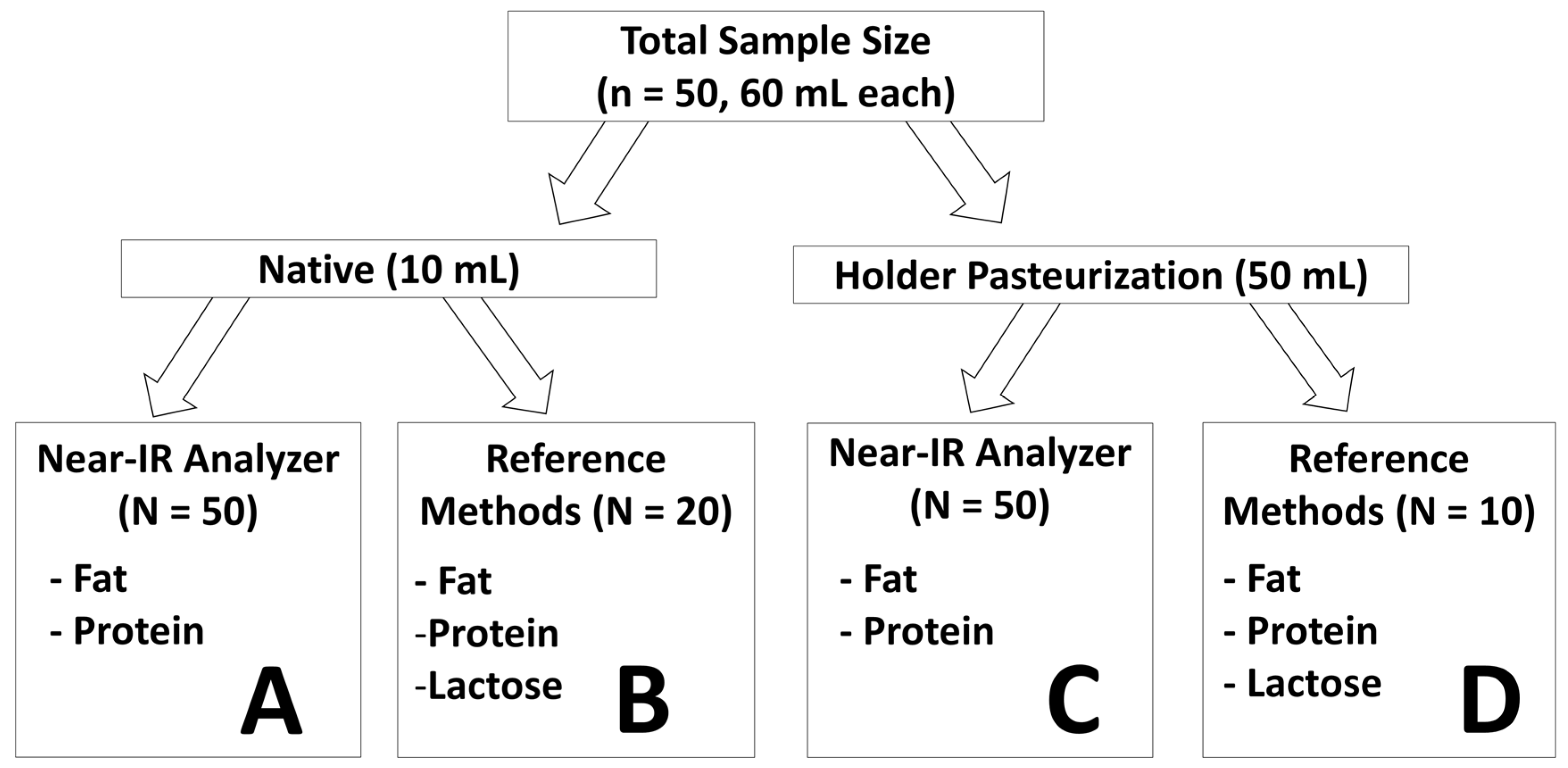{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
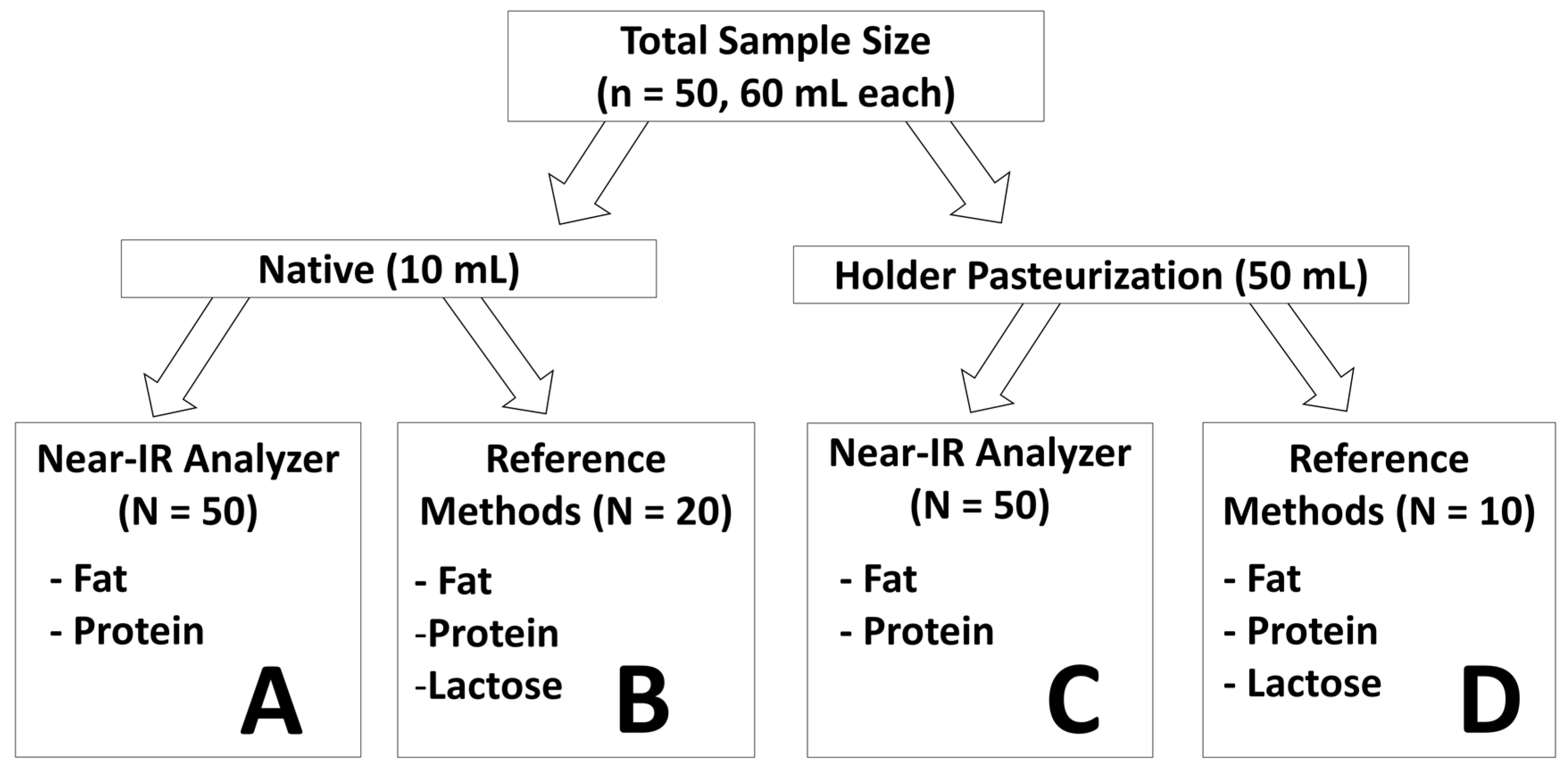
2.1. Study Design and Sample Collection
2.2. Measurement Methods for Fat, Protein, and Lactose in Breast Milk
2.3. Preparation of the Sample Set
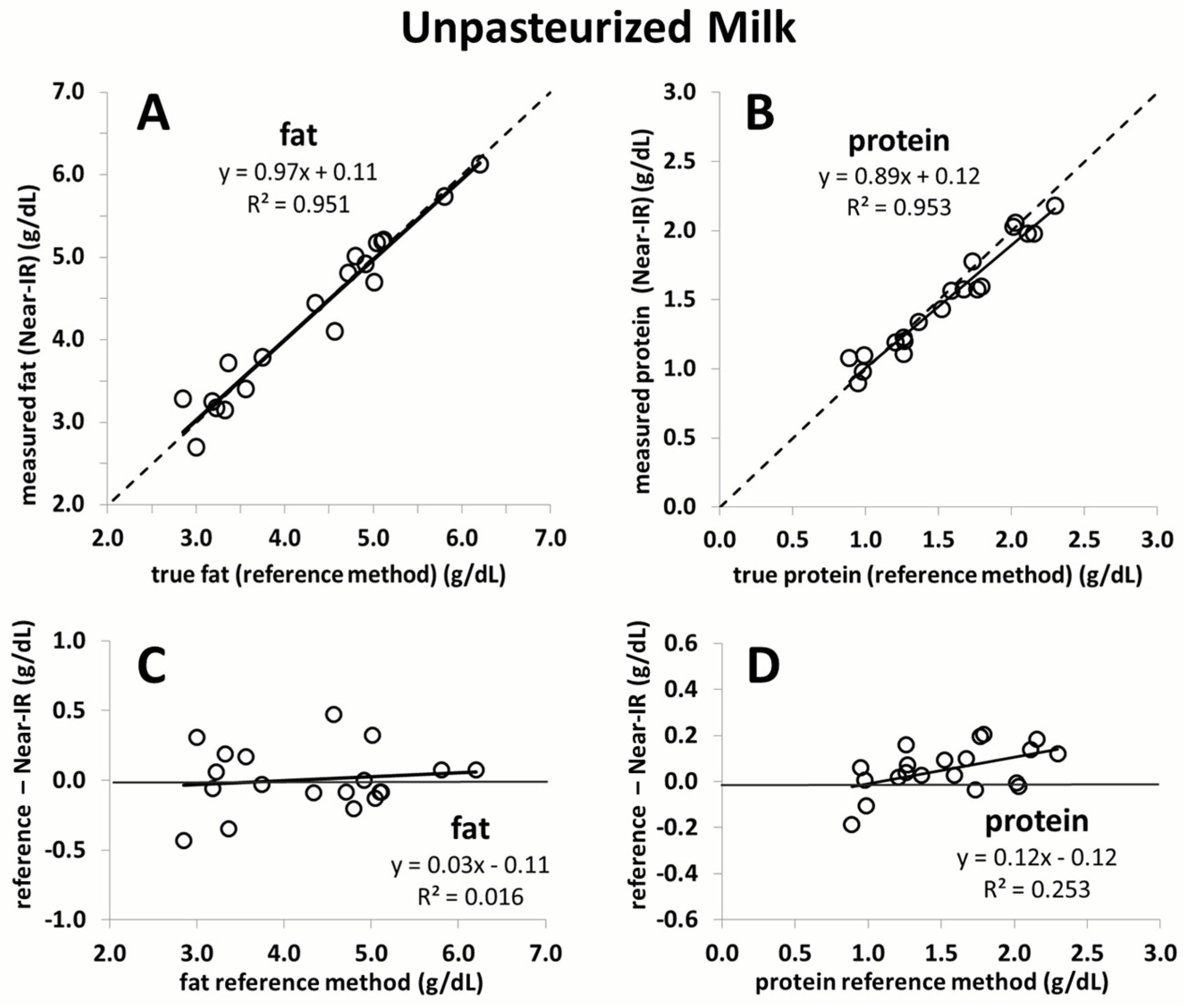
2.4. Component 1: Validation of the Near-IR Milk Analyzer
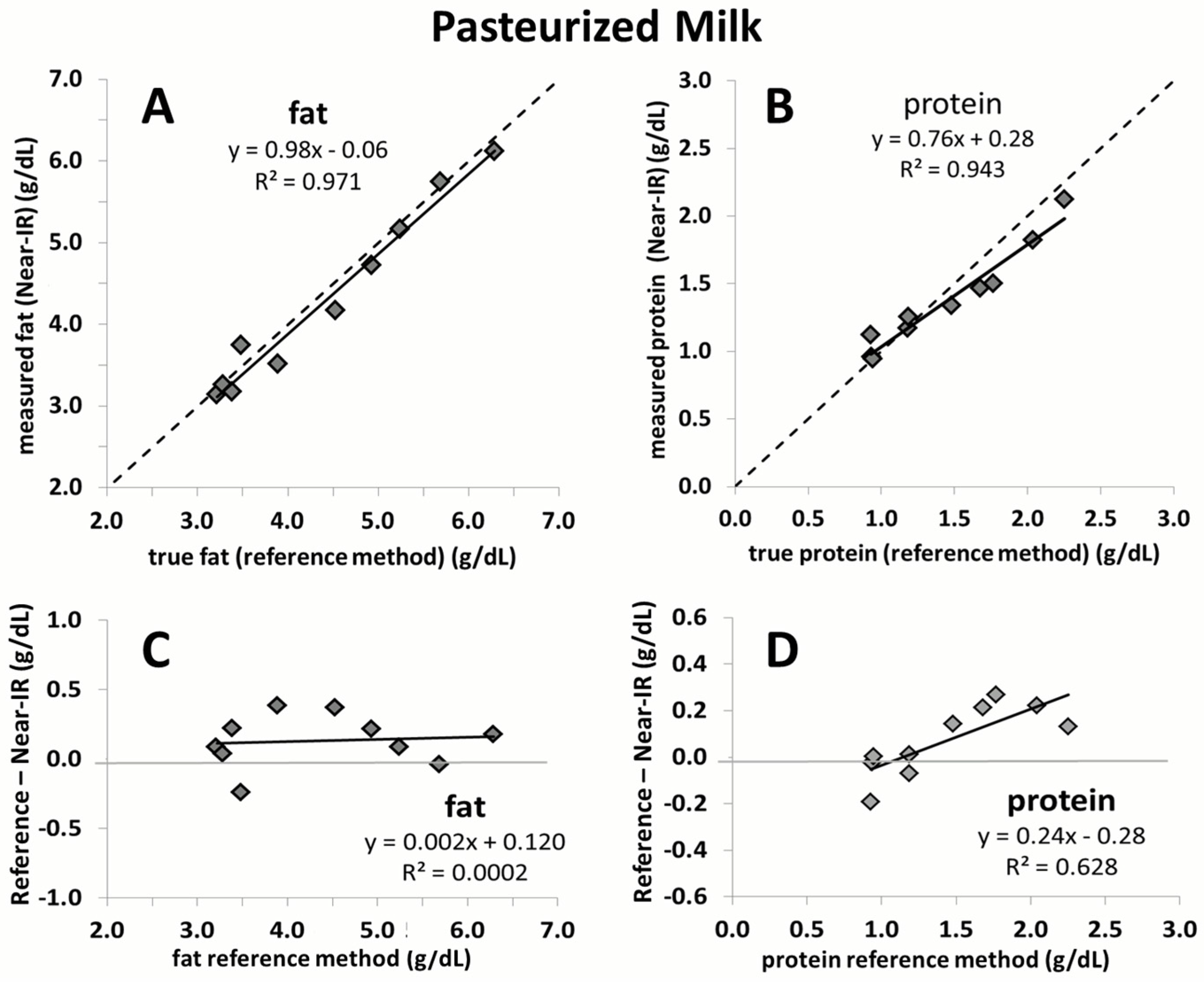
2.4.1. Component 2: Assessment of the Reliability in Pasteurized Breast Milk
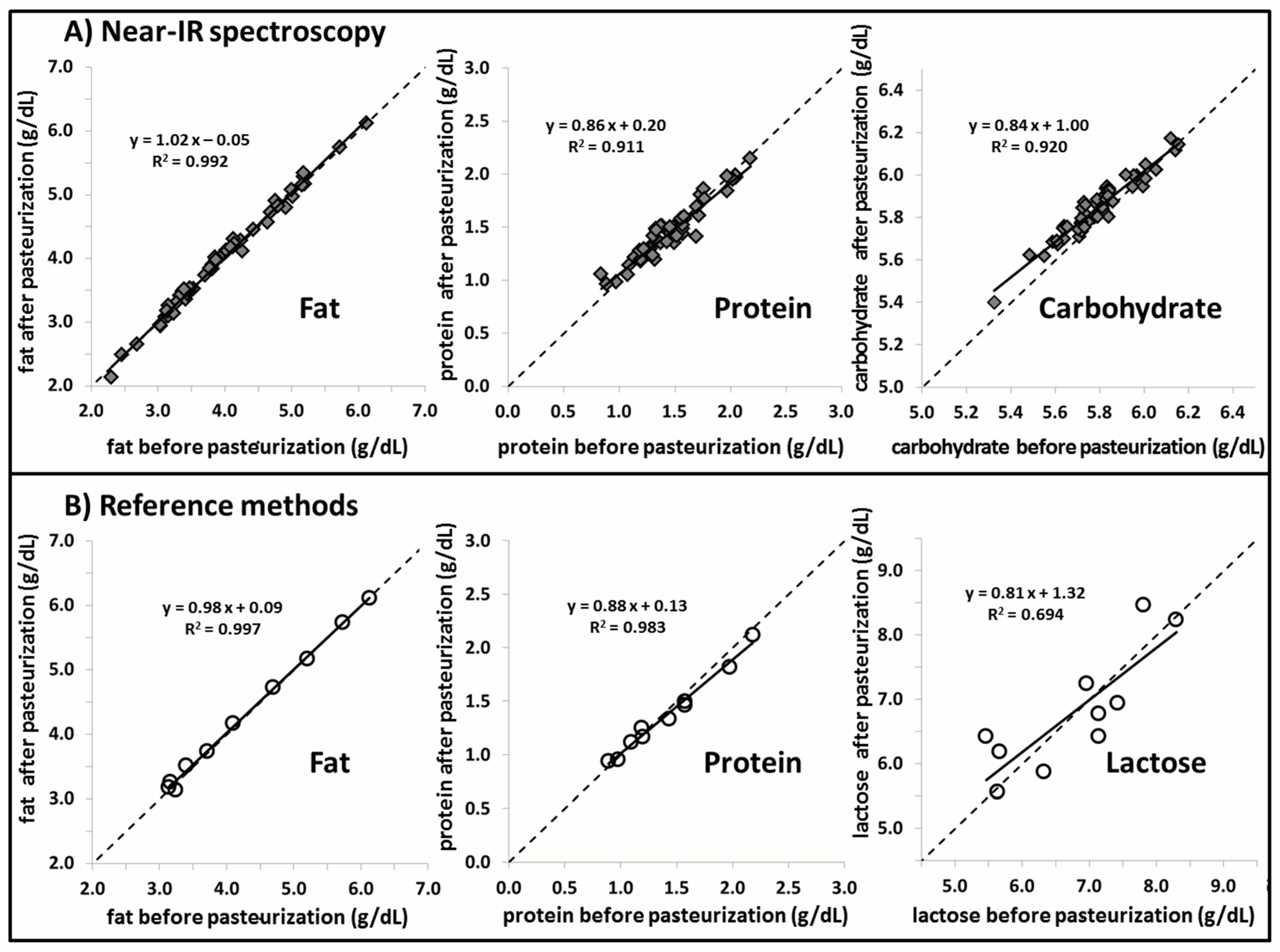
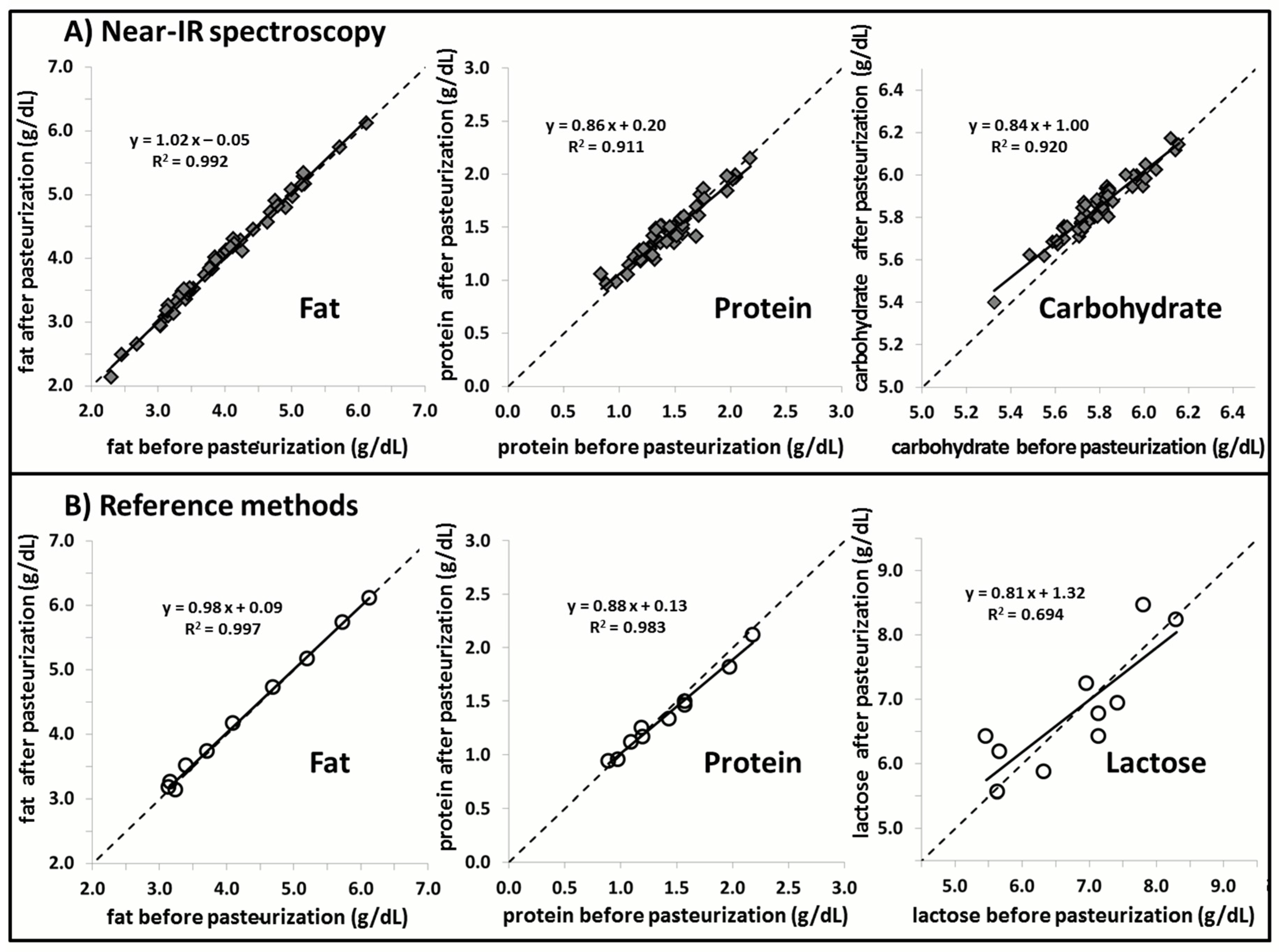
2.4.2. Component 3: Effect of Pasteurization
2.5. Statistical Analysis
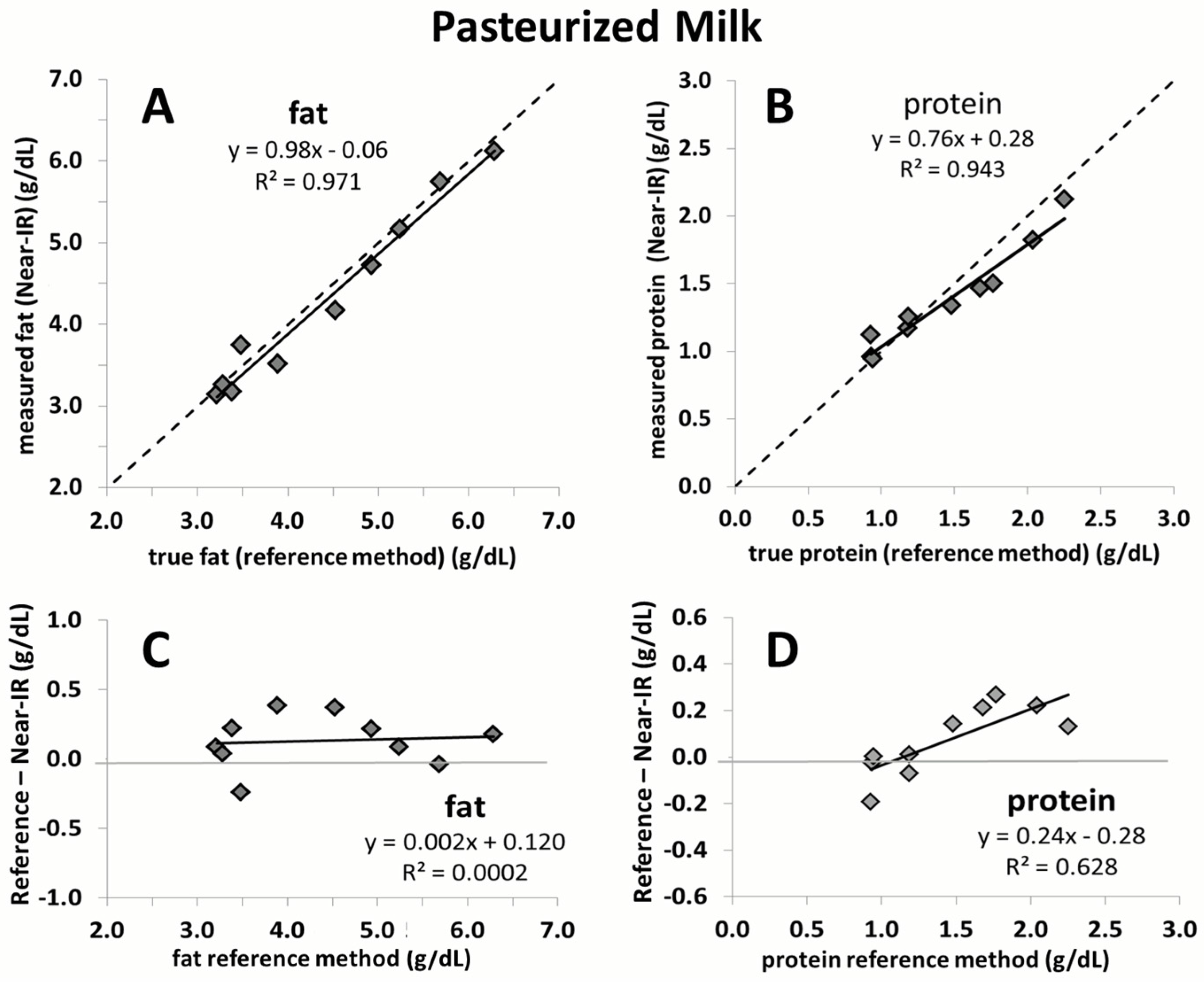
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Academy of Pediatrics Policy Statement. Breastfeeding and the use of human milk. Pediatrics 2012, 129, 827–841. [Google Scholar]
- Polberger, S.K.; Axelsson, I.A.; Raiha, N.C. Growth of very low birth weight infants on varying amounts of human milk protein. Pediatr. Res. 1989, 25, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.; Schulze, K.F.; Forsyth, M.; Dell, R.B.; Ramakrishnan, R.; Heird, W.C. Growth, nutrient retention, and metabolic response of low-birth-weight infants fed supplemented and unsupplemented preterm human milk. Am. J. Clin. Nutr. 1990, 52, 254–262. [Google Scholar] [PubMed]
- Clark, R.; Thomas, P.; Peabody, J. Extrauterine Growth Restriction Remains a Serious Problem in Prematurely Born Neonates. Pediatrics 2003, 111, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Rochow, N.; Fusch, G.; Choi, A.; Chessell, L.; Elliott, L.; McDonald, K.; Kuiper, E.; Purcha, M.; Turner, S.; Chan, E.; et al. Target Fortification of Breast Milk with Fat, Protein, and Carbohydrates for Preterm Infants. J. Pediatr. 2013, 163, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Fusch, G.; Rochow, N.; Choi, A.; Fusch, S.; Poeschl, S.; Ubah, A.; Lee, S.Y.; Raja, P.; Fusch, C. Rapid measurement of macronutrients in breast milk: How reliable are infrared milk analyzers? Clin. Nutr. 2014, 34, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Gartner, L.M.; Morton, J.; Lawrence, R.A.; Naylor, A.J.; O’Hare, D.; Schanler, R.J.; Eidelman, A.I. American Academy of Pediatrics Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2005, 115, 496–506. [Google Scholar] [PubMed]
- Vieira, A.A.; Soares, F.V.; Pimenta, H.P.; Abranches, A.D.; Moreira, M.E. Analysis of the influence of pasteurization, freezing/thawing, and offer processes on human milk’s macronutrient concentrations. Early Hum. Dev. 2011, 87, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Lepri, L.; Del Bubba, M.; Maggini, R.; Donzelli, G.; Galvan, P. Effect of pasteurization and storage on some components of pooled human milk. J. Chromatogr. B 1997, 704, 1–10. [Google Scholar] [CrossRef]
- Fidler, N.; Sauerwald, T.; Koletzko, B.; Demmelmair, H. Effects of Human Milk Pasteurization and Sterilization on Available Fat Content and Fatty Acid Composition. J. Pediatr. Gastroenterol. Nutr. 1998, 27, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Wardell, J.; Hill, C.; D’Souza, S. Effect of Pasteurization and of Freezing and Thawing Human Milk on its Triglyceride Content. Acta Paediatr. 1981, 70, 467–471. [Google Scholar] [CrossRef]
- Henderson, T.; Fay, T.; Hamosh, M. Effect of pasteurization on long chain polyunsaturated fatty acid levels and enzyme activities of human milk. J. Pediatr. 1998, 132, 876–878. [Google Scholar] [CrossRef]
- Choi, A.; Fusch, G.; Rochow, N.; Sheikh, N.; Fusch, C. Establishment of micromethods for macronutrient contents analysis in breast milk. Mater. Child Nutr. 2013, 34, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Fusch, G.; Choi, A.; Rochow, N.; Fusch, C. Quantification of lactose content in human and cow’s milk using UPLC-tandem mass spectrometry. J. Chromatog. B 2011, 879, 3759–3762. [Google Scholar] [CrossRef] [PubMed]
- Krouwer, J.S. Why Bland-Altman plots should use X, not (Y+X)/2 when X is a reference method. Stat. Med. 2008, 27, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Sauer, C.W.; Kim, J.H. Human milk macronutrient analysis using point-of-care near-infrared spectrophotometry. J. Perinatol. 2011, 31, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, J.T.; Gho, D.S.; Mirmiran, M.; German, J.B.; Underwood, M.A. Rapid measurement of human milk macronutrients in the neonatal intensive care unit: Accuracy and precision of Fourier transform mid-infrared spectroscopy. J. Hum. Lact. 2014, 30, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Billard, H.; Simon, L.; Desnots, E.; Sochard, A.; Boscher, C.; Riaublanc, A.; Alexandre-Gouabau, M.C.; Boquien, C.Y. Calibration Adjustment of the Mid-infrared Analyzer for an Accurate Determination of the Macronutrient Composition of Human Milk. J. Hum. Lact. 2015. [Google Scholar] [CrossRef] [PubMed]
- Menjo, A.; Mizurno, K.; Murase, M.; Nishida, Y.; Taki, M.; Itabashi, K.; Shimono, T.; Namba, K. Bedside analysis of human milk for adjustable nutrition strategy. Acta Pediatr. 2009, 98, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Casadio, Y.S.; Williams, T.M.; Lai, C.T.; Olsson, S.E.; Hepworth, A.R.; Harmann, P.E. Evaluation of a mid-infrared analyzer for the determination of the macronutrient composition of human milk. J. Hum. Lact. 2010, 26, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, D.; Fraga, M.; Gormaz, M.; Torres, E.; Vento, M. Comparison of mid-infrared transmission spectroscopy with biochemical methods for the determination of macronutrients in human milk. Mater. Child Nutr. 2012, 10, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Corvaglia, L.; Battistini, B.; Paoletti, V.; Aceti, A.; Capretti, M.G.; Faldella, G. Near-infrared reflectance analysis to evaluate the nitrogen and fat content of human milk in neonatal intensive care units. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F372–F375. [Google Scholar] [CrossRef] [PubMed]
- ISO 22662:2007 (IDF 198: 2007). Milk and Milk Products—Determination of Lactose Content by High-Performance Liquid Chromatography (Reference Method). Available online: http://www.iso.org/iso/iso_catalogue/catalogue_tc/catalogue_detail.htm?csnumber=36384 (accessed on 1 December 2015).
- Espinosa-Martos, I.; Montilla, A.; Segura, A.; Escuder, D.; Bustos, G.; Pallás, C.; Rodríguez, J.M.; Corzo, N.; Fernández, L. Bacteriological, Biochemical, and Immunological Modifications in Human Colostrum after Holder Pasteurisation. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Valentine, C.; Morrow, G.; Fernandez, S.; Gulati, P.; Bartholomew, D.; Long, D.; Welty, S.E.; Morrow, A.L.; Rogers, L.K. Docosahexaenoic Acid and Amino Acid Contents in Pasteurized Donor Milk are Low for Preterm Infants. J. Pediatr. 2010, 157, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.; Quigley, M.; Brocklehurst, P. Donor breast milk versus infant formula for preterm infants: Systematic review and meta-analysis. Arch. Dis. Child. Fetal Neonatal Ed. 2007, 92, F169–F175. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, K.; Rechtman, D.; Lee, M.; Montoya, A.; Medo, E. Macronutrient Analysis of a Nationwide Sample of Donor Breast Milk. J. Am. Diet. Assoc. 2009, 109, 137–140. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotrri, G.; Fusch, G.; Kwan, C.; Choi, D.; Choi, A.; Al Kafi, N.; Rochow, N.; Fusch, C. Validation of Correction Algorithms for Near-IR Analysis of Human Milk in an Independent Sample Set—Effect of Pasteurization. Nutrients 2016, 8, 119. https://doi.org/10.3390/nu8030119
Kotrri G, Fusch G, Kwan C, Choi D, Choi A, Al Kafi N, Rochow N, Fusch C. Validation of Correction Algorithms for Near-IR Analysis of Human Milk in an Independent Sample Set—Effect of Pasteurization. Nutrients. 2016; 8(3):119. https://doi.org/10.3390/nu8030119
Chicago/Turabian StyleKotrri, Gynter, Gerhard Fusch, Celia Kwan, Dasol Choi, Arum Choi, Nisreen Al Kafi, Niels Rochow, and Christoph Fusch. 2016. "Validation of Correction Algorithms for Near-IR Analysis of Human Milk in an Independent Sample Set—Effect of Pasteurization" Nutrients 8, no. 3: 119. https://doi.org/10.3390/nu8030119
APA StyleKotrri, G., Fusch, G., Kwan, C., Choi, D., Choi, A., Al Kafi, N., Rochow, N., & Fusch, C. (2016). Validation of Correction Algorithms for Near-IR Analysis of Human Milk in an Independent Sample Set—Effect of Pasteurization. Nutrients, 8(3), 119. https://doi.org/10.3390/nu8030119