Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline

Abstract
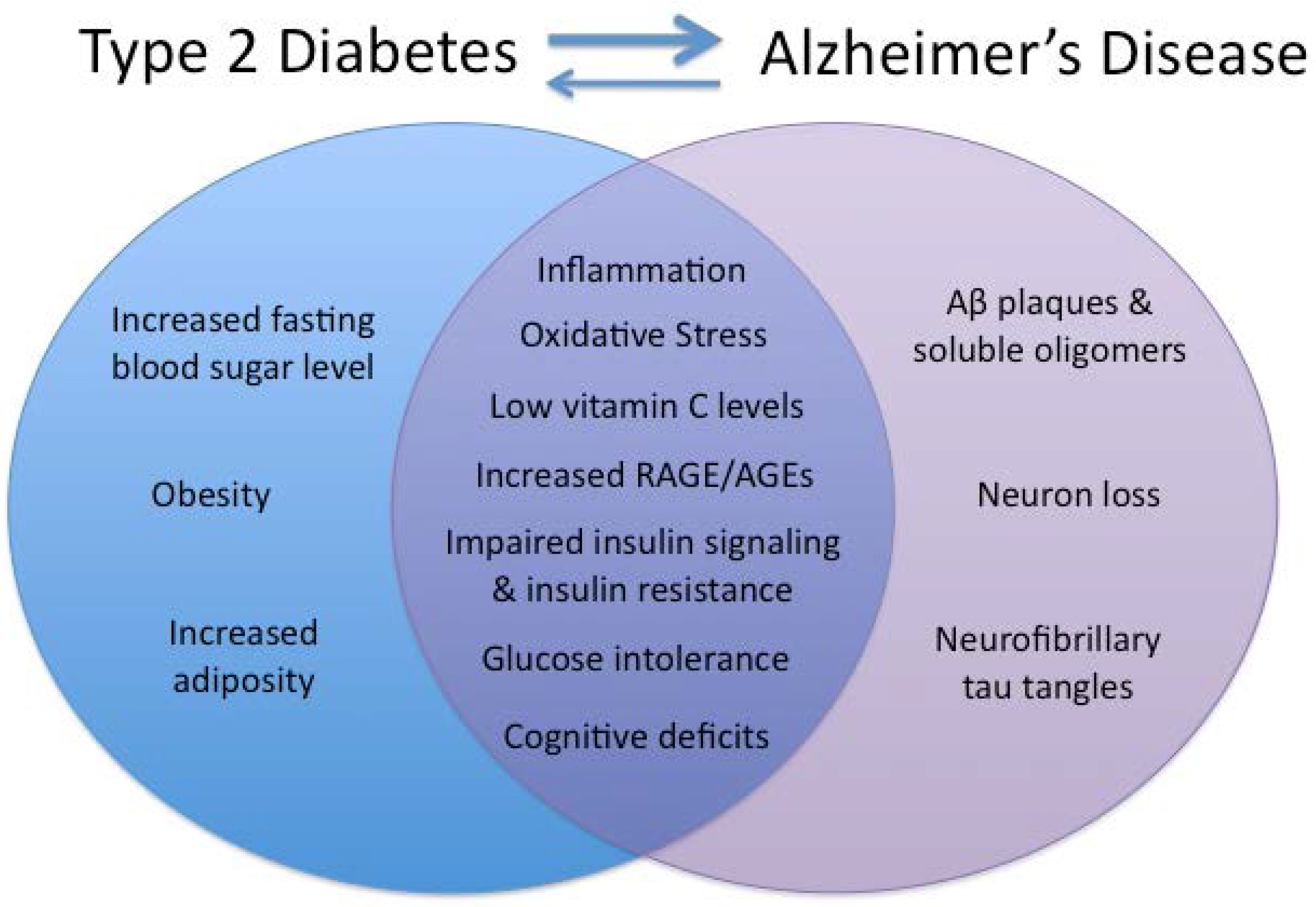
:1. Introduction

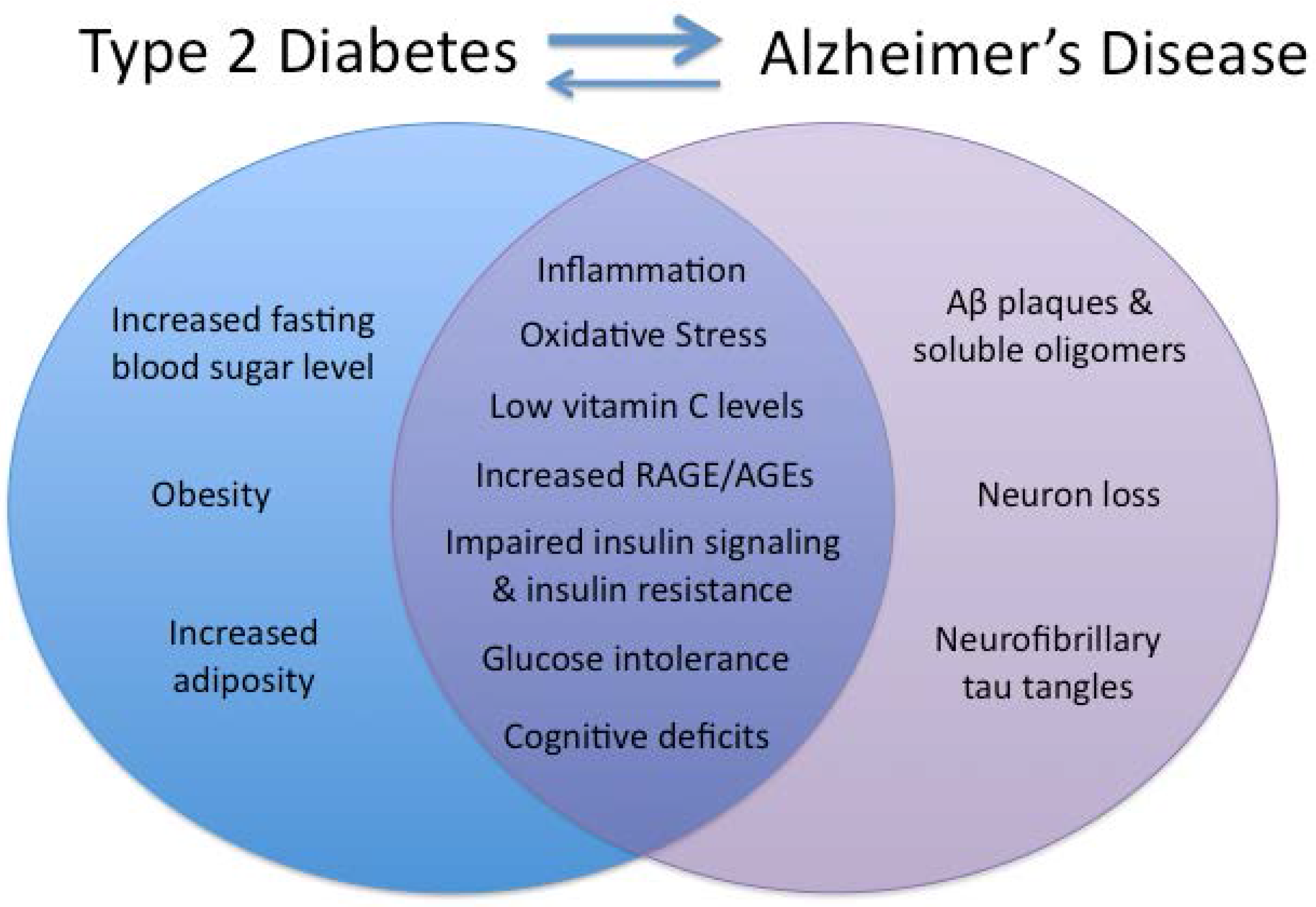{kind=link}
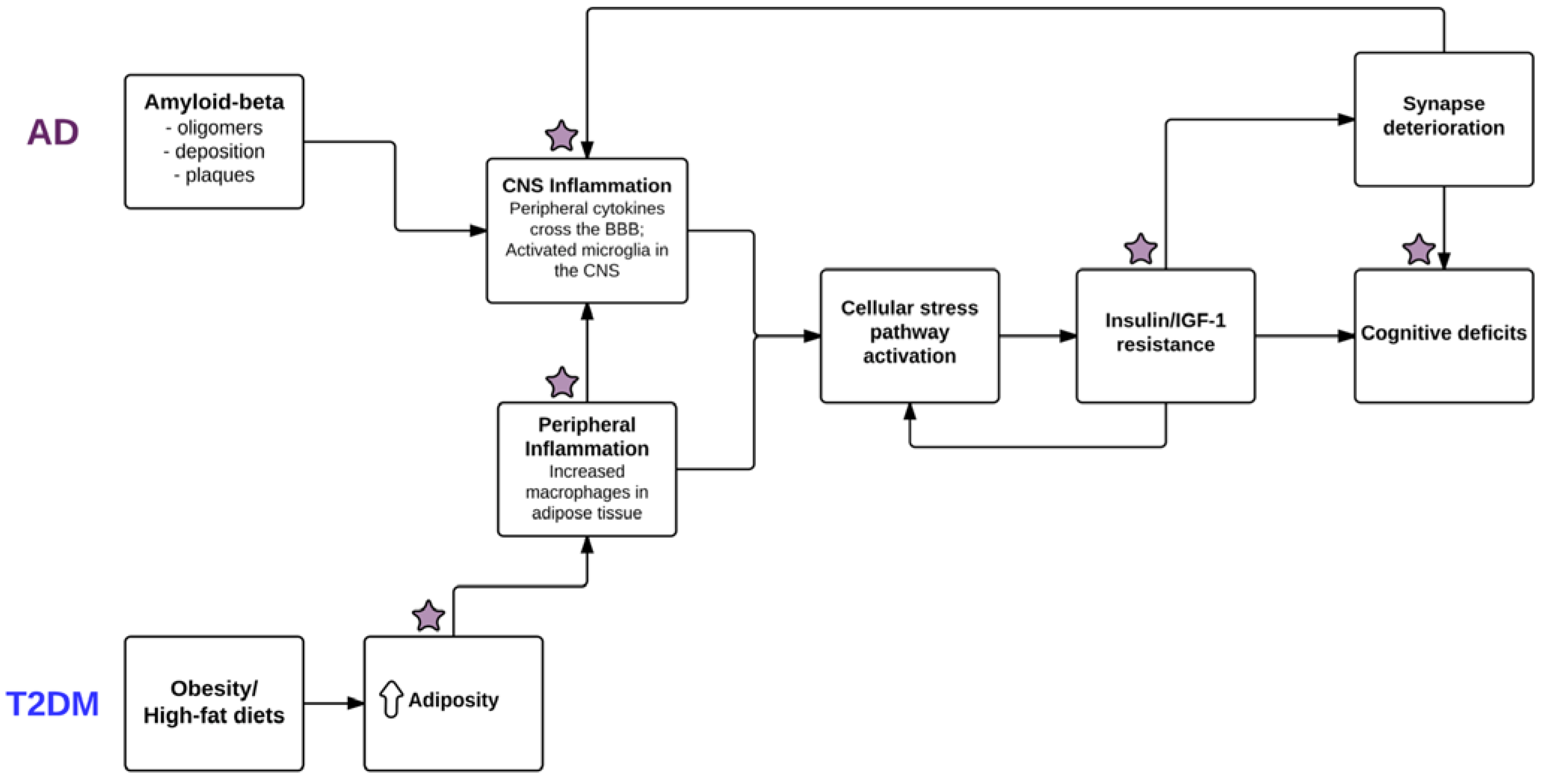{kind=link}
| Abbreviations | ||
|---|---|---|
| AD—Alzheimer’s disease | HFD—High fat diet | LRP-1—Low-density lipoprotein receptor-related protein 1 |
| AGEs—Advanced glycation end-products | IDE—Insulin degrading enzyme | MCP-1—Monocyte chemotactic protein 1 |
| APP—Amyloid precursor protein | IGF-1—Insulin growth factor 1 | MRI—Magnetic resonance imaging |
| BBB—Blood-brain barrier | IHC—Immunohistochemistry | MWM—Morris water-maze |
| BDNF—Brain-derived neurotrophic factor | IKK—I κ-B kinase | NMDA—N-Methyl-D-aspartate |
| CNS – Central nervous system | IL—Interleukin | PI3K—Phosphoinositide 3 kinase |
| CSF—Cerebrospinal fluid | INF-γ—Interferon γ | PKR—Protein kinase RNA-activated |
| EE—Environmental enrichment | i.p. —intraperitoneal | PS1—Presenilin 1 |
| EGCG—(-)-epigallocatechin-3-gallate | IR—Insulin resistance | RAGE—Receptor for advanced glycation end-products |
| GFAP—Glial fibrillary acidic protein | IRS—Insulin receptor substrate | T2DM—Type 2 diabetes mellitus |
| GLP-1—Glucagon-like peptide 1 | IV—Intravenous | TNF—Tumor necrosis factor |
| GLUT4—Glucose transporter 4 | JNK—c-Jun NH2-terminal kinase | WB—Western blotting |
| GTT—Glucose tolerance test | LFD—Low fat diet | |
2. Insulin and Insulin Resistance

3. Inflammation Is Related to Impaired Insulin Signaling in the Periphery and the CNS
4. High-Fat Diet Feeding, Obesity, and T2DM Can Induce Cognitive Impairments in Non-AD Humans and Animals, and Is Associated with Changes in the Brain
5. Mouse Models of AD Exhibit Greater Sensitivity to Obesity Phenotype and High-Fat Diet
Limitations of the Current Studies
6. Inflammation Is Related to Cognitive Deficits
6.1. Anti-Inflammatory and Pro-Cognitive Effects of Diet Reversal
6.2. Limitations of the Previous Studies
7. Discussion
Limitations of the Current Evidence Linking T2DM with AD
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.B.; Hayes, L.D.; Brown, K.; Hoo, E.C.; Ethier, K.A. CDC national health report: Leading causes of morbidity and mortality and associated behavioral risk and protective factors—United States, 2005–2013. MMWR Surveill. Summ. 2014, 63 (Suppl 4), 3–27. [Google Scholar] [PubMed]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based european prospective investigation into cancer and nutrition (EPIC)-potsdam study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Chandalia, M.; Abate, N. Metabolic complications of obesity: Inflated or inflamed? J. Diabetes Complic. 2007, 21, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Brands, A.M.; Van den Berg, E.; Manschot, S.M.; Biessels, G.J.; Kappelle, L.J.; De Haan, E.H.; Kessels, R.P. A detailed profile of cognitive dysfunction and its relation to psychological distress in patients with type 2 diabetes mellitus. J. Int. Neuropsychol. Soc. 2007, 13, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Awad, N.; Gagnon, M.; Messier, C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J. Clin. Exp. Neuropsychol. 2004, 26, 1044–1080. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Wilson, R.S. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 2004, 62, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.M.; Requejo, A.M.; Andres, P.; Lopez-Sobaler, A.M.; Quintas, M.E.; Redondo, M.R.; Navia, B.; Rivas, T. Dietary intake and cognitive function in a group of elderly people. Am. J. Clin. Nutr. 1997, 66, 803–809. [Google Scholar] [PubMed]
- Prickett, C.; Brennan, L.; Stolwyk, R. Examining the relationship between obesity and cognitive function: A systematic literature review. Obes. Res. Clin. Pract. 2015, 9, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.H.; Scholey, A.; Howe, P.R. Assessing premorbid cognitive ability in adults with type 2 diabetes mellitus—A review with implications for future intervention studies. Curr. Diabetes Rep. 2014, 14, 547. [Google Scholar] [CrossRef] [PubMed]
- Winocur, G.; Greenwood, C.E. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol. Aging 2005, 26 (Suppl 1), 46–49. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Wang, Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: A systematic review and meta-analysis. Obes. Rev. 2008, 9, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam study. Ann. Neurol. 1997, 42, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V. Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Dzwolak, W.; Winter, R. Amyloidogenic self-assembly of insulin aggregates probed by high resolution atomic force microscopy. Biophys. J. 2005, 88, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Holscher, C. The effect of ageing on neurogenesis and oxidative stress in the Appswe/PS1 deltaE9 mouse model of Alzheimer’s disease. Brain Res. 2012, 1449, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer disease and oxidative stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Stampfer-Kountchev, M.; Robatscher, P.; Veerhuis, R.; Eikelenboom, P.; Grubeck-Loebenstein, B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell. 2004, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged App SW transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef]
- Bales, K.R.; Du, Y.; Dodel, R.C.; Yan, G.M.; Hamilton-Byrd, E.; Paul, S.M. The NF-kappaB/Rel family of proteins mediates abeta-induced neurotoxicity and glial activation. Brain Res. Mol. Brain Res. 1998, 57, 63–72. [Google Scholar] [CrossRef]
- Aisen, P.S.; Davis, K.L. Inflammatory mechanisms in Alzheimer’s disease: Implications for therapy. Am. J. Psychiatry 1994, 151, 1105–1113. [Google Scholar] [PubMed]
- Folstein, M.F.; Whitehouse, P.J. Cognitive impairment of Alzheimer disease. Neurobehav. Toxicol. Teratol. 1983, 5, 631–634. [Google Scholar] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the national institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Resende, R.; Ferreiro, E.; Rego, A.C.; Pereira, C.F. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 2010, 11, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Haley, A.P.; Knight-Scott, J.; Simnad, V.I.; Manning, C.A. Increased glucose concentration in the hippocampus in early Alzheimer’s disease following oral glucose ingestion. J. Magn. Reson. Imaging 2006, 24, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Barberger-Gateau, P.; Letenneur, L.; Deschamps, V.; Peres, K.; Dartigues, J.F.; Renaud, S. Fish, meat, and risk of Dementia: Cohort study. BMJ 2002, 325, 932–933. [Google Scholar] [CrossRef] [PubMed]
- Kalmijn, S.; Feskens, E.J.; Launer, L.J.; Kromhout, D. Polyunsaturated fatty acids, antioxidants, and cognitive function in very old men. Am. J. Epidemiol. 1997, 145, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef] [PubMed]
- Devore, E.E.; Grodstein, F.; van Rooij, F.J.; Hofman, A.; Stampfer, M.J.; Witteman, J.C.; Breteler, M.M. Dietary antioxidants and long-term risk of Dementia. Arch. Neurol. 2010, 67, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B.; Sandoval, J. Amelioration of early cognitive deficits by aged garlic extract in Alzheimer’s transgenic mice. Phytother. Res. 2007, 21, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Chauhan, N.B.; Lahiri, D.K. Oxidative insults to neurons and synapse are prevented by aged garlic extract and S-Allyl-l-cysteine treatment in the neuronal culture and App-Tg mouse model. J. Neurochem. 2011, 117, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Su, C.; Li, R.; Wang, H.; Ren, Y.; Sun, H.; Yang, J.; Sun, J.; Shi, J.; Tian, J.; et al. Mechanisms and effects of curcumin on spatial learning and memory improvement in Appswe/PS1dE9 mice. J. Neurosci. Res. 2014, 92, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Klakotskaia, D.; Ajit, D.; Weisman, G.A.; Wood, W.G.; Sun, G.Y.; Serfozo, P.; Simonyi, A.; Schachtman, T.R. Beneficial effects of dietary EGCG and voluntary exercise on behavior in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014, 44. [Google Scholar] [CrossRef]
- Harrison, F.E.; Allard, J.; Bixler, R.; Usoh, C.; Li, L.; May, J.M.; McDonald, M.P. Antioxidants and cognitive training interact to affect oxidative stress and memory in APP/PSEN1 mice. Nutr. Neurosci. 2009, 12, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, Z.; Wang, R.; Zhang, X.; Zhang, J.; Dong, W.; Xu, B. Caloric restriction can improve learning ability in C57/Bl mice via regulation of the insulin-PI3K/Akt signaling pathway. Neurol. Sci. 2014, 35, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Mouton, P.R.; Chachich, M.E.; Quigley, C.; Spangler, E.; Ingram, D.K. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci. Lett. 2009, 464, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Halagappa, V.K.; Guo, Z.; Pearson, M.; Matsuoka, Y.; Cutler, R.G.; Laferla, F.M.; Mattson, M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2007, 26, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ho, L.; Qin, W.; Rocher, A.B.; Seror, I.; Humala, N.; Maniar, K.; Dolios, G.; Wang, R.; Hof, P.R.; et al. Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2005, 19, 659–661. [Google Scholar] [PubMed]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; et al. Prevalence of Dementia in the United States: The Aging, Demographics, and Memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Battistin, L.; Cagnin, A. Vascular cognitive disorder. A biological and clinical overview. Neurochem. Res. 2010, 35, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, M.; Vieweg, W.V. Possible role of vascular risk factors in Alzheimer’s disease and vascular Dementia. Curr. Pharm. Des. 2014, 20, 6007–6013. [Google Scholar] [CrossRef] [PubMed]
- Steffens, D.C.; Potter, G.G.; McQuoid, D.R.; MacFall, J.R.; Payne, M.E.; Burke, J.R.; Plassman, B.L.; Welsh-Bohmer, K.A. Longitudinal magnetic resonance imaging vascular changes, apolipoprotein E genotype, and development of Dementia in the neurocognitive outcomes of depression in the elderly study. Am. J. Geriatr. Psychiatry 2007, 15, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Wynne, K.; Stanley, S.; McGowan, B.; Bloom, S. Appetite control. J. Endocrinol. 2005, 184, 291–318. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.L.; King, M.G.; Baskin, D.G. Localization of insulin and type 1 IGF receptors in rat brain by in vitro autoradiography and in situ hybridization. Adv. Exp. Med. Biol. 1991, 293, 459–470. [Google Scholar] [PubMed]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Cardona-Gomez, G.P.; Mendez, P.; DonCarlos, L.L.; Azcoitia, I.; Garcia-Segura, L.M. Interactions of estrogens and insulin-like growth factor-I in the brain: Implications for neuroprotection. Brain Res. Brain Res. Rev. 2001, 37, 320–334. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Sanchez-Perez, A.; Trejo, J.L.; Martin-Aldana, J.A.; Cano Jaimez, M.; Pons, S.; Umanzor, C.A.; Menes, L.; White, M.F.; Burks, D.J. IRS-2 deficiency impairs NMDA receptor-dependent long-term potentiation. Cereb. Cortex 2012, 22, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B. Epidemiology of the insulin resistance syndrome. Curr. Diabetes Rep. 2003, 3, 73–79. [Google Scholar] [CrossRef]
- Stein, L.J.; Dorsa, D.M.; Baskin, D.G.; Figlewicz, D.P.; Porte, D., Jr.; Woods, S.C. Reduced effect of experimental peripheral hyperinsulinemia to elevate cerebrospinal fluid insulin concentrations of obese zucker rats. Endocrinology 1987, 121, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Brooks, S.J.; Kullberg, J.; Burgos, J.; Kempton, M.J.; Nordenskjold, R.; Nylander, R.; Kilander, L.; Craft, S.; Larsson, E.M.; et al. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care 2012, 35, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, H.; Sweat, V.; Hassenstab, J.; Polyakov, V.; Convit, A. Cognitive impairment in nondiabetic middle-aged and older adults is associated with insulin resistance. J. Clin. Exp. Neuropsychol. 2010, 32, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases CSF Aβ 42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Insulin resistance syndrome and Alzheimer’s disease: Age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol. Aging 2005, 26 (Suppl 1), 65–69. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.A.; Watson, G.S.; Montine, T.J.; Wang, Q.; Green, P.S.; Kulstad, J.J.; Cook, D.G.; Peskind, E.R.; Baker, L.D.; Goldgaber, D.; et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch. Neurol. 2005, 62, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Zhang, F.; Wu, J.; Hu, Y.; Liu, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance by activating JAK2/STAT3/SOCS-1 signaling pathway. Diabetes 2012, 61, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Intrahippocampal administration of amyloid-β (1–42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J. Alzheimers Dis. 2012, 30, 413–422. [Google Scholar] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J. Clin. Invest. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H.; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [PubMed]
- Holscher, C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014, 10, S47–S54. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Holscher, C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 2014, 86, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, Y.; Dai, C.L.; Liang, Z.; Run, X.; Iqbal, K.; Liu, F.; Gong, C.X. Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Aβ level and microglia activation in the brains of 3xTg-AD mice. Exp. Neurol. 2014, 261, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; O’Banion, M.K. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007, 184, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased blood-brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol. Aging 2013, 34, 2064–2070. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Bednar, I.; Mix, E.; Marie, S.; Paterson, D.; Ljungberg, A.; Morris, C.; Winblad, B.; Nordberg, A.; Zhu, J. Up-regulation of the inflammatory cytokines IFN-γ and IL-12 and down-regulation of IL-4 in cerebral cortex regions of Appswe transgenic mice. J. Neuroimmunol. 2002, 126, 50–57. [Google Scholar] [CrossRef]
- Zhao, W.Q.; de Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid β oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L. Treatment of Alzheimer’s disease: Current and future therapeutic approaches. Rev. Neurol. Dis. 2004, 1, 60–69. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Tsang, V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010, 24, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Winocur, G.; Greenwood, C.E.; Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Reagan, L.P.; McEwen, B.S. Memory impairment in obese zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 2005, 119, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Molteni, R.; Barnard, R.J.; Ying, Z.; Roberts, C.K.; Gomez-Pinilla, F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 2002, 112, 803–814. [Google Scholar] [CrossRef]
- Pannacciulli, N.; del Parigi, A.; Chen, K.; Le, D.S.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.F.; Elias, P.K.; Sullivan, L.M.; Wolf, P.A.; D’Agostino, R.B. Obesity, diabetes and cognitive deficit: The framingham heart study. Neurobiol. Aging 2005, 26 (Suppl 1), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Waldstein, S.R.; Katzel, L.I. Interactive relations of central versus total obesity and blood pressure to cognitive function. Int. J. Obes. (Lond) 2006, 30, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Dahl, A.K.; Hassing, L.B. Obesity and cognitive aging. Epidemiol. Rev. 2013, 35, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Fors, N.; Hall, J.; Marttala, K.; Stenborg, A. A comparison of three different methods to evaluate endothelium-dependent vasodilation in the elderly: The prospective investigation of the vasculature in uppsala seniors (PIVUS) study. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, H.; Wolf, O.T.; Sweat, V.; Tirsi, A.; Richardson, S.; Convit, A. Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. 2009, 1280, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Reijmer, Y.D.; van den Berg, E.; de Bresser, J.; Kessels, R.P.; Kappelle, L.J.; Algra, A.; Biessels, G.J. Accelerated cognitive decline in patients with type 2 diabetes: MRI correlates and risk factors. Diabetes Metab. Res. Rev. 2011, 27, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Reijmer, Y.D.; Leemans, A.; Brundel, M.; Kappelle, L.J.; Biessels, G.J. Disruption of the cerebral white matter network is related to slowing of information processing speed in patients with type 2 diabetes. Diabetes 2013, 62, 2112–2115. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Reijmer, Y.D. Brain changes underlying cognitive dysfunction in diabetes: What can we learn from MRI? Diabetes 2014, 63, 2244–2252. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Ratovitski, T.; van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef]
- Mody, N.; Agouni, A.; McIlroy, G.D.; Platt, B.; Delibegovic, M. Susceptibility to diet-induced obesity and glucose intolerance in the Appswe/PSEN1 (A246E) mouse model of Alzheimer’s disease is associated with increased brain levels of protein tyrosine phosphatase 1b (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia 2011, 54, 2143–2151. [Google Scholar] [PubMed]
- Dixit, S.; Bernardo, A.; Walker, J.M.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in App/PSEN1 and normally aging mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Reiserer, R.S.; Harrison, F.E.; Syverud, D.C.; McDonald, M.P. Impaired spatial learning in the Appswe + PSEN1DELTAE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav. 2007, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; D’Hooge, R.; Staufenbiel, M.; van Ginneken, C.; van Meir, F.; de Deyn, P.P. Age-dependent cognitive decline in the App23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Niedowicz, D.M.; Reeves, V.L.; Platt, T.L.; Kohler, K.; Beckett, T.L.; Powell, D.K.; Lee, T.L.; Sexton, T.R.; Song, E.S.; Brewer, L.D.; et al. Obesity and diabetes cause cognitive dysfunction in the absence of accelerated β-amyloid deposition in a novel murine model of mixed or vascular Dementia. Acta Neuropathol. Commun. 2014, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Tou, J.C.; Wade, C.E. Determinants affecting physical activity levels in animal models. Exp. Biol. Med. (Maywood) 2002, 227, 587–600. [Google Scholar] [PubMed]
- Peng, Y.; Liu, J.; Tang, Y.; Han, T.; Han, S.; Li, H.; Hou, C.; Long, J. High-fat-diet-induced weight gain ameliorates bone loss without exacerbating AβPP processing and cognition in female App/PS1 mice. Front. Cell. Neurosci. 2014, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Buckman, L.B.; Thompson, M.M.; Moreno, H.N.; Ellacott, K.L. Regional astrogliosis in the mouse hypothalamus in response to obesity. J. Comp. Neurol. 2013, 521, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Rosi, S.; Pert, C.B.; Ruff, M.R.; McGann-Gramling, K.; Wenk, G.L. Chemokine receptor 5 antagonist d-Ala-peptide T-amide reduces microglia and astrocyte activation within the hippocampus in a neuroinflammatory rat model of Alzheimer’s disease. Neuroscience 2005, 134, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.H.; Aid, S.; Zhang, Y.; Becker, K.G.; Bosetti, F. The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccharide in mice. Pharmacogen. J. 2009, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.Y.; Martin, R.G.; Duncan, R.E.; Choi, D.; Lu, S.Y.; Schroer, S.A.; Cai, E.P.; Luk, C.T.; Hopperton, K.E.; Domenichiello, A.F.; et al. Hepatocyte-specific deletion of Janus kinase 2 (JAK2) protects against diet-induced steatohepatitis and glucose intolerance. J. Biol. Chem. 2012, 287, 10277–10288. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Li, Y.; Ma, G.; Wang, Y.; Cai, Y.; Liu, S.; Chen, Y.; Li, J.; Xie, Y.; Liu, G.; et al. A functional polymorphism in the promoter region of microRNA-146a is associated with the risk of Alzheimer disease and the rate of cognitive decline in patients. PLoS ONE 2014, 9, e89019. [Google Scholar] [CrossRef] [PubMed]
- Barber, R. Inflammatory signaling in Alzheimer disease and depression. Cleve Clin. J. Med. 2011, 78 (Suppl 1), S47–S49. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, J.A.; Larson, E.B.; Brickell, K.; Crane, P.K.; Woltjer, R.; Montine, T.J.; Craft, S. Different patterns of cerebral injury in Dementia with or without diabetes. Arch. Neurol. 2009, 66, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Brachova, L.; Civin, W.H.; Rogers, J. Inflammation, a β deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol. 1996, 55, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Inzana, J.A.; Kung, M.; Shu, L.; Hamada, D.; Xing, L.P.; Zuscik, M.J.; Awad, H.A.; Mooney, R.A. Immature mice are more susceptible to the detrimental effects of high fat diet on cancellous bone in the distal femur. Bone 2013, 57, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.Y.; Ko, H.J.; Lichtman, E.I.; Lee, E.; Lawton, E.; Ong, H.; Yu, K.; Azuma, Y.; Friedline, R.H.; Lee, K.W.; et al. Short-term weight loss attenuates local tissue inflammation and improves insulin sensitivity without affecting adipose inflammation in obese mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E964–E976. [Google Scholar] [CrossRef] [PubMed]
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Hayashida, N.; Asada-Utsugi, M.; Watanabe, K.; Uemura, M.; Kihara, T.; et al. Exercise is more effective than diet control in preventing high fat diet-induced β-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J. Biol. Chem. 2012, 287, 23024–23033. [Google Scholar] [CrossRef] [PubMed]
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Asada, M.; Watanabe, K.; Hayashida, N.; Ihara, M.; Ito, H.; et al. Environmental enrichment ameliorated high-fat diet-induced Aβ deposition and memory deficit in App transgenic mice. Neurobiol. Aging 2012, 33, 1011.e11–1011.e23. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Argente-Arizon, P.; Ros, P.; Argente, J.; Chowen, J.A. Sex differences in adipose tissue: It is not only a question of quantity and distribution. Adipocyte 2013, 2, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Medrikova, D.; Jilkova, Z.M.; Bardova, K.; Janovska, P.; Rossmeisl, M.; Kopecky, J. Sex differences during the course of diet-induced obesity in mice: Adipose tissue expandability and glycemic control. Int. J. Obes. (Lond) 2012, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Grove, K.L.; Fried, S.K.; Greenberg, A.S.; Xiao, X.Q.; Clegg, D.J. A microarray analysis of sexual dimorphism of adipose tissues in high-fat-diet-induced obese mice. Int. J. Obes. (Lond) 2010, 34, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Dishman, R.K.; Berthoud, H.R.; Booth, F.W.; Cotman, C.W.; Edgerton, V.R.; Fleshner, M.R.; Gandevia, S.C.; Gomez-Pinilla, F.; Greenwood, B.N.; Hillman, C.H.; et al. Neurobiology of exercise. Obesity (Silver Spring) 2006, 14, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.K.; Ross, J.M.; Harrison, F.E.; Bernardo, A.; Reiserer, R.S.; Mobley, J.A.; McDonald, M.P. Differential proteomic and behavioral effects of long-term voluntary exercise in wild-type and App-overexpressing transgenics. Neurobiol. Dis. 2015, 78, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.M.; McGarry, N.B.; Kelly, A.M. Short-term environmental enrichment, in the absence of exercise, improves memory, and increases NGF concentration, early neuronal survival, and synaptogenesis in the dentate gyrus in a time-dependent manner. Hippocampus 2013, 23, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Koutsouraki, E.S.; Gasiorowski, K.; Leszek, J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: A new approach to therapy. CNS Neurol. Disord. Drug Targets 2013, 12, 870–881. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332-7357. https://doi.org/10.3390/nu7095341
Walker JM, Harrison FE. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients. 2015; 7(9):7332-7357. https://doi.org/10.3390/nu7095341
Chicago/Turabian StyleWalker, Jennifer M., and Fiona E. Harrison. 2015. "Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline" Nutrients 7, no. 9: 7332-7357. https://doi.org/10.3390/nu7095341
APA StyleWalker, J. M., & Harrison, F. E. (2015). Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients, 7(9), 7332-7357. https://doi.org/10.3390/nu7095341