Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals



Abstract
:
1. Introduction
2. Role of Nrf2 as Antioxidant and Chemoprotective Regulator
2.1. Antioxidant
2.1.1. ARE-Responsive Enzymes Associated with Glutathione
2.1.2. Other ARE-Responsive Antioxidant Enzymes
2.1.3. Other Proteins Relevant to Redox, not Necessarily ARE-Responsive
2.2. Detoxification and Neutralization of Carcinogens
ARE-Responsive Phase 2 Enzymes
2.3. Synthesis and Regeneration of NADPH
3. Regulation of Nrf2
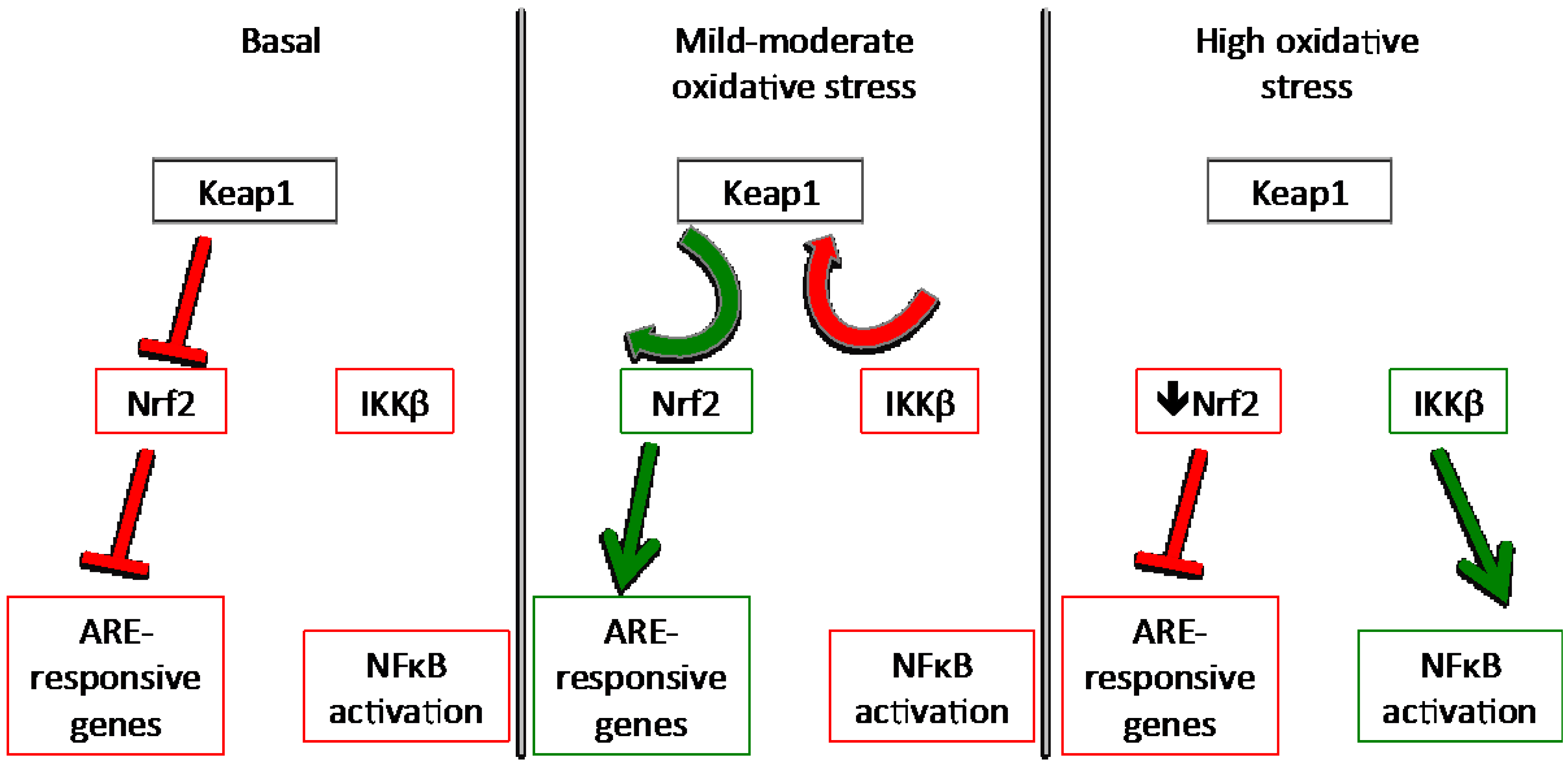
3.1. Keap1
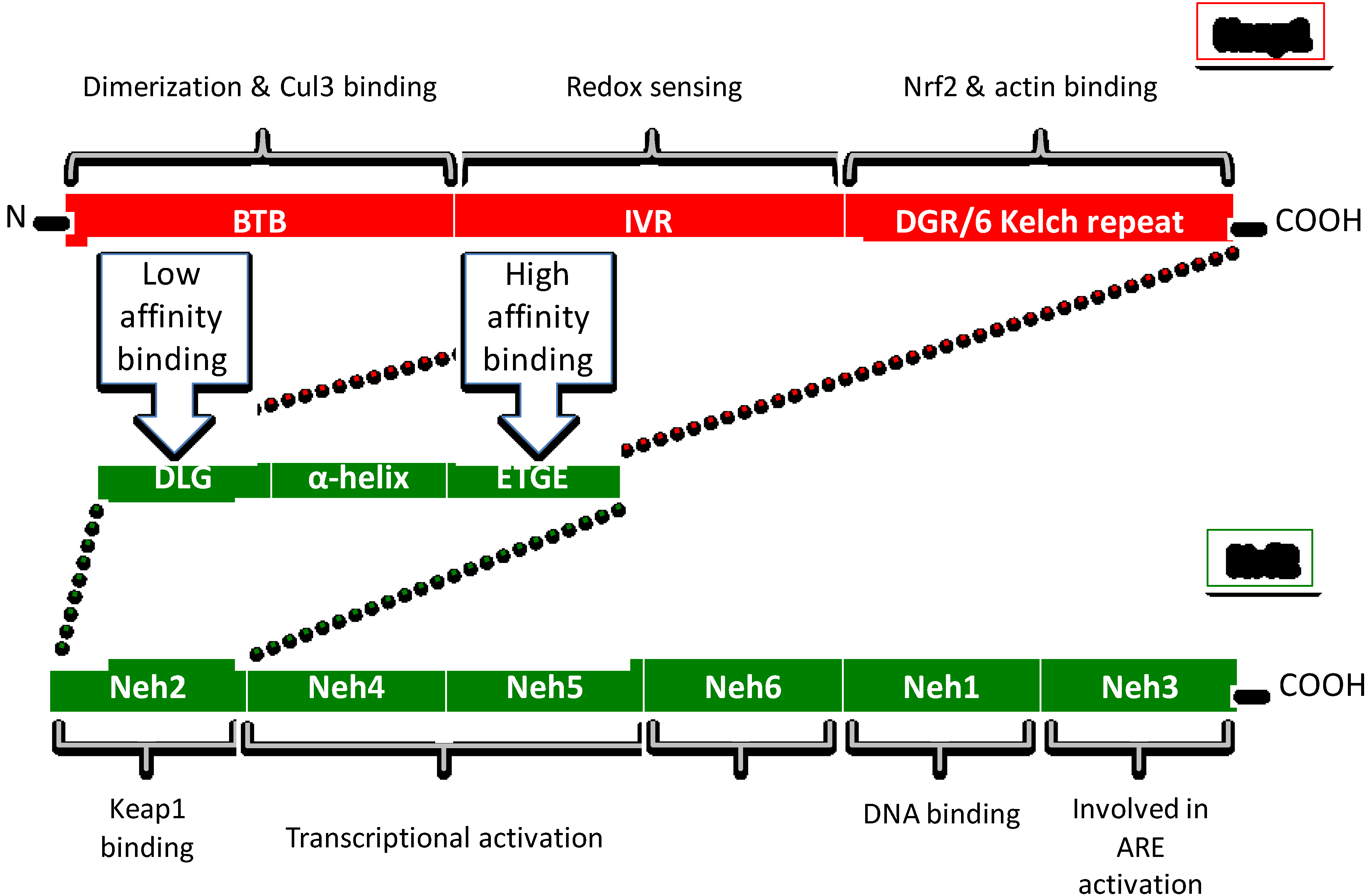
Redox Sensing Mechanisms of Keap1
3.2. Nrf2
3.3. NFκB
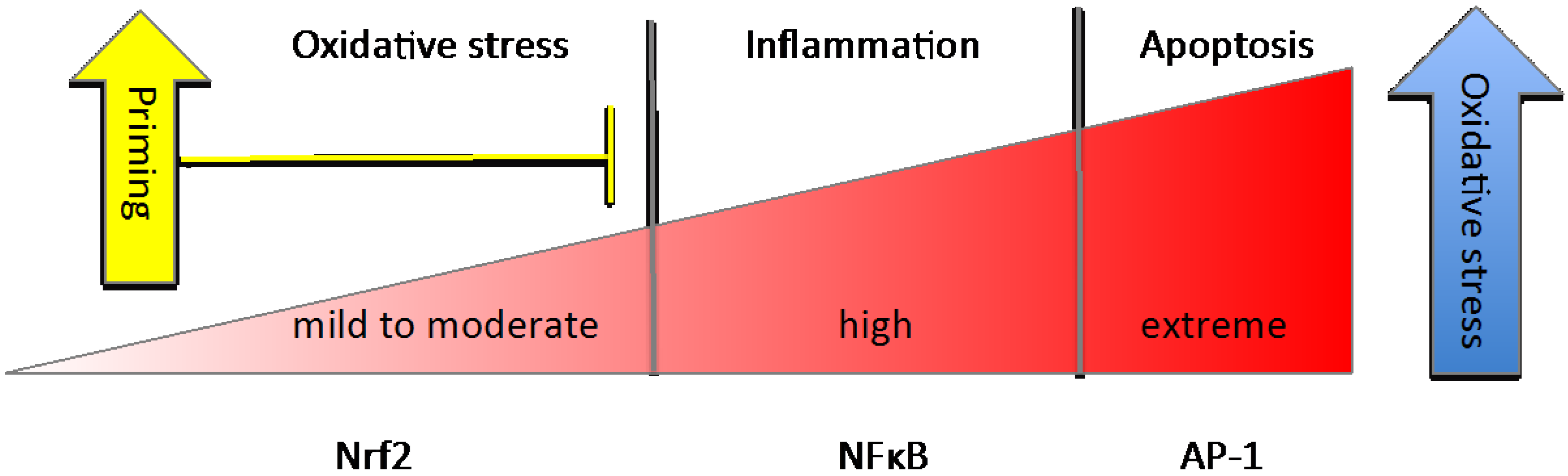
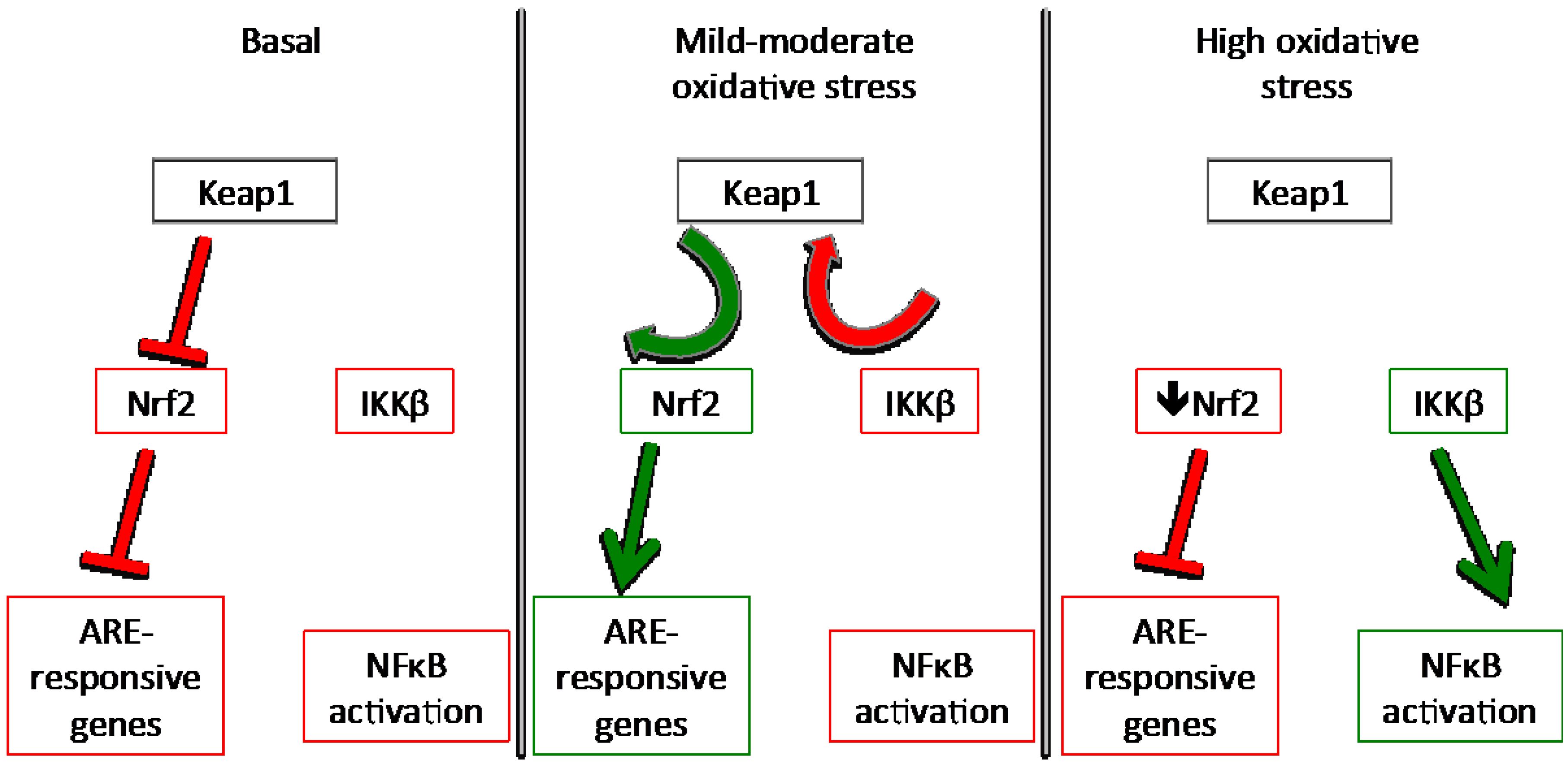
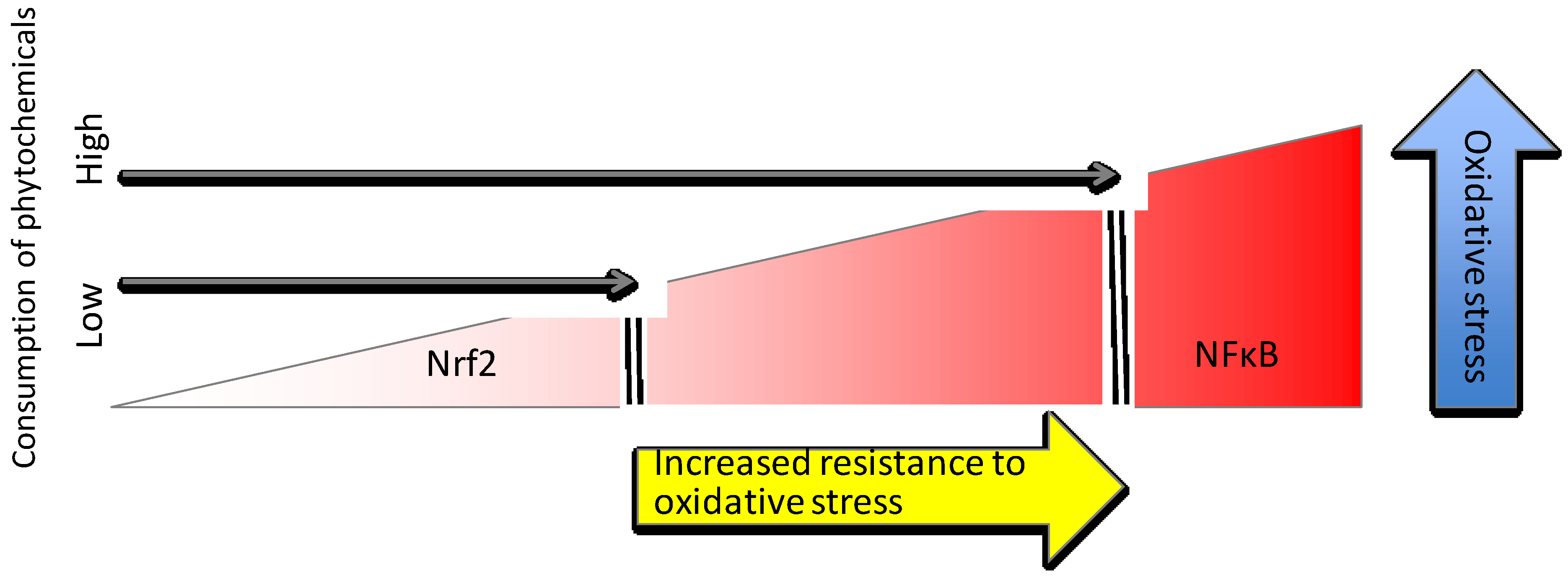
4. Nutrient Gene Interactions
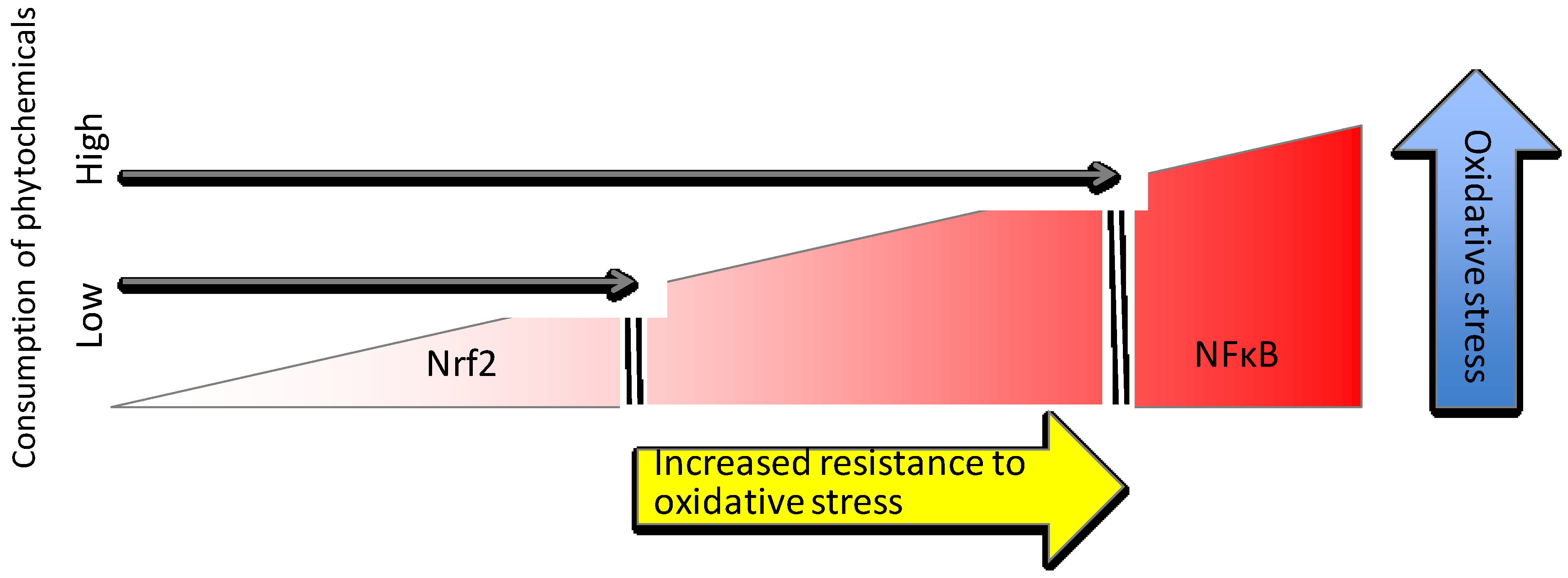
4.1. Phytochemical Activators of Nrf2
4.1.1. Mechanisms of Phytochemical Interaction with Keap1
4.1.2. Brassica Family Vegetables and Organosulfides
4.1.3. Other Fruit and Vegetable-Derived Compounds (Polyphenols, Carotenoids, Polyenes)
4.1.4. Herbs, Spices and Flavor Enhancers
4.2. Trace Minerals
4.2.1. Zinc
4.2.2. Selenium
5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
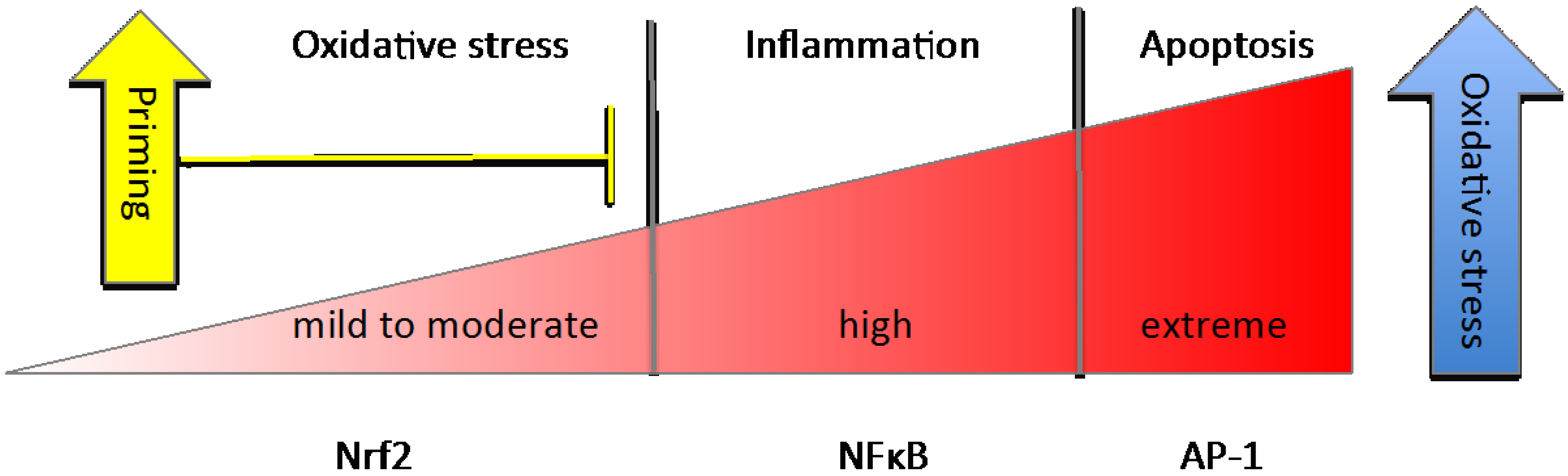
| Basal Conditions |
|
| Priming or Pre-Induction |
|
| Induction |
|
| Resolution or Next-Level Response |
|
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostaglandin. Other Lipid Mediators 2011, 96, 27–36. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-Deficient Mice Have an Increased Susceptibility to Dextran Sulfate Sodium-Induced Colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway: Identification of novel gene clusters for cell survival. J. Biol. Chem. 2002, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic Evidence that Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Nerland, D.E. The antioxidant/electrophile response element motif. Drug Metab. Rev. 2007, 39, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Shelly, C. Lu Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Jacob, C.; Giles, G.I.; Giles, N.M.; Sies, H. Sulfur and Selenium: The Role of Oxidation State in Protein Structure and Function. Angew. Chem. Int. Ed. 2003, 42, 4742–4758. [Google Scholar] [CrossRef]
- Matés, J.M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Krȩżel, A.; Maret, W. Dual Nanomolar and Picomolar Zn(II) Binding Properties of Metallothionein. J. Am. Chem. Soc. 2007, 129, 10911–10921. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 Play Distinct Roles in Activation of Antioxidant Response Element-dependent Genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.K.; Cook, J.L. Nrf2, a Cap‘n’Collar Transcription Factor, Regulates Induction of the Heme Oxygenase-1 Gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Kröncke, K.D.; Fehsel, K.; Schmidt, T.; Zenke, F.T.; Dasting, I.; Wesener, J.R.; Betterman, H.; Breunig, K.D.; Kolb-Bachofen, V. Nitric Oxide Destroys Zinc-Sulfur Clusters Inducing Zinc Release from Metallothionein and Inhibition of the Zinc Finger-Type Yeast Transcription Activator LAC9. Biochem. Biophys. Res. Commun. 1994, 200, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. USA 1994, 91, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Suschek, C.V.; Wetzel, W.; Kroncke, K.D.; Kolb-Bachofen, V. Nitric oxide-mediated protection of endothelial cells from hydrogen peroxide is mediated by intracellular zinc and glutathione. AJP Cell Physiol. 2009, 296, C811–C820. [Google Scholar] [CrossRef]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Krȩżel, A.; Maret, W. Thionein/metallothionein control Zn(II) availability and the activity of enzymes. J. Biol. Inorg. Chem. 2007, 13, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Hasse, H.; Rink, L. Zinc signals and immune function. BioFactors 2013, 40, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Vallee, B.L. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc. Natl. Acad. Sci. USA 1998, 95, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Maret, W.; Vallee, B.L. Selenium redox biochemistry of zinc—Sulfur coordination sites in proteins and enzymes. Proc. Natl. Acad. Sci. USA 1999, 96, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Jacob, C.; Vallee, B.L.; Fischer, E. Inhibitory sites in enzymes: Zinc removal and reactivation by thionein. Proc. Natl. Acad. Sci. USA 1999, 96, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; Dinkova-Kostova, A.T.; Holtzclaw, W.D. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv. Enzym. Regul. 2003, 43, 121–134. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Zhang, Q.-Y. The small intestine as a xenobiotic-metabolizing organ. Drug Metab. Dispos. 2003, 31, 1520–1524. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, M.G.; Lambert, G.H. Xenobiotic-induced hepatoxicity: Mechanisms of liver injury and methods of monitoring hepatic function. Clin. Chem. 1997, 43, 1512–1526. [Google Scholar] [PubMed]
- Yang, Y.M.; Noh, K.; Han, C.Y.; Kim, S.G. Transactivation of Genes Encoding for Phase II Enzymes and Phase III Transporters by Phytochemical Antioxidants. Molecules 2010, 15, 6332–6348. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; You, D.-J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.-I. Suppression of NF-kappaB signaling by Keap1 regulation of IKK-beta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Ashford, J.H.M.J.; Ashford, M.L.J. Nrf2 Orchestrates Fuel Partitioning for Cell Proliferation. Cell Metab. 2012, 16, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat 2011, 132, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator ofcytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 1999, 10, 17–24. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Wakabayashi, N. Keap1, the Sensor for Electrophiles and Oxidants that Regulates the Phase 2 Response, Is a Zinc Metalloprotein. Biochemistry 2005, 44, 6889–6899. [Google Scholar] [CrossRef] [PubMed]
- Velichkova, M.; Hasson, T. Keap1 in Adhesion Complexes. Cell Motil. Cytoskelet. 2003, 56, 109–119. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ Dimerization Function Is Required to Sequester Nrf2 in Cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar] [PubMed]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.-I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R. The antioxidant properties of zinc. J. Nutr. 2000, 130, 1447S–1454S. [Google Scholar] [PubMed]
- Eide, D.J. The oxidative stress of zinc deficiency. Metallomics 2011, 3, 1124. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Specific Patterns of Electrophile Adduction Trigger Keap1 Ubiquitination and Nrf2 Activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Nakayama, S.; Ana, E.; Nishiyama, T.; Ogura, K.; Hiratsuka, A. Activation of the Nrf2/ARE pathway via S-alkylation of cysteine 151 in the chemopreventive agent-sensor Keap1 protein by falcarindiol, a conjugated diacetylene compound. Toxicol. Appl. Pharmacol. 2010, 244, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Eggler, A.L.; Liu, D.; Liu, G.; Mesecar, A.D.; van Breemen, R.B. Sites of alkylation of human Keap1 by natural chemoprevention agents. J. Am. Soc. Mass Spectrom. 2007, 18, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2005, 26, 221–229. [Google Scholar] [CrossRef]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; McEvoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cell. 2007, 12, 1163–1178. [Google Scholar] [CrossRef]
- Woods, C.G.; Fu, J.; Xue, P.; Hou, Y.; Pluta, L.J.; Yang, L.; Zhang, Q.; Thomas, R.S.; Andersen, M.E.; Pi, J. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicol. Appl. Pharmacol. 2009, 238, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-F.; Kuo, H.-P.; Liu, M.; Chou, C.-K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.-T.; Huo, L.; Hsu, M.-C.; et al. Keap1 E3 ligase-mediated downregulation of NFkB signaling by targeting IKK-beta. Mol. Cell 2009, 36, 131–140. [Google Scholar]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Massiah, M.A.; Bozak, R.E.; Hicks, R.J.; Talalay, P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc. Natl. Acad. Sci. USA 2001, 98, 3404–3409. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.A.; Haskew-Layton, R.E.; Basso, M.; Hushpulian, D.M.; Payappilly, J.B.; Speer, R.E.; Ahn, Y.-H.; Rakhman, I.; Cole, P.A.; Pinto, J.T.; Ratan, R.R.; et al. Development of Neh2-Luciferase Reporter and Its Application for High Throughput Screening and Real-Time Monitoring of Nrf2 Activators. Chem. Biol. 2011, 18, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The Role of Keap1 in Cellular Protective Responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.-O.; Jeong, G.-S.; Jeong, S.-O.; Kim, H.S.; Kim, S.-A.; Kim, Y.-C.; Yoo, S.-J.; Kin, H.-D.; Chung, H.-T. Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp. Mol. Med. 2007, 39, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Murakami, A.; Aggarwal, B.B. Zerumbone abolishes NF-κB and IκBα kinase activation leading to suppression of antiapoptotic and metastatic gene expression, upregulation of apoptosis, and downregulation of invasion. Oncogene 2005, 24, 6957–6969. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Irie, K.; Murakami, A. In vitro covalent binding of zerumbone, a chemopreventive food factor. Biosci. Biotechnol. Biochem. 2009, 73, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, B.K.; Prasad, A.K.; Parmar, V.S.; Biswal, S.; Ghosh, B. Ethyl 3’,4’,5’-trimethoxythionocinnamate modulates NF-κB and Nrf2 transcription factors. Eur. J. Pharmacol. 2013, 700, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Purup, S.; Larsen, E.; Christensen, L.P. Differential Effects of Falcarinol and Related Aliphatic C 17-Polyacetylenes on Intestinal Cell Proliferation. J. Agric. Food Chem. 2009, 57, 8290–8296. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, D.; Lehmann, C.; Florian, S.; Barknowitz, G.; Haack, M.; Mewis, I.; Wiesner, M.; Schreiner, M.; Glatt, H.; Brigelius-Flohé, R.; et al. Glucosinolates from pak choi and broccoli induce enzymes and inhibit inflammation and colon cancer differently. Food Funct. 2014, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Lekli, I.; Ray, D.; Gangopadhyay, H.; Raychaudhuri, U.; Das, D.K. Comparison of the protective effects of steamed and cooked broccolis on ischaemia—Reperfusion-induced cardiac injury. Br. J. Nutr. 2010, 103, 815–823. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Institutes of Health ClinicalTrials.gov. Available online: http://clinicaltrials.gov (accessed on 30 January 2014).
- Philbrook, N.A.; Winn, L.M. Sub-chronic sulforaphane exposure in CD-1 pregnant mice enhances maternal NADPH quinone oxidoreductase 1 (NQO1) activity and mRNA expression of NQO1, glutathione S-transferase, and glutamate-cysteine ligase. Reprod. Toxicol. 2014, 43, 30–37. [Google Scholar] [CrossRef]
- Leoncini, E.; Malaguti, M.; Angeloni, C.; Motori, E.; Fabbri, D.; Hrelia, S. Cruciferous Vegetable Phytochemical Sulforaphane Affects Phase II Enzyme Expression and Activity in Rat Cardiomyocytes through Modulation of Akt Signaling Pathway. J. Food Sci. 2011, 76, H175–H181. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Bai, Y.; Sun, W.; Cui, W.; Xin, Y.; Wang, Y.; Tan, Y.; Miao, L.; Fu, Y.; Su, G.; Cai, L. Sulforaphane prevention of diabetes-induced aortic damage was associated with the up-regulation of Nrf2 and its down-stream antioxidants. Nutr. Metab. 2012, 9, 84. [Google Scholar] [CrossRef]
- Miao, X.; Wang, Y.; Sun, J.; Sun, W.; Tan, Y.; Cai, L.; Zheng, Y.; Su, G.; Liu, Q.; Wang, Y. Zinc protects against diabetes-induced pathogenic changes in the aorta: Roles of metallothionein and nuclear factor (erythroid-derived 2)-like 2. Cardiovasc. Diabetol. 2013, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Iori, R.; de Nicola, G.R.; Bramanti, P.; Mazzon, E. Anti-inflammatory and anti-apoptotic effects of (RS)-glucoraphanin bioactivated with myrosinase in murine sub-acute and acute MPTP-induced Parkinson’s disease. Bioorganic Med. Chem. 2013, 21, 5532–5547. [Google Scholar] [CrossRef]
- Galuppo, M.; Giacoppo, S.; de Nicola, G.R.; Iori, R.; Mazzon, E.; Bramanti, P. Journal of the Neurological Sciences. J. Neurol. Sci. 2013, 334, 88–96. [Google Scholar] [CrossRef] [PubMed]
- McWalter, G.K.; Higgins, L.G.; McLellan, L.I.; Henderson, C.J.; Song, L.; Thornalley, P.J.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription Factor Nrf2 Is Essential for Induction of NAD(P)H:Quinone Oxidoreductase 1, Glutathione S-Transferases, and Glutamate Cysteine Ligase by Broccoli Seeds and Isothiocyanates. J. Nutr. 2004, 134, 3499S–3506S. [Google Scholar] [PubMed]
- N’jai, A.U.; Kemp, M.Q.; Metzger, B.T.; Hanlon, P.R.; Robbins, M.; Czuyprynski, C.; Barnes, D.M. Spanish Black Radish (Raphanus Sativus L. Var. niger) Diet Enhances Clearance of DMBA and Diminishes Toxic Effects on Bone Marrow Progenitor Cells. Nutr. Cancer 2012, 64, 1038–1048. [Google Scholar]
- Brandt, J.Z.; Silveira, L.T.R.; Grassi, T.F.; Anselmo-Franci, J.A.; Fávaro, W.J.; Felisbino, S.L.; Barbisan, L.F.; Scarano, W.R. Indole-3-carbinol attenuates the deleterious gestational effects of bisphenol A exposure on the prostate gland of male F1 rats. Reprod. Toxicol. 2014, 43, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Lin, J.; Wu, D. Sulforaphane induces Nrf2 and protects against CYP2E1-dependent binge alcohol-induced liver steatosis. Biochim. Biophys. Acta 2014, 1840, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kleszczyński, K.; Ernst, I.M.A.; Wagner, A.E.; Kruse, N.; Zillikens, D.; Rimbach, G.; Fischer, T.W. Sulforaphane and phenylethyl isothiocyanate protect human skin against UVR-induced oxidative stress and apoptosis: Role of Nrf2-dependent gene expression and antioxidant enzymes. Pharmacol. Res. 2013, 78, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.T.; Bergström, P.; Hammarsten, O. Repeated Nrf2 stimulation using sulforaphane protects fibroblasts from ionizing radiation. Toxicol. Appl. Pharmacol. 2014, 276, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Seymour, E.M.; Bennink, M.R.; Bolling, S.F. Diet-relevant phytochemical intake affects the cardiac AhR and nrf2 transcriptome and reduces heart failure in hypertensive rats. J. Nutr. Biochem. 2013, 24, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.-S.; Tsai, M.-L.; Nagabhushanam, K.; Wang, Y.-J.; Wu, C.-H.; Ho, C.-T.; Pan, M.-H. Pterostilbene Is More Potent than Resveratrol in Preventing Azoxymethane (AOM)-Induced Colon Tumorigenesis via Activation of the NF-E2-Related Factor 2 (Nrf2)-Mediated Antioxidant Signaling Pathway. J. Agricul. Food Chem. 2011, 59, 2725–2733. [Google Scholar]
- Bishayee, A.; Bhatia, D.; Thoppil, R.J.; Darvesh, A.S.; Nevo, E.; Lansky, E.P. Pomegranate-mediated chemoprevention of experimental hepatocarcinogenesis involves Nrf2-regulated antioxidant mechanisms. Carcinogenesis 2011, 32, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Thoppil, R.J.; Darvesh, A.S.; Ohanyan, V.; Meszaros, J.G.; Bhatia, D. Pomegranate phytoconstituents blunt the inflammatory cascade in a chemically induced rodent model of hepatocellular carcinogenesis. J. Nutr. Biochem. 2013, 24, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Santos, V.; Bisen-Hersh, E.; Yu, Y.; Cabral, I.S.; Nardini, V.; Culbreth, M.; Teixeira da Rocha, J.B.; Barbosa, F., Jr.; Aschner, M. Anthocyanin-Rich Açaí (Euterpe oleracea Mart.) Extract Attenuates Manganese-Induced Oxidative Stress in Rat Primary Astrocyte Cultures. J. Toxicol. Environ. Health Part A 2014, 77, 390–404. [Google Scholar]
- Linnewiel, K.; Ernst, H.; Caris-Veyrat, C.; Ben-Dor, A.; Kampf, A.; Salman, H.; Danilenko, M.; Levy, J.; Sharoni, Y. Structure activity relationship of carotenoid derivatives in activation of the electrophile/antioxidant response element transcription system. Free Radic. Biol. Med. 2009, 47, 659–667. [Google Scholar] [PubMed]
- Ben-Dor, A.; Steiner, M.; Gheber, L.; Danilenko, M.; Dubi, N.; Linnewiel, K.; Zick, A.; Sharoni, Y.; Levy, J. Carotenoids activate the antioxidant response element transcription system. Mol. Cancer Ther. 2005, 4, 177–186. [Google Scholar] [PubMed]
- Ohnuma, T.; Komatsu, T.; Nakayama, S.; Nishiyama, T.; Ogura, K.; Hiratsuka, A. Induction of antioxidant and phase 2 drug-metabolizing enzymes by falcarindiol isolated from Notopterygium incisum extract, which activates the Nrf2/ARE pathway, leads to cytoprotection against oxidative and electrophilic stress. Arch. Biochem. Biophys. 2009, 488, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Ana, E.; Hoashi, R.; Takeda, Y.; Nishiyama, T.; Ogura, K.; Hiratsuka, A. Dietary diacetylene falcarindiol induces phase 2 drug-metabolizing enzymes and blocks carbon tetrachloride-induced hepatotoxicity in mice through suppression of lipid peroxidation. Biol. Pharm. Bull. 2011, 34, 371–378. [Google Scholar] [PubMed]
- Sahin, K.; Orhan, C.; Tuzcu, M.; Sahin, N.; Ali, S.; Bahcecioglu, I.H.; Guler, O.; Ozercan, I.; Ilhan, N.; Kucuk, O. Orally Administered Lycopene Attenuates Diethylnitrosamine-Induced Hepatocarcinogenesis in Rats by Modulating Nrf-2/HO-1 and Akt/mTOR Pathways. Nutr. Cancer 2014, 66, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-C.; Lii, C.-K.; Lin, A.-H.; Yeh, Y.-W.; Yao, H.-T.; Li, C.-C.; Liu, K.-L.; Chen, H.-W. Induction of glutathione synthesis and heme oxygenase 1 by the flavonoids butein and phloretin is mediated through the ERK/Nrf2 pathway and protects against oxidative stress. Free Rad. Biol. Med. 2011, 51, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.-L.; Liu, J.-M.; Zhao, G.-J.; Wang, L.; Liang, G.; Wu, B.; Li, M.-F.; Qiu, Q.-M.; Lu, Z.-Q. The reversal of paraquat-induced mitochondria- mediated apoptosis by cycloartenyl ferulate, the important role of Nrf2 pathway. Exp. Cell Res. 2013, 319, 2845–2855. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-Y.; Kim, S.-M.; Yuk, J.-E.; Kwon, O.-K.; Oh, S.-R.; Lee, H.-K.; Jeong, H.; Ahn, K.-S. Capsicum annuum L. Methanolic Extract Inhibits Ovalbumin-Induced Airway Inflammation and Oxidative Stress in a Mouse Model of Asthma. J. Med. Food 2011, 14, 1144–1151. [Google Scholar]
- Kavitha, K.; Thiyagarajan, P.; Nandhini, J.R.; Mishra, R.; Nagini, S. Chemopreventive effects of diverse dietary phytochemicals against DMBA-induced hamster buccal pouch carcinogenesis via the induction of Nrf2-mediated cytoprotective antioxidant, detoxification, and DNA repair enzymes. Biochimie 2013, 95, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Feng, Z.; Li, Y.; Wang, Y.; Wertz, K.; Weber, P.; Fu, Y.; Liu, J. Stimulation of GSH synthesis to prevent oxidative stress-induced apoptosis by hydroxytyrosol in human retinal pigment epithelial cells: Activation of Nrf2 and JNK-p62/SQSTM1 pathways. J. Nutr. Biochem. 2012, 23, 994–1006. [Google Scholar] [PubMed]
- Ho, C.-Y.; Weng, C.-J.; Jhang, J.-J.; Cheng, Y.-T.; Huang, S.-M.; Yen, G.-C. Diallyl sulfide as a potential dietary agent to reduce TNF-α- and histamine-induced proinflammatory responses in A7r5 cells. Mol. Nutr. Food Res. 2014, 58, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Parkin, K.L.; Nitteranon, V.; Liang, J.; Yang, W.; Li, Y.; Zhang, G.; Hu, Q. Isolation of quinone reductase (QR) inducing agents from ginger rhizome and their in vitro anti-inflammatory activity. Food Res. Int. 2011, 44, 1597–1603. [Google Scholar] [CrossRef]
- Liao, B.-C.; Hsieh, C.-W.; Liu, Y.-C.; Tzeng, T.-T.; Sun, Y.-W.; Wung, B.-S. Cinnamaldehyde inhibits the tumor necrosis factor-α-induced expression of cell adhesion molecules in endothelial cells by suppressing NF-κB activation: Effects upon IκB and Nrf2. Toxicol. Appl. Pharmacol. 2008, 229, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K.; Mimura, J.; Itoh, K.; Satoh, T.; Shimojo, Y.; Kitajima, C.; Maruyama, A.; Yamamoto, M.; Shirasawa, T. Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells. J. Biochem. 2010, 147, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Kosaka, K.; Maruyama, A.; Satoh, T.; Harada, N.; Yoshida, H.; Satoh, K.; Yamamoto, M.; Itoh, K. Nrf2 regulates NGF mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J. Biochem. 2011, 150, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Kröncke, K.D.; Buchczyk, D.P.; Sies, H. Role of copper, zinc, selenium and tellurium in the cellular defense against oxidative and nitrosative stress. J. Nutr. 2003, 133, 1448S–1451S. [Google Scholar] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, Z.; Brown, K.H.; Gibson, R.S.; Hotz, C.; King, J.C.; Lönnerdal, B.; Lopez de Romaña Forga, D.; Peerson, J.M.; Rivera, J.A.; Ruel, M.T.; et al. Estimated Risk of Zinc Deficiency by Country. Food Nutr. Bull. 2004, 25, S189–S195. [Google Scholar]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Thompson, R.B.; Stoddard, A.K.; Fierke, C.A. Measuring Picomolar Intracellular Exchangeable Zinc in PC-12 Cells Using a Ratiometric Fluorescence Biosensor. ACS Chem. Biol. 2006, 1, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metals on the move: Zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. Biometals 2011, 24, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Q.; Lu, Y.-F.; Pi, J. New insights into generalized hepatoprotective effects of oleanolic acid: Key roles of metallothionein and Nrf2 induction. Biochem. Pharmacol. 2008, 76, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Z.-X.; Zhang, W.; Bell, M.W.; Waalkes, M.P. Changes in hepatic gene expression in response to hepatoprotective levels of zinc. Liver Int. 2009, 29, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-X.; Sun, X.; Lambert, J.C.; Saari, J.T.; Kang, Y.J. Metallothionein-Independent Zinc Protection from Alcoholic Liver Injury. Am. J. Pathol. 2002, 160, 2267–2274. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.F.; Loo, G. Upregulation of haeme oxygenase-1 by zinc in HCT-116 cells. Free Radic. Res. 2012, 46, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.-N.; Chen, Y.; Cai, J.; Sternberg, P., Jr. Increased Glutathione Synthesis throughan ARE-Nrf2-Dependent Pathway by Zinc in the RPE: Implication for Protection against Oxidative Stress. Investig. Ophthamol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef]
- Tate, D.J., Jr.; Miceli, M.V.; Newsome, D.A. Zinc induces catalase expression in cultured fetal human retinal pigment epithelial cells. Curr. Eye Res. 1997, 16, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.J., Jr.; Miceli, M.V.; Newsome, D.A.; Alcock, N.W.; Oliver, P.D. Influence of zinc on selected cellular functions of cultured human retinal pigment epithelium. Curr. Eye Res. 1995, 14, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Ames, B.N. Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFkB, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc. Natl. Acad. Sci. USA 2002, 99, 16770–16775. [Google Scholar] [CrossRef] [PubMed]
- Omata, Y.; Salvador, G.A.; Supasai, S.; Keenan, A.H.; Oteiza, P.I. Decreased Zinc Availability Affects Glutathione Metabolism in Neuronal Cells and in the Developing Brain. Toxicol. Sci. 2013, 133, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Bröcker, M.J.; Ho, J.M.L.; Church, G.M.; Söll, D.; O’Donoghue, P. Recoding the Genetic Code with Selenocysteine. Angew. Chem. Int. Ed. 2013, 53, 319–323. [Google Scholar] [CrossRef]
- Müller, M.; Banning, A.; Brigelius-Flohé, R.; Kipp, A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010, 5, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Müller, M.; Lippmann, D.; Kipp, A.P. The Yin and Yang of Nrf2-Regulated Selenoproteins in Carcinogenesis. Int. J. Cell Biol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Krehl, S.; Loewinger, M.; Florian, S.; Kipp, A.P.; Banning, A.; Wessjohann, L.A.; Brauer, M.N.; Iori, R.; Esworthy, R.S.; Chu, F.F.; et al. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis 2012, 33, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, W.; Shan, Y.; Barrera, L.N.; Howie, A.F.; Beckett, G.J.; Wu, K.; Bao, Y. Synergy between sulforaphane and selenium in the up-regulation of thioredoxin reductase and protection against hydrogen peroxide-induced cell death in human hepatocytes. Food Chem. 2012, 133, 300–307. [Google Scholar] [CrossRef]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–10. [Google Scholar] [CrossRef]
- Gillies, P.J. Preemptive nutrition of pro-inflammatory states: A nutrigenomic model. Nutr. Rev. 2007, 65, S217–S220. [Google Scholar] [CrossRef] [PubMed]
- Szarc vel Szic, K.; Ndlovu, M.N.; Haegeman, G.; Vanden Berghe, W. Nature or nurture: Let food be your epigenetic medicine in chronic inflammatory disorders. Biochem. Pharmacol. 2010, 80, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stefanson, A.L.; Bakovic, M. Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals. Nutrients 2014, 6, 3777-3801. https://doi.org/10.3390/nu6093777
Stefanson AL, Bakovic M. Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals. Nutrients. 2014; 6(9):3777-3801. https://doi.org/10.3390/nu6093777
Chicago/Turabian StyleStefanson, Amanda L., and Marica Bakovic. 2014. "Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals" Nutrients 6, no. 9: 3777-3801. https://doi.org/10.3390/nu6093777
APA StyleStefanson, A. L., & Bakovic, M. (2014). Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals. Nutrients, 6(9), 3777-3801. https://doi.org/10.3390/nu6093777