Dysbiotic Events in Gut Microbiota: Impact on Human Health

Abstract
:
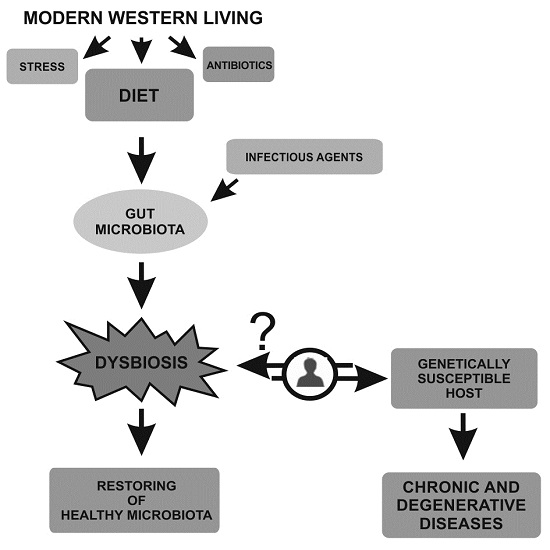{kind=link}
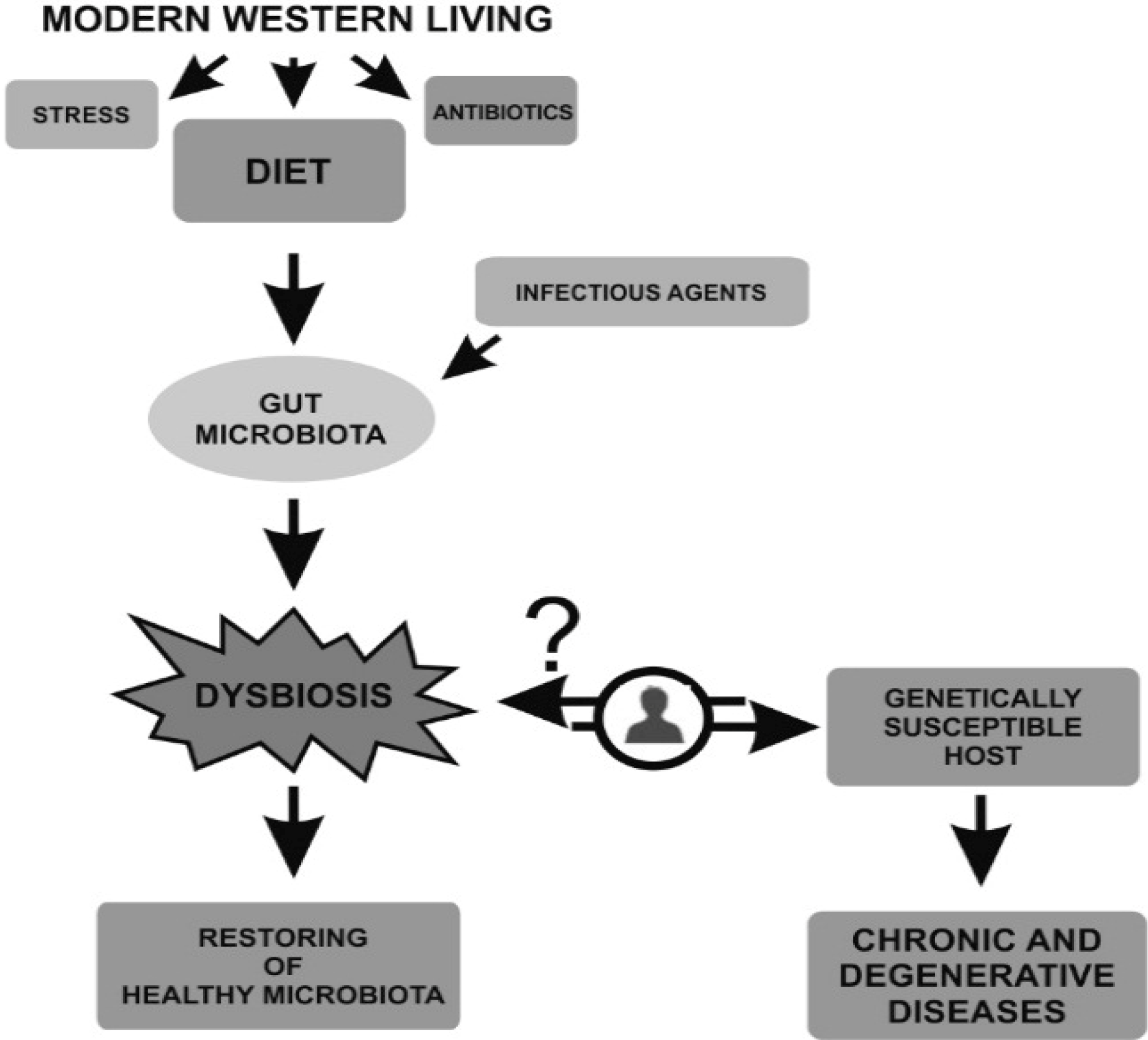{kind=link}
1. Introduction
2. Gut Microbiota
2.1. Composition
2.2. Metabolic Functions
2.3. Epithelium Barrier Integrity Effect
2.4. Defense Barrier
2.5. Host Defense Development
3. Dysbiosis
3.1. Dysbiosis in Inflammatory Bowel Disease (IBD)
3.2. Crohn’s Disease in Pediatric Patients
3.3. Mucosa Associated Microbiota
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Bik, E.M. Composition and function of the human-associated microbiota. Nutr. Rev. 2009, 67, S164–S171. [Google Scholar] [CrossRef] [PubMed]
- Hawrelak, J.A.; Myers, S.P. The causes of intestinal dysbiosis. Altern. Med. Rev. 2004, 9, 180–197. [Google Scholar] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Noverr, M.C.; Huffnagle, G.B. The “microflora hypothesis” of allergic diseases. Clin. Exp. Allergy 2005, 35, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Zhu, W.; Sartor, R.B.; Li, E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 2011, 19, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, C.P.; Neut, C.; Desreumaux, P.; Colombel, J.F. Dysbiosis in inflammatory bowel disease. Gut 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Lee, S.M.; Shen, Y.; Khosravi, A.; Mazmanian, S.K. Host-bacterial symbiosis in health and disease. Adv. Immunol. 2010, 107, 243–274. [Google Scholar] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinalmicrobial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Bäckhed, F. The gut microbiota masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Korecka, A.; Arulampalam, V. The gut microbiome: Scourge, sentinel or spectator? J. Oral Microbiol. 2012, 4, 1–14. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L., St.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. Human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ed Yong. Gut Microbial “Enterotypes” Become Less Clear-Cut. Communities of Gut Bacteria May Form a Spectrum Rather than Falling into Distinct Groups. Available online: http://www.nature.com/news/gut-microbial-enterotypes-become-less-clear-cut-1.10276 (accessed on 21 March 2012).
- Scanlan, P.D.; Shanahan, F.; Marchesi, J.R. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 2008, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Yachi, S.; Loreau, M. Biodiversity and ecosystem productivity in a fluctuating environment: The insurance hypothesis. Proc. Natl. Acad. Sci. USA 1999, 16, 1463–1468. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Vlachou, A.; Verbrugghe, K.; de Vuyst, L. Cross feeding between Bifidobacterium longum BB536 and acetate converting, butyrate-producing colon bacteria during growth on oligo fructose. Appl. Environ. Microbiol. 2006, 72, 7835–7841. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Zhang, M.; et al. Symbiotic gut microbes modulate human metabolic phenotypes. PNAS 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Mai, V. Dietary modification of the intestinal microbiota. Nutr. Rev. 2004, 62, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lunn, J.C.; Kuhnle, G.; Mai, V.; Frankenfeld, C.; Shuker, D.E.; Glen, R.C.; Goodman, J.M.; Pollock, J.R.; Bingham, S.A. The effect of haem in red and processed meat on the endogenous format ion of N-nitroso compounds in the upper gastrointestinal tract. Carcinogenesis 2007, 28, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Saemann, M.D.; Bohmig, G.A.; Zlabinger, G.J. Short-chain fatty acids: Bacterial mediators of a balanced host-microbial relationship in the human gut. Wien. Klin. Wochenschr. 2002, 114, 289–300. [Google Scholar] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Berndt, B.E.; Zhang, M.; Owyang, S.Y.; Cole, T.S.; Wang, T.W.; Luther, J.; Veniaminova, N.A.; Merchant, J.L.; Chen, C.C.; Huffnagle, G.B.; et al. Butyrate increases IL-23 production by stimulated dendritic cells. Am. J. Physiol. Gastrointest. Liver. Physiol. 2012, 303, 1384–1392. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 19, 446–450. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Neale, G.; Gibson, G.R.; Christl, S.U.; Cummings, J.H. Metabolism of dietary sulphate: Absorption and excretion in humans. Gut 1991, 132, 766–773. [Google Scholar] [CrossRef]
- Petersen, L.C. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim. Biophys. Acta 1977, 460, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Vermeiren, J.; Hindryckx, P.; van Nieuwenhuyse, G.; Laukens, D.; de Vos, M.; Boon, N.; van de Wiele, T. Intrarectal nitric oxide administration prevents cellular infiltration but not colonic injury during dextran sodium sulfate colitis. Dig. Dis. Sci. 2012, 57, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kasper, D.L. Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Curr. Opin. Immunol. 2010, 22, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.J.; Siegel, J.; Snellen, D.W. Transit time of epithelial cells in the small intestines of germ-free mice and ex-germfree mice associated with indigenous microorganisms. Appl. Environ. Microbiol. 1981, 42, 996–1001. [Google Scholar] [PubMed]
- Shirkey, T.W.; Siggers, R.H.; Goldade, B.G.; Marshall, J.K.; Drew, M.D.; Laarveld, B.; van Kessel, A.G. Effects of commensal bacteria on intestinal morphology and expression of proin-flammatory cytokines in the gnotobiotic pig. Exp. Biol. Med. 2006, 231, 1333–1345. [Google Scholar]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Leatham, M.P.; Banerjee, S.; Autieri, S.M.; Mercado-Lubo, R.; Conway, T.; Cohen, P.S. Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157: H7 growth in the streptomycin-treated mouse intestine. Infect. Immun. 2009, 77, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Van der Waaij, D.; Berghuis-de Vries, J.M.; Lekkerkerk, V. Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J. Hyg. (Lond.) 1971, 69, 405–411. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Joncquel Chevalier-Curt, M.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Hoffmann, C.; Abt, M.C.; Du, Y.; Kobuley, D.; Kirn, T.J.; Bushman, F.D.; Artis, D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010, 3, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Reis, B.S.; Mucida, D. The role of the intestinal context in the generation of tolerance and inflammation. Clin. Dev. Immunol. 2012, 2012, 157948. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Haverson, K.; Rehakova, Z.; Sinkora, J.; Sver, L.; Bailey, M. Immune development in jejunal mucosa after colonization with selected commensal gut bacteria: A study in germ-free pigs. Vet. Immunol. Immunopathol. 2007, 119, 243–253. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Scully, P.; O’Mahony, D.; Murphy, S.; O’Brien, F.; Lyons, A.; Sherlock, G.; MacSharry, J.; Kiely, B.; Shanahan, F.; et al. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-kappaB activation. PLoS Pathog. 2008, 4, 1–10. [Google Scholar]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lécuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; de Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Santeliz, J.; Karp, C.L. The germless theory of allergic disease: Revisiting the hygiene hypothesis. Nat. Rev. Immunol. 2001, 1, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Eun, C.S.; Mishima, Y.; Wohlgemuth, S.; Liu, B.; Bower, M.; Carroll, I.M.; Sartor, R.B. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10/mice. Infect Immun. 2014, 82, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K.; Pearce, K. Dysbiosis of Gut Microbiota (DOGMA)—A novel theory for the development of Polycystic Ovarian Syndrome. Med. Hypotheses 2012, 79, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 4, 55–60. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Denizot, J.; Dreux, N.; Robin, F.; Billard, E.; Bonnet, R.; Darfeuille-Michaud, A.; Barnich, N. Western diet induces dysbiosis with increased E. coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 2014, 63, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.Y.; Devkota, S.; Moscoso, D.; Chang, E.B.; Leone, V.A. The role of diet in triggering human inflammatory disorders in the modern age. Microbes Infect. 2013, 15, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Marlow, G.; Ellett, S.; Ferguson, I.R.; Zhu, S.; Karunasinghe, N.; Jesuthasan, A.C.; Han, D.Y.; Fraser, A.G.; Ferguson, L.R. Transcriptomics to study the effect of a Mediterranean-inspired diet on inflammation in Crohn’s disease patients. Hum. Genomics 2013, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Nord, C.E. The effect of antimicrobial agents on the ecology of the human intestinal microflora. Vet. Microbiol. 1993, 35, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Gismondo, M.R. Antibiotic impact on intestinal microflora. Gastroenterol. Int. 1998, 11, 29–30. [Google Scholar]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Rafii, F.; Sutherland, J.B.; Cerniglia, C.E. Effects of treatment with antimicrobial agents on the human colonic microflora. Ther. Clin. Risk Manag. 2008, 4, 1343–1358. [Google Scholar] [PubMed]
- Hurley, B.W.; Nguyen, C.C. The spectrum of pseudomembranous enterocolitis and antibiotic-associated diarrhea. Arch. Intern. Med. 2002, 162, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unraveling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, D.K. Lessons from genetic models of inflammatory bowel disease. Acta Gastroenterol. Belg. 1997, 60, 163–165. [Google Scholar] [PubMed]
- Sartor, R.B. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. Am. J. Gastroenterol. 1997, 12, 5S–11S. [Google Scholar]
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.W.; Barrett, J.C.; Parkes, M.; Satsangi, J. New IBD genetics: Common pathways with other diseases. Gut 2011, 60, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 10, 11–17. [Google Scholar]
- Rivas, M.A.; Beaudoin, M.; Gardet, A.; Stevens, C.; Sharma, Y.; Zhang, C.K.; Boucher, G.; Ripke, S.; Ellinghaus, D.; Burtt, N.; et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011, 43, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 31, 599–603. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; de la Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Achkar, J.P.; Klei, L.; de Bakker, P.I.; Bellone, G.; Rebert, N.; Scott, R.; Lu, Y.; Regueiro, M.; Brzezinski, A.; Kamboh, M.I.; et al. Amino acid position 11 of HLA-DRβ1 is a major determinant of chromosome 6p association with ulcerative colitis. Genes Immun. 2012, 13, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Cominelli, F. Immunopathogenesis of inflammatory bowel disease: Current concepts. Curr. Opin. Gastroenterol. 2007, 23, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Mannon, P.J.; Hornung, R.L.; Yang, Z.; Yi, C.; Groden, C.; Friend, J.; Yao, M.; Strober, W.; Fuss, I.J. Suppression of inflammation in ulcerative colitis by interferon-β-1a is accompanied by inhibition of IL-13 production. Gut 2011, 60, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Sarra, M.; Pallone, F.; Macdonald, T.T.; Monteleone, G. IL-23/IL-17 axis in IBD. Inflamm. Bowel Dis. 2010, 16, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Rolhion, N.; Hofman, P.; Darfeuille-Michaud, A. The endoplasmic reticulum stress response chaperone: Gp96, a host receptor for Crohn disease-associated adherent-invasive Escherichia coli. Gut Microbes 2011, 2, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 5, 743–756. [Google Scholar] [CrossRef]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Schulzke, J.D.; Bojarski, C.; Zeissig, S.; Heller, F.; Gitter, A.H.; Fromm, M. Disrupted barrier function through epithelial cell apoptosis. Ann. N. Y. Acad. Sci. 2006, 1072, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.; Makharia, G.K.; Ahuja, V.; Kalaivani, M.; Joshi, Y.K. Intestinal permeability and its association with the patient and disease characteristics in Crohn’s disease. World J. Gastroenterol. 2008, 14, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J. Immunopathogenesis of inflammatory bowel disease. Med. Sci. 2010, 26, 405–410. [Google Scholar]
- D’Incà, R.; Annese, V.; di Leo, V.; Latiano, A.; Quaino, V.; Abazia, C.; Vettorato, M.G.; Sturniolo, G.C. Increased intestinal permeability and NOD2 variants in familial and sporadic Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 15, 1455–1461. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Turner, J.R. The tight junction in inflammatory disease: Communication breakdown. Curr. Opin. Pharmacol. 2009, 9, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 16, 1–18. [Google Scholar]
- Lopez-Siles, M.; Khan, T.M.; Duncan, S.H.; Harmsen, H.J.; Garcia-Gil, L.J.; Flint, H.J. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl. Environ. Microbiol. 2012, 78, 420–428. [Google Scholar] [CrossRef] [PubMed]
- El-Zaatari, F.A.; Naser, S.A.; Hulten, K.; Burch, P.; Graham, D.Y. Characterization of Mycobacterium paratuberculosis p36 antigen and its seroreactivities in Crohn’s disease. Curr. Microbiol. 1999, 39, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Naser, S.A.; Ghobrial, G.; Romero, C.; Valentine, J.F. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet 2004, 364, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.H.; Wiedmann, M.; McDonough, P.; Kim, S.G.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Maier, L.; Hardt, W.D. “Blooming” in the gut: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Winter, M.G.; Xavier, M.N.; Thiennimitr, P.; Poon, V.; Keestra, A.M.; Laughlin, R.C.; Gomez, G.; Wu, J.; Lawhon, S.D.; et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013, 339, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Bäumler, A.J. Dysbiosis in the inflamed intestine: Chance favors the prepared microbe. Gut Microbes 2014, 5, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [PubMed]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Blanco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.M.; Bringer, A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sitaraman, S.V.; Babbin, B.A.; Gerner-Smidt, P.; Ribot, E.M.; Garrett, N.; Alpern, J.A.; Akyildiz, A.; Theiss, A.L.; Nusrat, A.; et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab. Investig. 2007, 87, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Glasser, A.L.; Darfeuille-Michaud, A. Abnormalities in the handling of intracellular bacteria in Crohn’s disease: A link between infectious etiology and host genetic susceptibility. Arch. Immunol. Ther. Exp. 2008, 56, 237–244. [Google Scholar] [CrossRef]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Ryan, P.; Kelly, R.G.; Lee, G.; Collins, J.K.; O’Sullivan, G.C.; O’Connell, J.; Shanahan, F. Bacterial DNA within granulomas of patients with Crohn’s disease—Detection by laser capture microdissection and PCR. Am. J. Gastroenterol. 2004, 99, 1539–1543. [Google Scholar] [CrossRef]
- Flanagan, P.; Campbell, B.J.; Rhodes, J.M. Bacteria in the pathogenesis of inflammatory bowel disease. Biochem. Soc. Trans. 2011, 39, 1067–1072. [Google Scholar] [CrossRef]
- Barnich, N.; Carvalho, F.A.; Glasser, A.L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest. 2007, 117, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Darfeuille-Michaud, A. The interaction of Crohn’s disease-associated Escherichia coli to Peyer’s patches of the intestinal mucosa involves long polar fimbriae. Med. Sci. 2011, 27, 572–573. [Google Scholar]
- Schippa, S.; Iebba, V.; Totino, V.; Santangelo, F.; Lepanto, M.; Alessandri, C.; Nuti, F.; Viola, F.; di Nardo, G.; Cucchiara, S.; et al. A potential role of Escherichia coli pathobionts in the pathogenesis of pediatric inflammatory bowel disease. Can. J. Microbiol. 2012, 58, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Mobley, H.L. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr. Opin. Microbiol. 2012, 15, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Peyretaillade, E.; Claret, L.; de Vallée, A.; Dossat, C.; Vacherie, B.; el Zineb, H.; Segurens, B.; Barbe, V.; Sauvanet, P.; et al. Complete genome sequence of Crohn’s disease-associated adherent-invasive E. coli strain LF82. PLoS One 2010, 5, e12714. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Conte, M.P.; Lepanto, M.S.; di Nardo, G.; Santangelo, F.; Aloi, M.; Totino, V.; Checchi, M.P.; Longhi, C.; Cucchiara, S.; et al. Microevolution in fimH gene of mucosa-associated Escherichia coli strains isolated from pediatric patients with inflammatory bowel disease. Infect. Immun. 2012, 80, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Russell, R.K.; Reiff, C.; Louis, P.; McIntosh, F.; Berry, S.H.; Mukhopadhya, I.; Bisset, W.M.; Barclay, A.R.; Bishop, J.; et al. Microbiota of de-novo pediatric IBD: Increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am. J. Gastroenterol. 2012, 107, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.P.; Schippa, S.; Zamboni, I.; Penta, M.; Chiarini, F.; Seganti, L.; Osborn, J.; Falconieri, P.; Borrelli, O.; Cucchiara, S. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 2006, 55, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Santangelo, F.; Totino, V.; Nicoletti, M.; Gagliardi, A.; de Biase, R.V.; Cucchiara, S.; Nencioni, L.; Conte, M.P.; Schippa, S. Higher prevalence and abundance of Bdellovibrio bacteriovorus in the human gut of healthy subjects. PLoS One 2013, 8, 1–9. [Google Scholar]
- Lee, A.; Griffiths, T.A.; Parab, R.S.; King, R.K.; Dubinsky, M.C.; Urbanski, S.J.; Wrobel, I.; Rioux, K.P. Association of Mycobacterium avium subspecies paratuberculosis with Crohn Disease in pediatric patients. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 170–174. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schippa, S.; Conte, M.P. Dysbiotic Events in Gut Microbiota: Impact on Human Health. Nutrients 2014, 6, 5786-5805. https://doi.org/10.3390/nu6125786
Schippa S, Conte MP. Dysbiotic Events in Gut Microbiota: Impact on Human Health. Nutrients. 2014; 6(12):5786-5805. https://doi.org/10.3390/nu6125786
Chicago/Turabian StyleSchippa, Serena, and Maria Pia Conte. 2014. "Dysbiotic Events in Gut Microbiota: Impact on Human Health" Nutrients 6, no. 12: 5786-5805. https://doi.org/10.3390/nu6125786
APA StyleSchippa, S., & Conte, M. P. (2014). Dysbiotic Events in Gut Microbiota: Impact on Human Health. Nutrients, 6(12), 5786-5805. https://doi.org/10.3390/nu6125786