Impact of Soy Isoflavones on the Epigenome in Cancer Prevention

Abstract
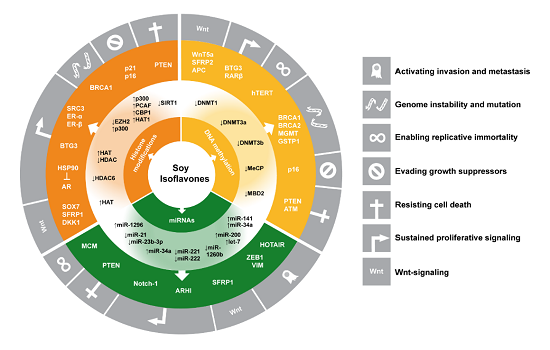
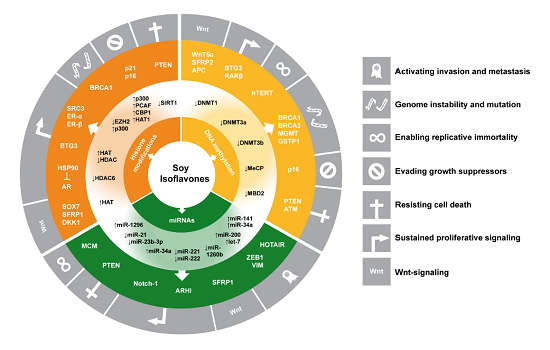
:
1. Introduction
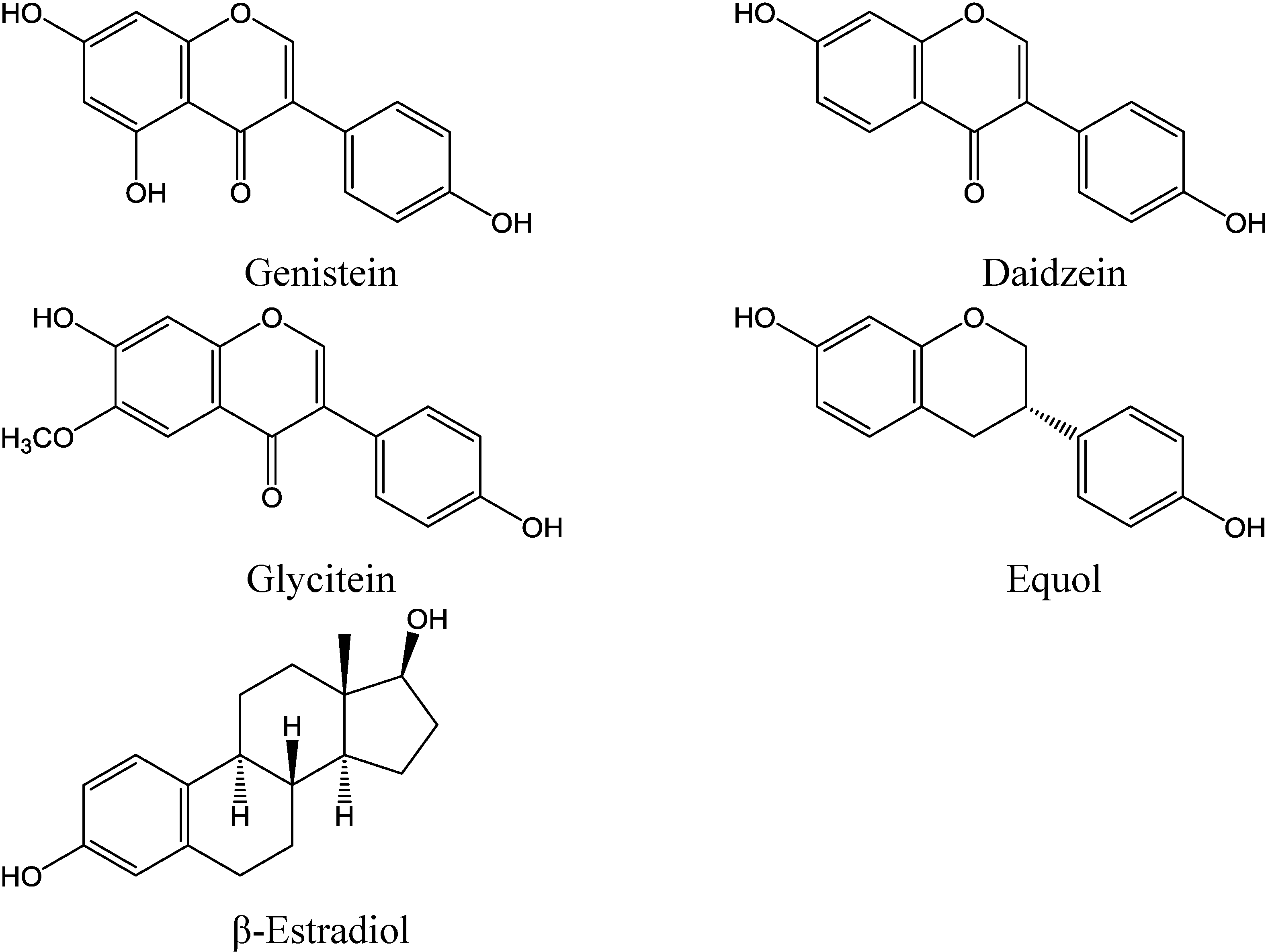
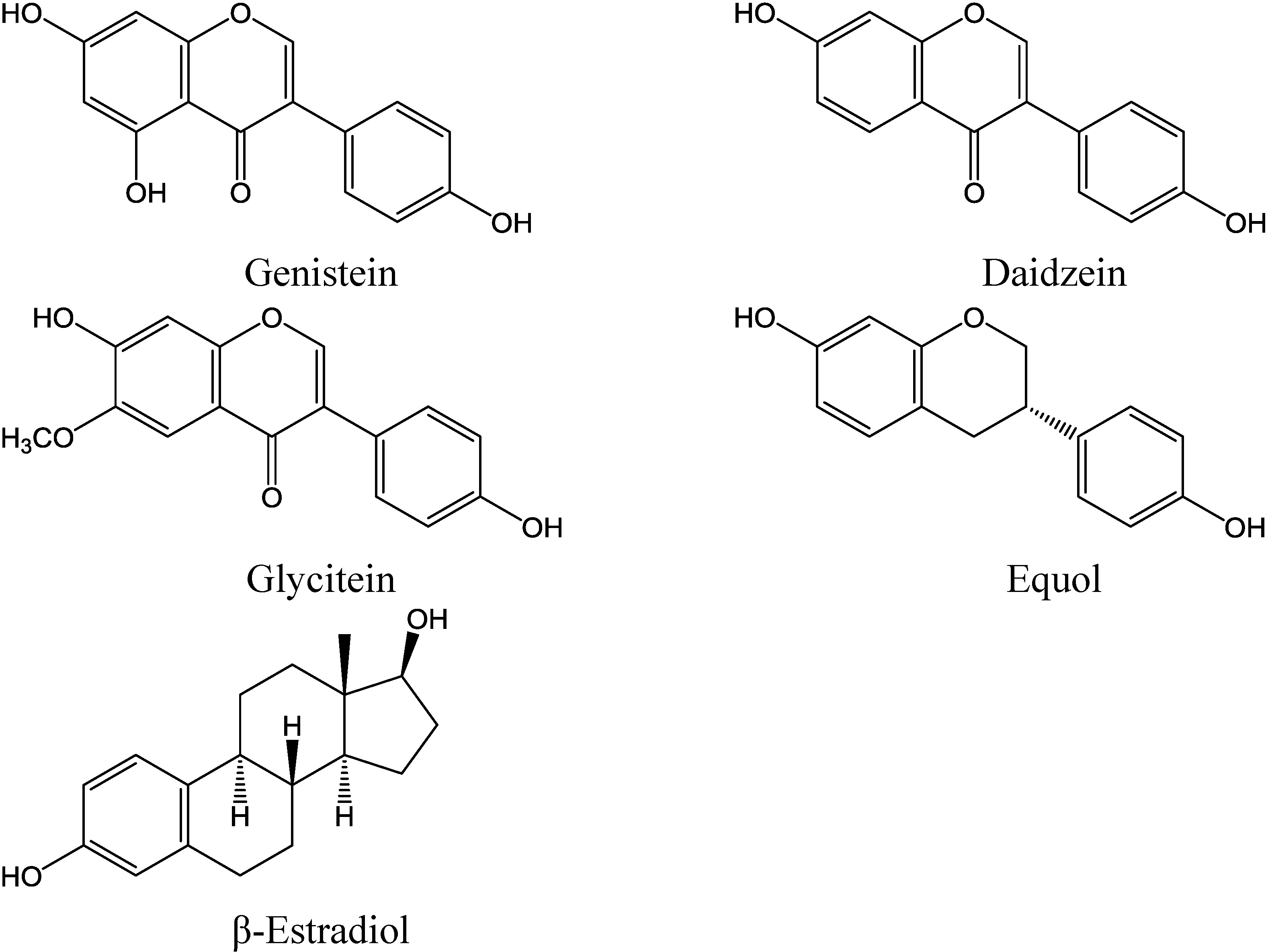
1.1. Isoflavones as Cancer Preventive Agents
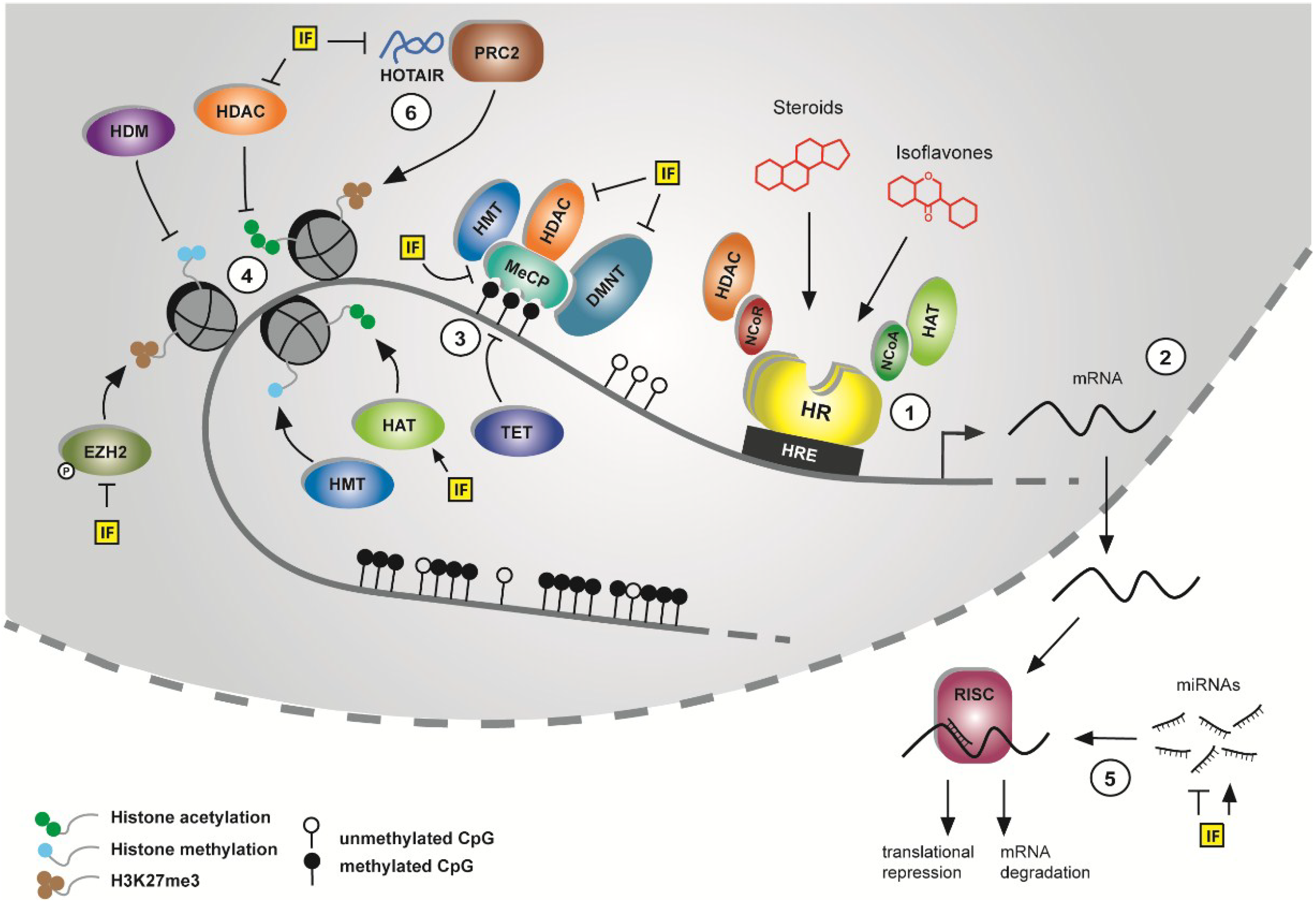
1.2. Epigenetic Mechanisms as Targets in Cancer Prevention
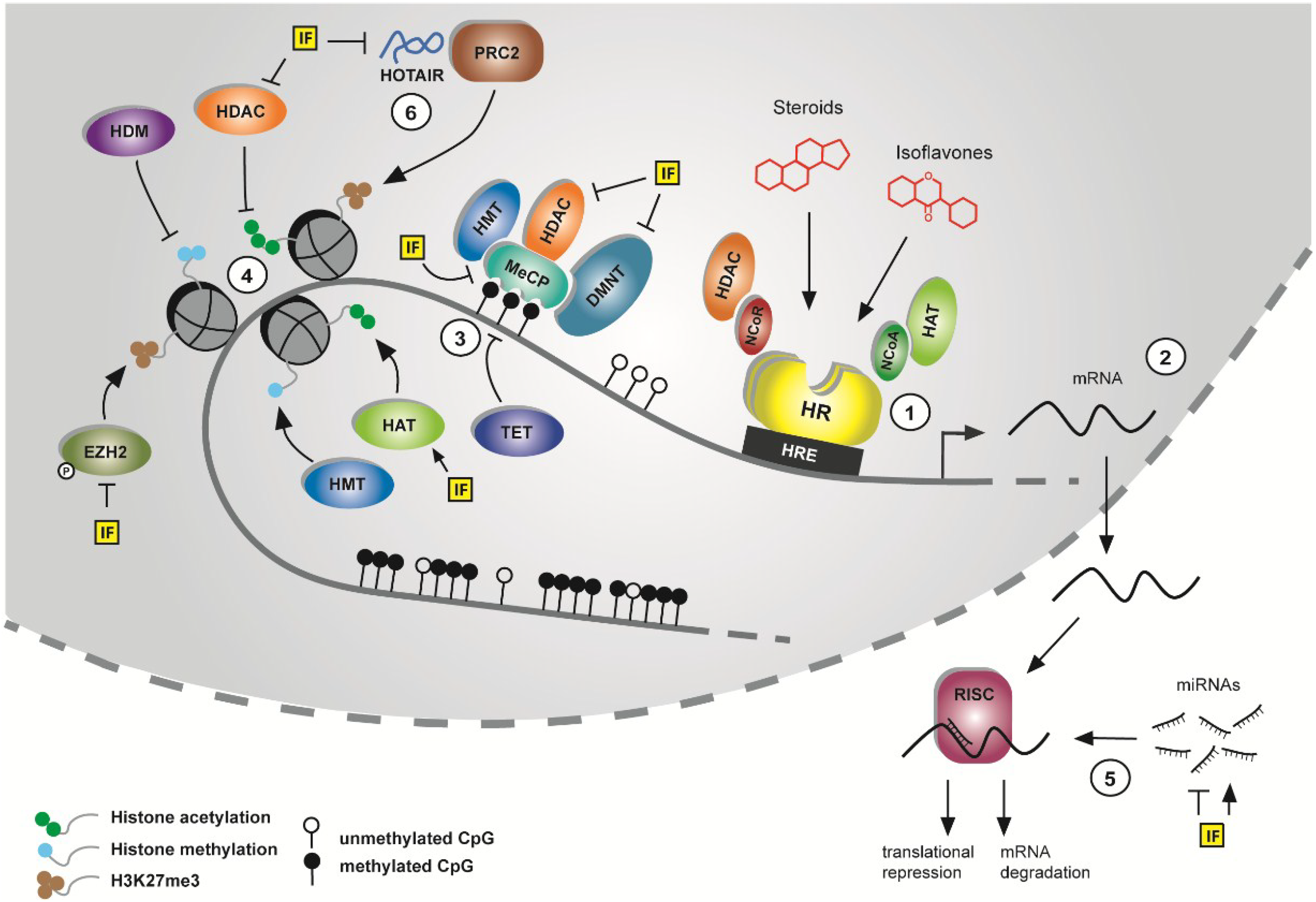
1.2.1. DNA Methylation
1.2.2. Histone Modifications
1.2.3. Regulation of Gene Expression by Noncoding (micro) RNAs
2. Overview of Epigenetic Mechanisms Influenced by Soy Isoflavones according to Organ Type
2.1. Breast
2.1.1. IF Effects on Histone Modifications and DNA Methylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds and Concentration/Dose Tested | Treatment Time | Cell Lines—In vivo Models | Genes Regulated and Underlying Mechanisms | Methods Used—Comments | First Author, Year [Reference] |
|---|---|---|---|---|---|
| GEN, DAI, AglyMax, Equol at 12.5 nM–12.8 μM | in vitro with recombinant proteins | ↑ ac. histones, via ER-α or ER-β-mediated activation of co-activator SRC2 and recruitment of HAT p300. Effect on ER-β >ER-α ER-α: E2 >equol >GEN >AglyMax >DAI ER-β: E2 >equol = GEN = AglyMax = DAI | In vitro HAT activity assay, co-incubation of purified chromatin and proteins, radioactive detection by SDS-PAGE | Hong, 2004 [60] | |
| GEN 10 nM | 40–60 days | MCF7 after long-term genistein treatment (LTGT) | ↓ H3 expression and acetylation response after HDACi treatment ↓ growth response to HDACi TSA and apicidin | MTT assay Western blotting | Jawaid, 2010 [61] |
| GEN 5, 10, 25, 50 μM In vivo: GEN 250 mg/kg diet for 2 weeks prior to xenograft, alone and in comb. with TAM | 1, 2, 3 day 6 weeks | MDA-MB-231 in vitro and as xenografts in vivo, C3(1) SV40 TAg mice | ↑ ER-α expression (MDA-MB-231) ↑ sensitivity to E2 and TAM, ↑ PGR mRNA ↑ acH3, H3K9ac, acH4 at the ER-α promoter, especially in combination with TSA ↓ HDAC activity ↓ cell growth ↓ xenograft growth, esp. in comb with TAM ↓ tumor growth, ↓ PCNA staining, ↑ ER-α ↓ DNMT1 and HDAC1 mRNA | RT-qPCR Western blotting IHC | Li, 2013 [62] |
| GEN 2.5–400 μM In vivo: AIN-93G diet with GEN 250 mg/kg | 3 days, 7 weeks | HMEC transformed with SV40, hTERT (SH) and Ha-Ras (SHR) in vitro and as xenograft in vivo | ↑ apoptosis ↑ p21, p16 expression ↓ Bmi-1, c-Myc expression ↑ acH3 ↓ H3K27me3, H3K9me3 at p21 and p16 promoter ↑ H3K4me3, H3K9me3, H3K27me3 HMT activity (SHR cells) ↓ xenograft growth, ↓ tumor weight, ↓ PCNA ↑ p21 mRNA, ↓ c-Myc mRNA | MTT assay Flow cytometry Western blotting IHC ChIP-PCR HDAC activity assay (Active Motif) HMT activity assay (Epigentek) RT-qPCR | Li, 2013 [63] |
| GEN 18.5 μM DAI 78.5 μM Equol 12.8 μM | 2 days | MCF-7 MDA-MB-231 | ↓ H3K27me3, ↓ H3K9me3, ↓ H3K4me3, ↑ H4K8ac, ↑ H3K4ac at selected gene promoters (EZH2, BRCA1, ER-α, ER-β, SRC3, p300) ↓ EZH2 (HMT) staining, ↑ p300 (HAT) expression | ChIP-PCR Immunocytochemistry | Dagdemir, 2013 [64] |
| GEN 3.125 μM MCF-7 (once in 1 week), MCF-10a (once/week for 2 weeks) MDA-MB-468 (3/week for 1 week) | 1–2 weeks | MCF-7 MDA-MB-468 MCF-10a | ↓ GSTP1 promoter methylation (MDA-MB-468) ↑ expression (MDA-MB-468) ↓ RARβ2 and HIN1 promoter methylation (MCF-10a) | MSP, RT-qPCR | King-Batoon, 2008 [65] |
| GEN 50 μM (MCF-10aT), GEN 100 μM (MCF-7) | 0, 1, 2, 3 day | MCF-7 MCF-10aT (T24 Ha-Ras transformed) | ↓ DNMT1, 3a, 3b, c-Myc protein expression ↑ E2F-1 protein expression ↓ methylation at E2F-1 binding site ↓ c-Myc- and ↑ E2F-1 promoter binding ↓ hTERT mRNA expression | BS, Luciferase Assay, RT-qPCR, Western blotting, ChIP-PCR, ChIP-BS | Li, 2009 [66] |
| GEN 18.5 μM DAI 78.5 μM | 2 days | MCF-7 MDA-MB-231 MCF-10a | ↓ BRCA1 (exon1) and BRCA2 (exon2) methylation, ↓ anti-5-mC and MeCP2 fluorescence signal ↑ expression BRCA1, BRCA2 | MeDIP/PCR, IHC | Bosviel, 2012 [67] |
| Equol 2 μM | 3 weeks (each for 2 days) | MCF-7 MDA-MB-231 MCF-10a | ↓ BRCA1 and BRCA2 promoter methylation (not in MCF-10a), ↑ expression in the nuclei (BRCA1) and the cytoplasm (BRCA2) | qPCR-based quantitative analysis of methylated alleles (QAMA) | Bosviel, 2012 [68] |
| GEN 60, 100 μM | 1, 2, 3 day | MCF-7 MDA-MB-231 | ↓ Cell viability and global DNA methylation ↓ DNMT activity, DNMT1 mRNA/protein expression ↓ promoter methylation and ↑ mRNA expression ATM, APC, PTEN, SERPINB5 | SuperSense DNA Methylation Kit, EpiQuik DNMT Activity Assay Kit (Epigentek), MSP, RT-qPCR, Western blotting | Xie, 2014 [69] |
| In vivo | |||||
| Soy extract with 70.5% GEN, 27% DAI, 1.3% GLY Low dose: 37.2 mg/day High dose: 128.8 mg/day | 28 days | American women, samples of mammary ductoscopy | ↔ p16, RASSF1, RARβ2, ER, CCND2 methylation; RARβ2 methylation correlated with GEN serum levels (high dose group) and in low dose group for CCND2 | qMS-PCR | Qin, 2009 [70] |
2.1.2. Ongoing Projects on Genome-Wide Methylome Profiling
2.2. Uterus and Ovaries
2.2.1. Uterus and Endometrium
| Organ | Compounds and Concentration/Dose Tested | Incubation Time | Cell Lines—In Vivo Models | Genes Regulated and Underlying Mechanisms | Methods Used—Comments | First Author, Year [Reference] |
|---|---|---|---|---|---|---|
| Uterus | GEN 50 mg/kg bw by s.c. injection | PND 1–5 | CD-1 mice (ovx and intact) | 10 differentially methylated regions ovx: ↑ Nsbp1 promoter methylation and ↓ Nsbp1 expression intact: ↓ Nsbp1 promoter methylation and ↑ expression | Methylation sensitive restriction fingerprinting (MSRF), BS, RT-qPCR | Tang, 2008 [73] |
| GEN In vivo: 60, 200 mg/kg diet In vitro: 10 μM | 7 days | C57BL-6JJmsSlc mice, primary endometrial cells | ↓ SF-1 promoter/1st exon methylation, most pronounced at luminal side of uteri ↑ SF-1 mRNA expression and downstream targets (Cyp11a1, StAR, Cyp17a1, Cyp19a1) ↔ proliferation | BS, High resolution melting assay (Roche), colony forming assay RT-qPCR | Matsukura, 2011 [74] | |
| GEN In vivo: 50 mg/kg bw by s.c. injection | PND 10–12 | Eker rats | ↑ PI3K/Akt signaling ↑ EZH2 phosphorylation ↓ H3K27me3 levels ↑ hypersensitivity of ER-responsive genes in neonatal uteri and adult myometrium ↑ uterine tumor incidence and multiplicity | RT-qPCR Western blotting | Greathouse, 2012 [22] | |
| (Liver, Pancreas) | In vivo: 2% soy germ extract (Soylife) in the diet | Prenatal until PNW 6 | C3H mice | ↔ DNA methylation of skeletal α-actin, ER‑ɑ, c-fos; ↓ gender differences in methylation levels | semi-quantitative bisulfite PCR sequencing | Guerrero-Bosagna, 2008 [75] |
| Cervix | GEN 20 μM | 6 days | SiHa | ↓ RARβ2 promoter methylation, ↑ RARβ2 expression ↑ apoptosis | MSP | Jha, 2010 [76] |
| Ovaries | GEN 5 μM | 2 days | UL-3A, UL-3B (from one patient during cancer progression) | ↑ in UL-3A: miR-122a, -137, -196a, -204, -206, -217, -331, -449b, -454, -501, -515, -578 ↑ in UL-3B: miR-517c, -7 ↑ in UL-3A and 3B: miR-135, -765 | miRNA microarray | Parker, 2009 [77] |
| GEN 25, 50, 100, 200 μM | 1–3 days | SKOV3 | ↓ miR-27a expression ↓ cell growth and migration ↑ SPRY2 expression | RT-qPCR Western blotting | Xu, 2013 [78] | |
| (Uveal Melanoma) | GEN 10, 25, 50, 100, 200 μM; GEN 25, 50, 100 mg/kg bw i.p. | 1.5–3 days, daily for 30 days | C918, C918 xenografts | ↓ miR-27a expression ↑ ZBTB10 expression ↓ xenograft tumor growth | RT-qPCR | Sun, 2009 [79] |
2.2.2. Cervix
2.2.3. Ovaries
2.3. Urogenital System
2.3.1. Prostate
| Organ | Compounds and Concentration/Dosetested | Incubation Time | Cell lines—In Vivo Models | Genes Regulated and Underlying Mechanisms | Methods Used—Comments | First Author, Year [Reference] |
|---|---|---|---|---|---|---|
| Prostate | GEN 1, 10, 25, 50 μM | 3 days (applied every day) | LNCaP | ↓ HDAC6 protein expression ↑ acetylation of HSP90 promotes dissociation and degradation of AR ↓ AR-mediated signaling, PSA levels | RT-qPCR Immunocytochemistry IP Western blotting HDAC6 siRNA | Basak, 2008 [87] |
| GEN 10, 25 μM | 4 days (applied every day) | LNCaP, DuPro | ↑ p300, PCAF, CBP, HAT1 expression ↑ acH3, acH4, H3K4me2 at p21 and p16 promoter ↑ p21, p16INK4a expression ↓ cyclin A2, B2, E2 expression ↑ cell cycle arrest, apoptosis (DuPro) | RT-PCR ChIP-PCR Western blotting flow cytometry | Majid, 2008 [88] | |
| GEN 1, 10, 25, 50 μM GEN 50 μM in comb. with TSA 300 nM for 1day | 1, 2, 3 day | LNCaP, PC-3 | ↑ PTEN, CYLD, p53, FOXO3a expression ↓ Akt signaling ↓ SIRT1 activity/expression ↑ acH3K9 and ↓ H3K9me2 at PTEN, CYLD and FOXO3a promoter | RT-PCR Western blotting BS ChIP-PCR | Kikuno, 2008 [89] | |
| GEN (10, 25), 50 μM | 3 days | LNCaP, PC-3, RWPE-1 prostate epithelial cells | ↑ BTG3 expression ↓ BTG3 promoter methylation ↓ DNMT 1, 3a, 3b levels ↓ MBD2 binding activity ↑ HAT activity, ↔ HDAC activity ↑ acH3, acH4, H3K4me2, H3K4me3 ↔ H3K9me2, H3K9me3 | RT-PCR BS EpiQuik DNMT, MBD2, HAT, HDAC activity kits (Epigentek) ChIP-PCR | Majid, 2010 [90] | |
| GEN In vivo: 300 mg/kg diet | 2–4 weeks | C57BL/6J male mice | 3/900 regions differentially methylated | BstUI/HpaII digestion; mouse differential methylation hybridization (mDMH) | Day, 2002 [91] | |
| GEN 10, 20 μM | 6 days | LNCaP, PC-3 | ↓ RARβ2 promoter methylation, ↑ RARβ2 mRNA expression | MSP, RT-qPCR | Fang, 2005 [92] | |
| GEN 40 μM, DAI 110 μM | 2 days | LNCaP, DU 145, PC-3 | ↓ GSTP1, EPHB2 (PC-3) and RASSF1A (PC-3 and LNCaP) promoter methylation ↔ BRCA1 GEN >DAI ↑ nuclear protein expression of GSTP1 and EPHB2 (DU 145) | MSP, IHC | Vardi, 2010 [93] | |
| GEN 40 μM DAI 110μM | 2 days | DU 145 PC-3 | ↓ BRCA1, GSTP1, EPB2 promoter methylation (GEN), ↑ BRCA1, GSTP1, EPB2 expression (PC-3) ↓ BRCA1, GSTP1 expression (DU 145) | Methylation Profiler Methylation Kit (Qiagen), Western blotting | Adjakly, 2011 [94] | |
| GEN 20 μM | 6 days | DU 145 PC-3 LNCaP, ARCaP-E, ARCaP-M | ↔ APC, SOX7, SFRP1, WIF1 promoter methylation ↔ genome-wide DNA methylation (27 k) ↑ H3K9ac at APC, SOX7, SFRP1, SFRP2, DKK, WIF1 promoter, ↑ HAT1 expression ↑ SOX7, SFRP1 mRNA expression ↓ proliferation, ↑ apoptosis in comb. with vorinostat (HDAC inhibitor) ↑ DNA repair genes BRCA1, BARD1, RAD23B, XRCC2 ↓ expression of BIRC7, SLUG, HES1, TGFB1I1 | MSP, Illumina 27 k, BS ChIP-PCR Western blotting RT-qPCR Whole genome expression profiling | Phillip, 2012 [95] | |
| GEN 40 μM DAI 100 μM | 2 days | DU 145 PC-3 LNCaP | ↓ miR-125a, -125b, -15b, -320, -155, -208b, -211, -376a, -411, -520g, -542-5p ↑ miR-548b, -15a | RT-qPCR | Rabiau, 2011 [96] | |
| GEN 25 μM (in comb. with 5 μM decitabine and TSA) | 4 days | DU 145 PC-3 PWR-1E | ↑ miR-145 expression ↑ TNFSF10 expression ↑ apoptosis and ↓ CDK6 in miR-145-overexpressing cells | RT-qPCR mRNA microarray Western blotting | Zaman, 2010 [97] | |
| GEN 25, 50 μM | 4 days | LNCaP PC-3 | ↑ miR-1296 levels ↓ expression of MCM genes ↓ CDK2, CDK7, CDT1 expression ↓ cells in S-phase | RT-qPCR Western blotting | Majid, 2010 [98] | |
| GEN 50 μM | 4 days | PC-3 LNCaP DU 145 patient samples | ↓ miR-221, miR-222 levels ↑ ARHI expression ↓ proliferation, ↑apoptosis in cells overexpressing ARHI | RT-qPCR Western blotting | Chen, 2011 [99] | |
| GEN 25 μM | 4 days | PC-3, DU 145, RWPE-1 | ↓ miR-151a-5p ↑ SOX17, ARHGDIA in miR-151a-5p precursor transfected cells | RT-qPCR Western blotting | Chiyomaru, 2012 [100] | |
| G2535 (equivalent to 20 μM GEN) | 5 days | LNCaP, PC-3, VCap, V4-2B, ARCaP-M | ↓ methylation at miR-29a and miR-1256 promoter ↑ miR-29a and miR-1256 ↓ TRIM68 and PGK-1 ↓ cell growth and invasion | Illumina 450k array MSP microarray Luc-Pair miR Luciferase assay, Western blotting | Li, 2012 [101] | |
| GEN 25, 50 μM | 4 days | PC-3 DU 145 RWPE‑1 patient samples, xenografts | ↑ miR-574-3p ↓ RAC1, EGFR, p300 in miR-574-3p precursor transfected cells ↓ proliferation, ↑apoptosis in cells overexpressing miR-574-3p | RT-qPCR Western blotting 5’UTR luciferase reporter assay | Chiyomaru, 2013 [102] | |
| GEN 25 μM | 4 days | PC-3 DU 145 Xenografts | ↑ miR-34a ↓ HOTAIR expression ↓ proliferation, ↑apoptosis | microarray RT-qPCR Dual-luciferase reporter assay | Chiyomaru, 2013 [103] | |
| GEN 25 μM | 4 days | PC-3 DU 145 RWPE-1 | ↓ miR-1260b expression ↑ SFRP1, Smad4 expression ↓ CpG methylation at SFRP1 promoter ↓ H3K9me2, H3K9me3, H3K27me3 at SFRP1 and Smad4 genes ↓ proliferation, ↓invasion, ↓migration ↑ apoptosis | microarray BS, ChIP-PCR, RT-qPCR Western blotting | Hirata, 2014 [104] | |
| Kidney | GEN 10, 25, 50 μM | 3 days | A498, ACHN, HEK-293, HK-2 non-malignant immortalized renal cells | ↓ cell growth, cell cycle progression ↑ BTG3 expression ↓ BTG3 promoter methylation ↓ DNMT activity, DNMT 3b levels ↓ MBD2 binding activity ↑ HAT activity, ↓ HDAC activity ↑ acH3, acH4, H3K4me2, H3K4me3 ↔ H3K9me2, H3K9me3 | flow cytometry MTT assay RT-qPCR BS EpiQuik DNMT, MBD2, HAT, HDAC activity kits (Epigentek) ChIP-PCR | Majid, 2009 [105] |
| GEN 25 μM | 4 days | A-498 in vitro and in vivo | ↓ miR-21 expression ↓ xenograft growth (pretreated with 25 μM GEN for 4 days) | RT-qPCR | Zaman, 2012 [106] | |
| GEN 25, 50 μM | 4 days | A-498, ACHN, Caki-1, -2 | ↓ miR-23b-3p ↑ PTEN in miR-23b-3p knockdown cells ↓ proliferation, ↓ invasion in miR-23b-3p knockdown cells | RT-qPCR Western blotting | Zaman, 2012 [107] | |
| GEN 25 μM | 4 days | RCC patient samples, A-498, 786-O, Caki-2 | ↓ miR-1260b expression ↓ Wnt-signaling ↑ viability, ↑ invasion ↓ apoptosis in miR-1260b transfected cells | microarray RT-qPCR TOPflash luciferase assay | Hirata, 2013 [108] | |
| GEN 25 μM | 4 days | 786-O ACHN HK-2 | ↑ miR-141 levels ↓ HOTAIR expression ↓ ABL2 expression ↑ PCDH10 in pre-miR-141 transfected cells ↑ Snail in anti-miR-141 transfected cells ↓ proliferation in HOTAIR knockdown cells ↑ proliferation in pre-miR-141 expressing cells | RT-qPCR Luciferase reporter assay | Chiyomaru, 2014 [109] |
2.3.2. Kidney
2.4. Gastrointestinal Tract
2.4.1. Esophagus
| Organ | Compounds and Concentration/Dose Tested | Incubation Time | Cell Lines—In Vivo Models | Genes Regulated and Underlying Mechanisms | Methods Used—Comments | First Author, Year [Reference] |
|---|---|---|---|---|---|---|
| Esophagus | GEN 2, 5, 10, 20 μM DAI 5, 10, 20 μM | 1, 2, 4, 6 day | KYSE 150, KYSE 510 | ↓ RARβ2, p16, MGMT promoter methylation ↑ RARβ2, p19, MGMT expression ↓ DNMT and HDAC activity (weak) ↓ cell growth ↔ DNMT and MBD2 mRNA expression | MSP RT-PCR, RT-qPCR DNMT activity assay with [3H]-SAM HDAC Kit (Upstate) | Fang, 2005 [92] |
| Stomach | GEN 10, 25, 50 μM | 3 days | AGS | ↓ PCDH17 promoter methylation ↑ PCDH17 expression | BS, RT-qPCR | Yang, 2012 [117] |
| Colon | GEN 25 μM | 3 days | HCT 116, HT-29 | ↓ RARβ2 promoter methylation | BS | Spurling, 2008 [118] |
| Novasoy Extract 200 μg/mL, GEN 75 μM | 4 days | SW1116, SW480, DLD-1 | ↓ WNT5a promoter methylation, ↑ WNT5a expression (in SW1116 GEN) ↓ cell viability | MSP, Methylation-sensitive restriction enzyme PCR (MSREP), BS | Wang, 2010 [119] | |
| GEN 75 μM | 4 days | DLD-1 | ↓ SFRP2 promoter methylation ↑ SFRP2 expression ↓ cell proliferation ↑ apoptosis | MSP, RT-qPCR | Zhang, 2011 [120] | |
| GEN 50, 75 μM | 2 days, 4 days | SW480, DLD-1, HCT-15, HT-29, RKO, SW48 | ↔ DKK1 promoter methylation, ↑ DKK1 expression (SW480) ↓ proliferation, ↑ cell cycle arrest in G2/M phase (SW480) ↓ cyclinD1 mRNA, ↔ p21, cMyc mRNA ↑ acH3 at DKK1 promoter ↔ acH4, H3K4me2 | MSP, BS RT-qPCR DKK1 knockdown and overexpression, ChIP-PCR | Wang, 2012 [121] | |
| SPI (140 mg/kg GEN) Casein protein plus 140 mg/kg GEN; AOM challenge | Prenatal until PNW 14 | Sprague-Dawley rats | ↓ AOM-induced promoter demethylation ↓ AOM-induced expression of Sfrp2, Sfrp5 and Wnt5a ↓ RNA PolII binding to Sfrp2 (GEN), Sfrp5 and Wnt5a promoter ↓ AcH3, H3K9me3, H3S10P at Sfrp2, Sfrp5, Wnt5a promoter ↑ nuclear HDAC3 expression | MSP, BS RT-qPCR ChIP-PCR Western blotting | Zhang, 2013 [122] | |
| GEN 10–200 μM | 1 day | HT-29 | ↓ HDAC1 expression and activity (IC50 97 μM) ↔ no cytotoxicity in the presence of catalase to remove artifactual hydrogen peroxide | HDAC assay kit (Cayman) Sulforhodamin B staining WST-1 (water soluble tetrazolium) assay Western blotting | Groh, 2013 [123] | |
| Pancreas | G2535 (equivalent to 10 μM GEN) | 3 weeks | AsPC-1, MiaPaCa-2, Panc-1 L3.6pl, Colo357, BxPC-3, HPAC | altered expression of 67/711 miRs ↑ miR-200 and let-7 family members ↓ EMT phenotype and markers ↑ sensitivity to gemcitabine in gem. resistant cells | miRNA microarray RT-qPCR Western blotting | Li, 2009 [124] |
| G2535 (equivalent to 25 μM GEN) | 2 days | Colo357, Panc-1, HPDE | ↑ miR-146a expression ↓ EGFR, IRAK-1, NF-κB and MTA-2 expression ↓ cell invasion | miRNA microarray RT-qPCR Western blotting | Li, 2010 [125] | |
| GEN 60 μM | 16 h, 3 days, 7 days | AsPC-1, MiaPaCa-2 | ↑ miR-34a expression ↓ Notch-1 expression ↓ cell growth ↑ apoptosis | RT-qPCR Western blotting | Xia, 2012 [126] | |
| GEN 60 μM | 3 days | AsPC-1, BxPC-3 | ↓ miR-223 expression ↑ Fbw7 protein expression ↓ cell growth ↑ apoptosis ↓ migration and invasion | MTT assay FACS for apoptosis detection RT-qPCR Western blotting | Ma, 2013 [127] | |
| GEN | n.a. | n.a. | ↓ miR-27a ↓ cell growth ↑ apoptosis ↓ invasion | MTT assay RT-qPCR Western blotting | Xia, 2014 [128] (abstract only) | |
| Equol 10, 100 μg | PND 1–10 | Sprague-Dawley rats | ↑ methylation at the c-Ha-Ras gene | methylation sensitive restriction digestion, Southern blotting | Lyn-Cook, 1995 [80] |
2.4.2. Stomach
2.4.3. Colon
2.4.4. Pancreas
2.5. Further in Vivo Studies on Epigenetic Effects of IF
| Organ | Compounds and Concentration/Dose Tested | Incubation Time | Cell lines—In Vivo Models | Genes Regulated, Underlying Mechanisms | Methods Used—Comments | First AutorYear [Reference] |
|---|---|---|---|---|---|---|
| Leukemia | GEN 0.1, 1, 10 μM In vivo: GEN 0.5% (w/w) in the diet | 2 days, whole lifespan | HL-60, L1210, L1210/ARA-C; CD2F1 male mice | ↑ loss of clonogenicity ↓ p57 promoter methylation ↔ 5-Aza-CdR incorporation ↑ survival time (mice) | Colony forming Assay, [6-3H]5-Aza-CdR incorporation, MSP, UPLC | Raynal, 2008 [139] |
| Neuro-blastoma | GEN In vivo: 2 mg/mouse/day | 15 days | SK-N-SH Neuroblastoma xenografts | ↓ xenograft tumor size and frequency ↓ CHD5 promoter methylation ↑ CHD5 mRNA expression ↓ DNTM3b mRNA expression | BS, RT-qPCR, Western blotting | Li, 2012 [140] |
| Agouti mice | GEN In vivo: 250 mg/kg diet | prenatal until PND 21 | Avymice (tail clips, liver, brain, kidney | ↑ methylation of CpG sites in the cryptic promoter region of Avy IAP shifting coat color to pseudo agouti (less obese) ↔ coat color ↔ coat color | BS | Dolinoy, 2006 [141] Badger, 2008 [142] Rosenfeld, 2013 [143] |
| ESC | GEN 5 μM | 4 days | CGE, E14Tg2a | 149 differentially methylated regions: 54 hyper- and 95 hypomethylated ↑ Ucp1 and Sytl1 demethylation during ESC differentiation after initial de novo methylation | MspI fragment based DNA methylation typing (MFMT), BS | Sato, 2011 [144] |
| Blood, Bone marrow | GEN In vivo: 270 mg/kg diet | prenatal until PND 0 | 123/SvJ:C57BL/6J mice | ↑ methylation in repetitive elements in adult mice (not in fetal mice) ↓ HDAC6, p21, cyclin D1, PCNA, and IGF2 expression in adult mice ↑ switch from primitive erythroid to definite erythroid lineage (blood cell development) | Methylation-sensitive McrBC real time PCR assay, Agilent Microarray 4x44k | Vanhees, 2011 [145] |
| Water flea | GEN In culture media 4.7 mg/L (3/week for 3 weeks) | 21 days, multi-generational | Daphnia magna | ↓ reproduction in F0 ↓ body length in F0, F1 and F2 ↔ global DNA methylation | UPLC | Vandegehuchte, 2010 [146] |
| Adipose tissue, muscle, (liver, blood) | TAD TAD + IF equivalent to 180 mg/person/day | 8 weeks, plus change of chow for 8 weeks | Cynomolgus monkeys | Methylation of HOXA5, HOXB1, HOXA11, NTRK3, PLAG12A, ABCG5, TBX5 differed significantly in muscle or fat tissue ↓ fasting insulin levels ↑ improved insulin sensitivity | Illumina 27k array PyroSequencing | Howard, 2011 [147] |
| PBMCs | soy rich diet (~230 mg IF/day) | 4 weeks | Heavy smokers | ↑ Line-1 methylation ↓ inter-individual methylation variability ↔ MTHFR, MLH1, RASSF1A, CDN2A, ARF | PyroSequencing | Scoccianti, 2011 [148] |
2.5.1. Anti-Cancer Treatment of Leukemia and Neuroblastoma in Mouse Models
2.5.2. Developmental/Multi-Generational Effects in Various Models
2.5.3. Short-Term Intervention in Adult Non-Human Primates and Humans
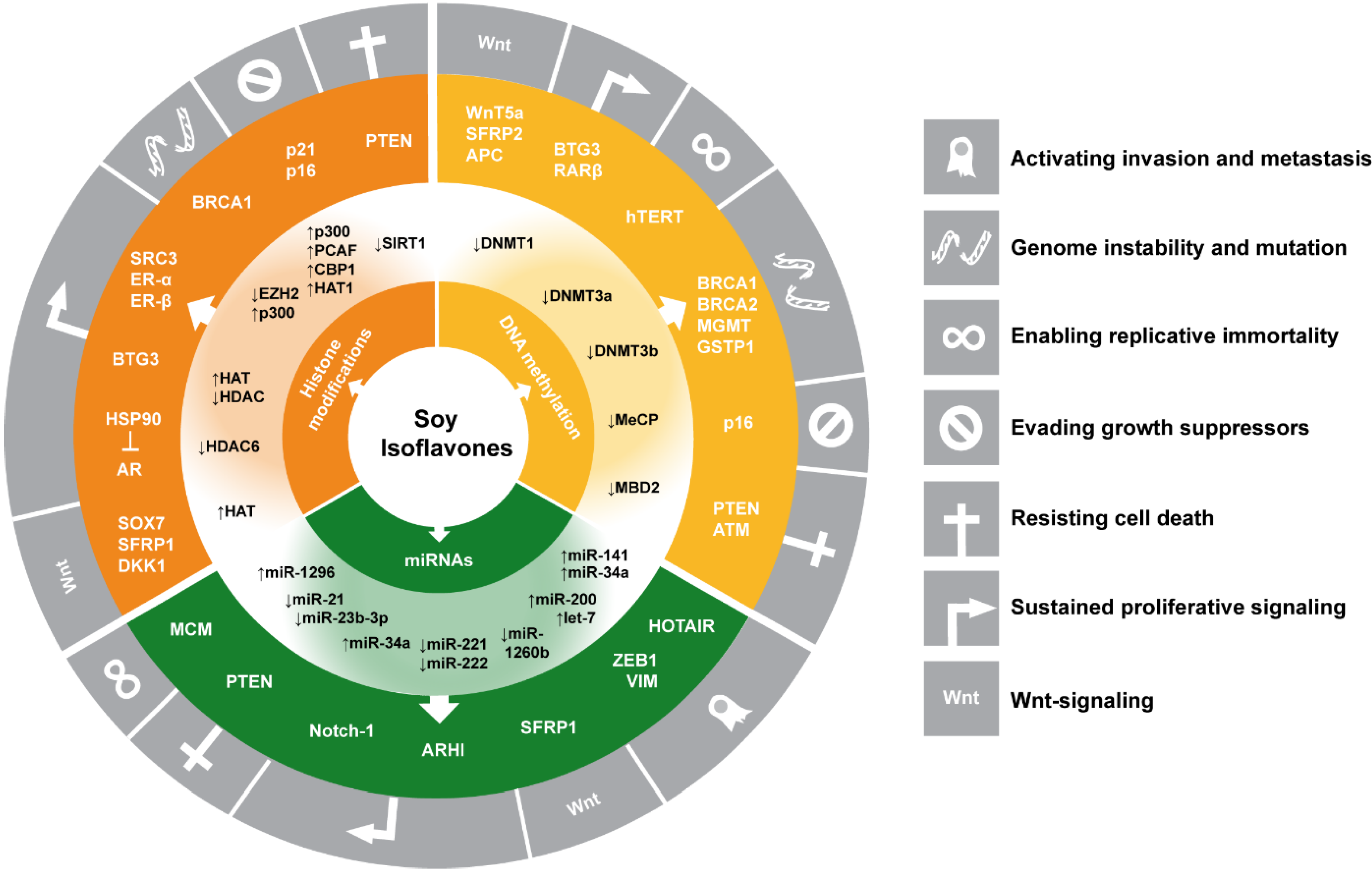
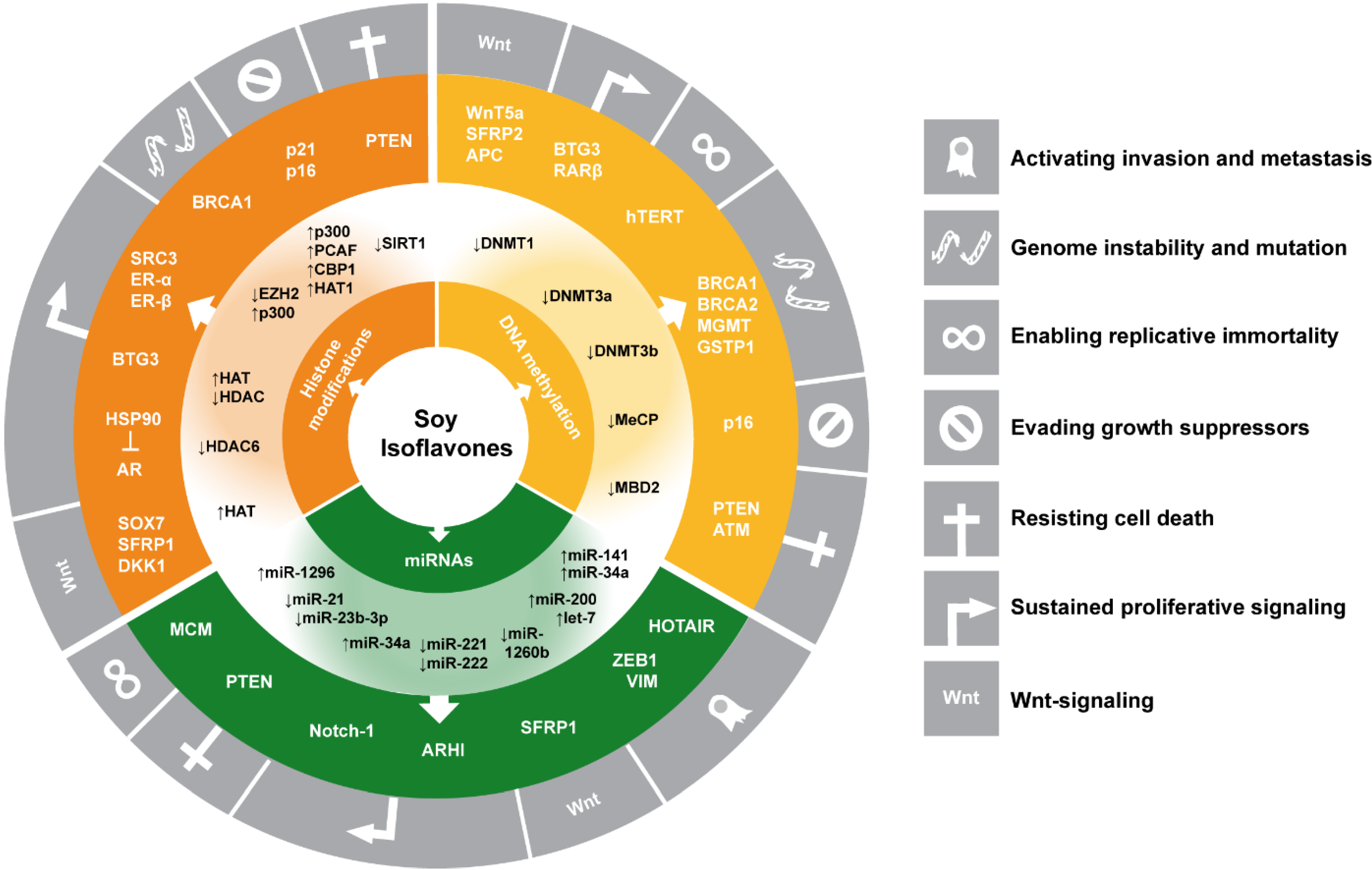
3. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IARC. World Cancer Report 2014, 1st ed.; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2014. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; AICR: Washington, DC, USA, 2007. [Google Scholar]
- Bingham, S.A.; Atkinson, C.; Liggins, J.; Bluck, L.; Coward, A. Phyto-oestrogens: Where are we now? Br. J. Nutr. 1998, 79, 393–406. [Google Scholar]
- Chen, Z.; Zheng, W.; Custer, L.J.; Dai, Q.; Shu, X.O.; Jin, F.; Franke, A.A. Usual dietary consumption of soy foods and its correlation with the excretion rate of isoflavonoids in overnight urine samples among Chinese women in Shanghai. Nutr. Cancer 1999, 33, 82–87. [Google Scholar]
- Seow, A.; Shi, C.Y.; Franke, A.A.; Hankin, J.H.; Lee, H.P.; Yu, M.C. Isoflavonoid levels in spot urine are associated with frequency of dietary soy intake in a population-based sample of middle-aged and older Chinese in Singapore. Cancer Epidemiol. Biomark. Prev. 1998, 7, 135–140. [Google Scholar]
- Cassidy, A.; Faughnan, M. Phyto-oestrogens through the life cycle. Proc. Nutr. Soc. 2000, 59, 489–496. [Google Scholar]
- Keinan-Boker, L.; Peeters, P.; Mulligan, A.; Navarro, C.; Slimani, N.; Mattisson, I.; Lundin, E.; McTaggart, A.; Allen, N.; Overvad, K.; et al. Soy product consumption in 10 European countries: The European Prospective Investigation into Cancer and Nutrition (EPIC) study. Public Health Nutr. 2002, 5, 1217–1226. [Google Scholar]
- Clarke, D.B.; Barnes, K.A.; Castle, L.; Rose, M.; Wilson, L.A.; Baxter, M.J.; Price, K.R.; DuPont, M.S. Levels of phytoestrogens, inorganic trace-elements, natural toxicants and nitrate in vegetarian duplicate diets. Food Chem. 2003, 81, 287–300. [Google Scholar]
- UK Ministry of Agriculture Fisheries and Food (MAFF UK). Plant oestrogens in soya-based infant formulae. Food Surveill. Inf. Sheet 1998, 167, 1–8. [Google Scholar]
- Steiner, C.; Arnould, S.; Scalbert, A.; Manach, C. Isoflavones and the prevention of breast and prostate cancer: New perspectives opened by nutrigenomics. Br. J. Nutr. 2008, 99, 78–108. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Available online: http://globocan.iarc.fr (accessed on 22 July 2014).
- Ziegler, R.G.; Hoover, R.N.; Pike, M.C.; Hildesheim, A.; Nomura, A.M.Y.; West, D.W.; Wu-Williams, A.H.; Kolonel, L.N.; Horn-Ross, P.L.; Rosenthal, J.F.; et al. Migration Patterns and Breast Cancer Risk in Asian-American Women. J. Natl. Cancer Inst. 1993, 85, 1819–1827. [Google Scholar]
- Tham, D.M.; Gardner, C.D.; Haskell, W.L. Potential Health Benefits of Dietary Phytoestrogens: A Review of the Clinical, Epidemiological, and Mechanistic Evidence. J. Clin. Endocrinol. Metab. 1998, 83, 2223–2235. [Google Scholar]
- Mori, M.; Masumori, N.; Fukuta, F.; Nagata, Y.; Sonoda, T.; Sakauchi, F.; Ohnishi, H.; Nojima, M.; Tsukamoto, T. Traditional Japanese diet and prostate cancer. Mol. Nutr. Food Res. 2009, 53, 191–200. [Google Scholar]
- Warri, A.; Saarinen, N.M.; Makela, S.; Hilakivi-Clarke, L. The role of early life genistein exposures in modifying breast cancer risk. Br. J. Cancer 2008, 98, 1485–1493. [Google Scholar]
- De Assis, S.; Hilakivi-Clarke, L. Timing of Dietary Estrogenic Exposures and Breast Cancer Risk. Ann. N. Y. Acad. Sci. 2006, 1089, 14–35. [Google Scholar]
- Hilakivi-Clarke, L. Nutritional modulation of terminal end buds: Its relevance to breast cancer prevention. Curr. Cancer Drug Target 2007, 7, 465–474. [Google Scholar]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar]
- Fang, H.; Tong, W.; Shi, L.M.; Blair, R.; Perkins, R.; Branham, W.; Hass, B.S.; Xie, Q.; Dial, S.L.; Moland, C.L.; et al. Structure—Activity Relationships for a Large Diverse Set of Natural, Synthetic, and Environmental Estrogens. Chem. Res. Toxicol. 2001, 14, 280–294. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar]
- Greathouse, K.L.; Bredfeldt, T.; Everitt, J.I.; Lin, K.; Berry, T.; Kannan, K.; Mittelstadt, M.L.; Ho, S.M.; Walker, C.L. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol. Cancer Res. 2012, 10, 546–557. [Google Scholar]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar]
- Goelz, S.E.; Vogelstein, B.; Hamilton, S.R.; Feinberg, A.P. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985, 228, 187–190. [Google Scholar]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar]
- Stefanska, B.; Suderman, M.; Machnes, Z.; Bhattacharyya, B.; Hallett, M.; Szyf, M. Transcription onset of genes critical in liver carcinogenesis is epigenetically regulated by methylated DNA-binding protein MBD2. Carcinogenesis 2013, 34, 2738–2749. [Google Scholar]
- Mayol, G.; Martín-Subero, J.; Ríos, J.; Queiros, A.; Kulis, M.; Suñol, M.; Esteller, M.; Gómez, S.; Garcia, I.; de Torres, C.; et al. DNA hypomethylation affects cancer-related biological functions and genes relevant in neuroblastoma pathogenesis. PLoS One 2012, 7, e48401. [Google Scholar]
- Stefanska, B.; Huang, J.; Bhattacharyya, B.; Suderman, M.; Hallett, M.; Han, Z.-G.; Szyf, M. Definition of the Landscape of Promoter DNA Hypomethylation in Liver Cancer. Cancer Res. 2011, 71, 5891–5903. [Google Scholar]
- Pakneshan, P.; Szyf, M.; Rabbani, S.A. Hypomethylation of Urokinase (uPA) Promoter in Breast and Prostate Cancer: Prognostic and Therapeutic Implications. Curr. Cancer Drug Targets 2005, 5, 471–488. [Google Scholar]
- Rauch, T.A.; Wu, X.; Zhong, X.; Riggs, A.D.; Pfeifer, G.P. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 671–678. [Google Scholar]
- Sato, N.; Fukushima, N.; Matsubayashi, H.; Goggins, M. Identification of maspin and S100P as novel hypomethylation targets in pancreatic cancer using global gene expression profiling. Oncogene 2003, 23, 1531–1538. [Google Scholar]
- Kopelovich, L.; Crowell, J.A.; Fay, J.R. The epigenome as a target for cancer chemoprevention. J. Natl. Cancer Inst. 2003, 95, 1747–1757. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar]
- Fullgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar]
- Upadhyay, A.K.; Cheng, X. Dynamics of histone lysine methylation: Structures of methyl writers and erasers. Prog. Drug Res. 2011, 67, 107–124. [Google Scholar]
- Laugesen, A.; Helin, K. Chromatin Repressive Complexes in Stem Cells, Development, and Cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar]
- miRBase: The microRNA Database. Available online: http://mirbase.org (accessed on 19 July 2014).
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. (Berl.) 2013, 91, 791–801. [Google Scholar]
- Rietjens, I.M.C.M.; Sotoca, A.M.; Vervoort, J.; Louisse, J. Mechanisms underlying the dualistic mode of action of major soy isoflavones in relation to cell proliferation and cancer risks. Mol. Nutr. Food Res. 2013, 57, 100–113. [Google Scholar]
- Fang, M.; Chen, D.; Yang, C.S. Dietary Polyphenols May Affect DNA Methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar]
- Grayson, M. Breast cancer. Nature 2012, 485, S49. [Google Scholar]
- National Cancer Institute (Bethesda, MD, USA): SEER Cancer Statistics Factsheets: All Cancer Sites. Available online: http://seer.cancer.gov/statfacts/html/all.html (accessed on 23 August 2014).
- Yager, J.D.; Davidson, N.E. Estrogen Carcinogenesis in Breast Cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar]
- Maxmen, A. The hard facts. Nature 2012, 485, S50–S51. [Google Scholar]
- Nagata, C.; Mizoue, T.; Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Wakai, K.; Inoue, M.; Tsugane, S.; Sasazuki, S.; et al. Soy Intake and Breast Cancer Risk: An Evaluation Based on a Systematic Review of Epidemiologic Evidence Among the Japanese Population. Jpn. J. Clin. Oncol. 2014, 44, 282–295. [Google Scholar]
- Messina, M. A Brief Historical Overview of the Past Two Decades of Soy and Isoflavone Research. J. Nutr. 2010, 140, 1350S–1354S. [Google Scholar]
- Taylor, C.K.; Levy, R.M.; Elliott, J.C.; Burnett, B.P. The effect of genistein aglycone on cancer and cancer risk: A review of in vitro, preclinical, and clinical studies. Nutr. Rev. 2009, 67, 398–415. [Google Scholar]
- Fritz, H.; Seely, D.; Flower, G.; Skidmore, B.; Fernandes, R.; Vadeboncoeur, S.; Kennedy, D.; Cooley, K.; Wong, R.; Sagar, S.; et al. Soy, red clover, and isoflavones and breast cancer: A systematic review. PLoS One 2013, 8, e81968. [Google Scholar]
- Magee, P.J.; Rowland, I. Soy products in the management of breast cancer. Curr. Opin. Clin. Nutr. Metab. Care 2012, 6, 586–591. [Google Scholar]
- D’Adamo, C.R.; Sahin, A. Soy foods and supplementation: A review of commonly perceived health benefits and risks. Altern. Ther. Health Med. 2014, 20, 39–51. [Google Scholar]
- Powles, T.J. Anti-oestrogenic prevention of breast cancer—The make or break point. Nat. Rev. Cancer 2002, 2, 787–794. [Google Scholar]
- Hong, T.; Nakagawa, T.; Pan, W.; Kim, M.Y.; Kraus, W.L.; Ikehara, T.; Yasui, K.; Aihara, H.; Takebe, M.; Muramatsu, M.; et al. Isoflavones stimulate estrogen receptor-mediated core histone acetylation. Biochem. Biophys. Res. Commun. 2004, 317, 259–264. [Google Scholar]
- Jawaid, K.; Crane, S.R.; Nowers, J.L.; Lacey, M.; Whitehead, S.A. Long-term genistein treatment of MCF-7 cells decreases acetylated histone 3 expression and alters growth responses to mitogens and histone deacetylase inhibitors. J. Steroid Biochem. Mol. Biol. 2010, 120, 164–171. [Google Scholar]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic reactivation of estrogen receptor-alpha (ERalpha) by genistein enhances hormonal therapy sensitivity in ERalpha-negative breast cancer. Mol. Cancer 2013, 12, 9. [Google Scholar]
- Li, Y.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic regulation of multiple tumor-related genes leads to suppression of breast tumorigenesis by dietary genistein. PLoS One 2013, 8, e54369. [Google Scholar]
- Dagdemir, A.; Durif, J.; Ngollo, M.; Bignon, Y.J.; Bernard-Gallon, D. Histone lysine trimethylation or acetylation can be modulated by phytoestrogen, estrogen or anti-HDAC in breast cancer cell lines. Epigenomics 2013, 5, 51–63. [Google Scholar]
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ. Mol. Mutagen. 2008, 49, 36–45. [Google Scholar]
- Li, Y.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int. J. Cancer 2009, 125, 286–296. [Google Scholar]
- Bosviel, R.; Dumollard, E.; Dechelotte, P.; Bignon, Y.J.; Bernard-Gallon, D. Can soy phytoestrogens decrease DNA methylation in BRCA1 and BRCA2 oncosuppressor genes in breast cancer? OMICS 2012, 16, 235–244. [Google Scholar]
- Bosviel, R.; Durif, J.; Dechelotte, P.; Bignon, Y.J.; Bernard-Gallon, D. Epigenetic modulation of BRCA1 and BRCA2 gene expression by equol in breast cancer cell lines. Br. J. Nutr. 2012, 108, 1187–1193. [Google Scholar]
- Xie, Q.; Bai, Q.; Zou, L.-Y.; Zhang, Q.-Y.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.-D.; Mi, M.-T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014, 53, 422–431. [Google Scholar]
- Qin, W.; Zhu, W.; Shi, H.; Hewett, J.E.; Ruhlen, R.L.; MacDonald, R.S.; Rottinghaus, G.E.; Chen, Y.C.; Sauter, E.R. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr. Cancer 2009, 61, 238–244. [Google Scholar]
- Sonnet, M.; Baer, C.; Rehli, M.; Weichenhan, D.; Plass, C. Enrichment of methylated DNA by methyl-CpG immunoprecipitation. Methods Mol. Biol. 2013, 971, 201–212. [Google Scholar]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar]
- Tang, W.Y.; Newbold, R.; Mardilovich, K.; Jefferson, W.; Cheng, R.Y.; Medvedovic, M.; Ho, S.M. Persistent hypomethylation in the promoter of nucleosomal binding protein 1 (Nsbp1) correlates with overexpression of Nsbp1 in mouse uteri neonatally exposed to diethylstilbestrol or genistein. Endocrinology 2008, 149, 5922–5931. [Google Scholar]
- Matsukura, H.; Aisaki, K.; Igarashi, K.; Matsushima, Y.; Kanno, J.; Muramatsu, M.; Sudo, K.; Sato, N. Genistein promotes DNA demethylation of the steroidogenic factor 1 (SF-1) promoter in endometrial stromal cells. Biochem. Biophys. Res. Commun. 2011, 412, 366–372. [Google Scholar]
- Guerrero-Bosagna, C.M.; Sabat, P.; Valdovinos, F.S.; Valladares, L.E.; Clark, S.J. Epigenetic and phenotypic changes result from a continuous pre and post natal dietary exposure to phytoestrogens in an experimental population of mice. BMC Physiol. 2008, 8, 17. [Google Scholar]
- Jha, A.K.; Nikbakht, M.; Parashar, G.; Shrivastava, A.; Capalash, N.; Kaur, J. Reversal of hypermethylation and reactivation of the RARbeta2 gene by natural compounds in cervical cancer cell lines. Folia Biol. (Praha) 2010, 56, 195–200. [Google Scholar]
- Parker, L.P.; Taylor, D.D.; Kesterson, J.; Metzinger, D.S.; Gercel-Taylor, C. Modulation of microRNA associated with ovarian cancer cells by genistein. Eur. J. Gynaecol. Oncol. 2009, 30, 616–621. [Google Scholar]
- Xu, L.; Xiang, J.; Shen, J.; Zou, X.; Zhai, S.; Yin, Y.; Li, P.; Wang, X.; Sun, Q. Oncogenic MicroRNA-27a is a target for genistein in ovarian cancer cells. Anticancer Agents Med. Chem. 2013, 13, 1126–1132. [Google Scholar]
- Sun, Q.; Cong, R.; Yan, H.; Gu, H.; Zeng, Y.; Liu, N.; Chen, J.; Wang, B. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol. Rep. 2009, 22, 563–567. [Google Scholar]
- Lyn-Cook, B.D.; Blann, E.; Payne, P.W.; Bo, J.; Sheehan, D.; Medlock, K. Methylation profile and amplification of proto-oncogenes in rat pancreas induced with phytoestrogens. Proc. Soc. Exp. Biol. Med. 1995, 208, 116–119. [Google Scholar]
- Bosch, F.X.; Broker, T.R.; Forman, D.; Moscicki, A.B.; Gillison, M.L.; Doorbar, J.; Stern, P.L.; Stanley, M.; Arbyn, M.; Poljak, M.; et al. Comprehensive control of human papillomavirus infections and related diseases. Vaccine 2013, 31 (Suppl. 8), 1–31. [Google Scholar]
- Karius, T.; Schnekenburger, M.; Dicato, M.; Diederich, M. MicroRNAs in cancer management and their modulation by dietary agents. Biochem. Pharmacol. 2012, 83, 1591–1601. [Google Scholar]
- Costello, A.J.; Corcoran, N.M. Development, applied and surgical development of the prostate. In Prostate Cancer: A Comprehensive Perspective; Tewari, A., Ed.; Springer Verlag: London, UK, 2013; pp. 3–18. [Google Scholar]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar]
- Kimura, T. East meets West: Ethnic differences in prostate cancer epidemiology between East Asians and Caucasians. Chin. J. Cancer 2012, 3, 421–429. [Google Scholar]
- Jian, L. Soy, isoflavones, and prostate cancer. Mol. Nutr. Food Res. 2009, 53, 217–226. [Google Scholar]
- Basak, S.; Pookot, D.; Noonan, E.J.; Dahiya, R. Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol. Cancer Ther. 2008, 7, 3195–3202. [Google Scholar]
- Majid, S.; Kikuno, N.; Nelles, J.; Noonan, E.; Tanaka, Y.; Kawamoto, K.; Hirata, H.; Li, L.C.; Zhao, H.; Okino, S.T.; et al. Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res. 2008, 68, 2736–2744. [Google Scholar]
- Kikuno, N.; Shiina, H.; Urakami, S.; Kawamoto, K.; Hirata, H.; Tanaka, Y.; Majid, S.; Igawa, M.; Dahiya, R. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int. J. Cancer 2008, 123, 552–560. [Google Scholar]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer 2010, 116, 66–76. [Google Scholar]
- Day, J.K.; Bauer, A.M.; DesBordes, C.; Zhuang, Y.; Kim, B.E.; Newton, L.G.; Nehra, V.; Forsee, K.M.; MacDonald, R.S.; Besch-Williford, C.; et al. Genistein alters methylation patterns in mice. J. Nutr. 2002, 132, 2419S–2423S. [Google Scholar]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar]
- Vardi, A.; Bosviel, R.; Rabiau, N.; Adjakly, M.; Satih, S.; Dechelotte, P.; Boiteux, J.P.; Fontana, L.; Bignon, Y.J.; Guy, L.; et al. Soy Phytoestrogens Modify DNA Methylation of GSTP1, RASSF1A, EPH2 and BRCA1 Promoter in Prostate Cancer Cells. In Vivo 2010, 24, 393–400. [Google Scholar]
- Adjakly, M.; Bosviel, R.; Rabiau, N.; Boiteux, J.P.; Bignon, Y.J.; Guy, L.; Bernard-Gallon, D. DNA methylation and soy phytoestrogens: Quantitative study in DU-145 and PC-3 human prostate cancer cell lines. Epigenomics 2011, 3, 795–803. [Google Scholar]
- Phillip, C.J.; Giardina, C.K.; Bilir, B.; Cutler, D.J.; Lai, Y.H.; Kucuk, O.; Moreno, C.S. Genistein cooperates with the histone deacetylase inhibitor vorinostat to induce cell death in prostate cancer cells. BMC Cancer 2012, 12, 145. [Google Scholar]
- Rabiau, N.; Trraf, H.K.; Adjakly, M.; Bosviel, R.; Guy, L.; Fontana, L.; Bignon, Y.J.; Bernard-Gallon, D.J. miRNAs differentially expressed in prostate cancer cell lines after soy treatment. In Vivo 2011, 25, 917–921. [Google Scholar]
- Zaman, M.S.; Chen, Y.; Deng, G.; Shahryari, V.; Suh, S.O.; Saini, S.; Majid, S.; Liu, J.; Khatri, G.; Tanaka, Y.; et al. The functional significance of microRNA-145 in prostate cancer. Br. J. Cancer 2010, 103, 256–264. [Google Scholar]
- Majid, S.; Dar, A.A.; Saini, S.; Chen, Y.; Shahryari, V.; Liu, J.; Zaman, M.S.; Hirata, H.; Yamamura, S.; Ueno, K.; et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010, 70, 2809–2818. [Google Scholar]
- Chen, Y.; Zaman, M.S.; Deng, G.; Majid, S.; Saini, S.; Liu, J.; Tanaka, Y.; Dahiya, R. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev. Res. (Phila.) 2011, 4, 76–86. [Google Scholar]
- Chiyomaru, T.; Yamamura, S.; Zaman, M.S.; Majid, S.; Deng, G.; Shahryari, V.; Saini, S.; Hirata, H.; Ueno, K.; Chang, I.; et al. Genistein suppresses prostate cancer growth through inhibition of oncogenic microRNA-151. PLoS One 2012, 7, e43812. [Google Scholar]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Dyson, G.; Sarkar, F.H. Epigenetic deregulation of miR-29a and miR-1256 by isoflavone contributes to the inhibition of prostate cancer cell growth and invasion. Epigenetics 2012, 7, 940–949. [Google Scholar]
- Chiyomaru, T.; Yamamura, S.; Fukuhara, S.; Hidaka, H.; Majid, S.; Saini, S.; Arora, S.; Deng, G.; Shahryari, V.; Chang, I.; et al. Genistein up-regulates tumor suppressor microRNA-574-3p in prostate cancer. PLoS One 2013, 8, e58929. [Google Scholar]
- Chiyomaru, T.; Yamamura, S.; Fukuhara, S.; Yoshino, H.; Kinoshita, T.; Majid, S.; Saini, S.; Chang, I.; Tanaka, Y.; Enokida, H.; et al. Genistein inhibits prostate cancer cell growth by targeting miR-34a and oncogenic HOTAIR. PLoS One 2013, 8, e70372. [Google Scholar]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Tanaka, Y.; Tabatabai, Z.L.; Dahiya, R. Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Br. J. Cancer 2014, 110, 1645–1654. [Google Scholar]
- Majid, S.; Dar, A.A.; Ahmad, A.E.; Hirata, H.; Kawakami, K.; Shahryari, V.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Khatri, G.; et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 2009, 30, 662–670. [Google Scholar]
- Zaman, M.S.; Shahryari, V.; Deng, G.; Thamminana, S.; Saini, S.; Majid, S.; Chang, I.; Hirata, H.; Ueno, K.; Yamamura, S.; et al. Up-regulation of microRNA-21 correlates with lower kidney cancer survival. PLoS One 2012, 7, e31060. [Google Scholar]
- Zaman, M.S.; Thamminana, S.; Shahryari, V.; Chiyomaru, T.; Deng, G.; Saini, S.; Majid, S.; Fukuhara, S.; Chang, I.; Arora, S.; et al. Inhibition of PTEN gene expression by oncogenic miR-23b-3p in renal cancer. PLoS One 2012, 7, e50203. [Google Scholar]
- Hirata, H.; Ueno, K.; Nakajima, K.; Tabatabai, Z.L.; Hinoda, Y.; Ishii, N.; Dahiya, R. Genistein downregulates onco-miR-1260b and inhibits Wnt-signalling in renal cancer cells. Br. J. Cancer 2013, 108, 2070–2078. [Google Scholar]
- Chiyomaru, T.; Fukuhara, S.; Saini, S.; Majid, S.; Deng, G.; Shahryari, V.; Chang, I.; Tanaka, Y.; Enokida, H.; Nakagawa, M.; et al. Long non-coding RNA HOTAIR is targeted and regulated by miR-141 in human cancer cells. J. Biol. Chem. 2014, 289, 12550–12565. [Google Scholar]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.-J.; Stehr, H.; Rausch, T.; et al. Integrative Genomic Analyses Reveal an Androgen-Driven Somatic Alteration Landscape in Early-Onset Prostate Cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar]
- Weiss, M.; Plass, C.; Gerhauser, C. Role of lncRNAs in prostate cancer development and progression. Biol. Chem. 2014, 395, 1275–1290. [Google Scholar]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar]
- Dalai, I.; Missiaglia, E.; Barbi, S.; Butturini, G.; Doglioni, C.; Falconi, M.; Scarpa, A. Low expression of ARHI is associated with shorter progression-free survival in pancreatic endocrine tumors. Neoplasia 2007, 9, 181–183. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Yang, Y.; Liu, J.; Li, X.; Li, J.C. PCDH17 gene promoter demethylation and cell cycle arrest by genistein in gastric cancer. Histol. Histopathol. 2012, 27, 217–224. [Google Scholar]
- Spurling, C.C.; Suhl, J.A.; Boucher, N.; Nelson, C.E.; Rosenberg, D.W.; Giardina, C. The short chain fatty acid butyrate induces promoter demethylation and reactivation of RARbeta2 in colon cancer cells. Nutr. Cancer 2008, 60, 692–702. [Google Scholar]
- Wang, Z.; Chen, H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res. 2010, 30, 4537–4545. [Google Scholar]
- Zhang, Y.; Chen, H. Genistein attenuates WNT signaling by up-regulating sFRP2 in a human colon cancer cell line. Exp. Biol. Med. (Maywood) 2011, 236, 714–722. [Google Scholar]
- Wang, H.; Li, Q.; Chen, H. Genistein affects histone modifications on Dickkopf-related protein 1 (DKK1) gene in SW480 human colon cancer cell line. PLoS One 2012, 7, e40955. [Google Scholar]
- Zhang, Y.; Li, Q.; Chen, H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis 2013, 34, 1756–1763. [Google Scholar]
- Groh, I.A.; Chen, C.; Luske, C.; Cartus, A.T.; Esselen, M. Plant polyphenols and oxidative metabolites of the herbal alkenylbenzene methyleugenol suppress histone deacetylase activity in human colon carcinoma cells. J. Nutr. Metab. 2013, 2013, 821082. [Google Scholar]
- Li, Y.; VandenBoom, T.G., 2nd; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar]
- Li, Y.; Vandenboom, T.G., 2nd; Wang, Z.; Kong, D.; Ali, S.; Philip, P.A.; Sarkar, F.H. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010, 70, 1486–1495. [Google Scholar]
- Xia, J.; Duan, Q.; Ahmad, A.; Bao, B.; Banerjee, S.; Shi, Y.; Ma, J.; Geng, J.; Chen, Z.; Rahman, K.M.; et al. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr. Drug Targets 2012, 13, 1750–1756. [Google Scholar]
- Ma, J.; Cheng, L.; Liu, H.; Zhang, J.; Shi, Y.; Zeng, F.; Miele, L.; Sarkar, F.H.; Xia, J.; Wang, Z. Genistein down-regulates miR-223 expression in pancreatic cancer cells. Curr. Drug Targets 2013, 14, 1150–1156. [Google Scholar]
- Xia, J.; Cheng, L.; Mei, C.; Ma, J.; Shi, Y.; Zeng, F.; Wang, Z.; Wang, Z. Genistein Inhibits Cell Growth and Invasion Through Regulation of MiR-27a in Pancreatic Cancer Cells. Curr. Pharm. Des. 2014, 20, 5348–5353. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar]
- Bo, H.; Zhang, S.; Gao, L.; Chen, Y.; Zhang, J.; Chang, X.; Zhu, M. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer 2013, 13, 496. [Google Scholar]
- Cheng, R.; Sun, B.; Liu, Z.; Zhao, X.; Qi, L.; Li, Y.; Gu, Q. Wnt5a Suppresses Colon Cancer by Inhibiting Cell Proliferation and Epithelial–Mesenchymal Transition. J. Cell. Physiol. 2014, 229, 1908–1917. [Google Scholar]
- Zhang, Y.; Li, Q.; Zhou, D.; Chen, H. Genistein, a soya isoflavone, prevents azoxymethane-induced up-regulation of WNT/beta-catenin signalling and reduces colon pre-neoplasia in rats. Br. J. Nutr. 2013, 109, 33–42. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar]
- Zhang, J.; Ma, L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar]
- Raynal, N.J.M.; Charbonneau, M.; Momparler, L.F.; Momparler, R.L. Synergistic Effect of 5-Aza-2’-Deoxycytidine and Genistein in Combination Against Leukemia. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2008, 17, 223–230. [Google Scholar]
- Li, H.; Xu, W.; Huang, Y.; Huang, X.; Xu, L.; Lv, Z. Genistein demethylates the promoter of CHD5 and inhibits neuroblastoma growth in vivo. Int. J. Mol. Med. 2012, 30, 1081–1086. [Google Scholar]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar]
- Badger, T.M.; Ronis, M.J.; Wolff, G.; Stanley, S.; Ferguson, M.; Shankar, K.; Simpson, P.; Jo, C.H. Soy protein isolate reduces hepatosteatosis in yellow Avy/a mice without altering coat color phenotype. Exp. Biol. Med. (Maywood) 2008, 233, 1242–1254. [Google Scholar]
- Rosenfeld, C.S.; Sieli, P.T.; Warzak, D.A.; Ellersieck, M.R.; Pennington, K.A.; Roberts, R.M. Maternal exposure to bisphenol A and genistein has minimal effect on Avy/a offspring coat color but favors birth of agouti over nonagouti mice. Proc. Natl. Acad. Sci. USA 2013, 110, 537–542. [Google Scholar]
- Sato, N.; Yamakawa, N.; Masuda, M.; Sudo, K.; Hatada, I.; Muramatsu, M. Genome-wide DNA methylation analysis reveals phytoestrogen modification of promoter methylation patterns during embryonic stem cell differentiation. PLoS One 2011, 6, e19278. [Google Scholar]
- Vanhees, K.; Coort, S.; Ruijters, E.J.; Godschalk, R.W.; van Schooten, F.J.; Barjesteh van Waalwijk van Doorn-Khosrovani, S. Epigenetics: Prenatal exposure to genistein leaves a permanent signature on the hematopoietic lineage. FASEB J. 2011, 25, 797–807. [Google Scholar]
- Vandegehuchte, M.B.; Lemière, F.; Vanhaecke, L.; vanden Berghe, W.; Janssen, C.R. Direct and transgenerational impact on Daphnia magna of chemicals with a known effect on DNA methylation. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2010, 151, 278–285. [Google Scholar]
- Howard, T.D.; Ho, S.M.; Zhang, L.; Chen, J.; Cui, W.; Slager, R.; Gray, S.; Hawkins, G.A.; Medvedovic, M.; Wagner, J.D. Epigenetic changes with dietary soy in cynomolgus monkeys. PLoS One 2011, 6, e26791. [Google Scholar]
- Scoccianti, C.; Ricceri, F.; Ferrari, P.; Cuenin, C.; Sacerdote, C.; Polidoro, S.; Jenab, M.; Hainaut, P.; Vineis, P.; Herceg, Z. Methylation patterns in sentinel genes in peripheral blood cells of heavy smokers: Influence of cruciferous vegetables in an intervention study. Epigenetics 2011, 6, 1114–1119. [Google Scholar]
- Bernal, A.J.; Jirtle, R.L. Epigenomic disruption: The effects of early developmental exposures. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 938–944. [Google Scholar]
- Brennan, K.; Garcia-Closas, M.; Orr, N.; Fletcher, O.; Jones, M.; Ashworth, A.; Swerdlow, A.; Thorne, H.; Investigators, K.C.; Riboli, E.; et al. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72, 2304–2313. [Google Scholar]
- Wang, X.; Zhu, H.; Snieder, H.; Su, S.; Munn, D.; Harshfield, G.; Maria, B.L.; Dong, Y.; Treiber, F.; Gutin, B.; et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010, 8, 87. [Google Scholar]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar]
- Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlen, S.E.; Greco, D.; Soderhall, C.; Scheynius, A.; Kere, J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS One 2012, 7, e41361. [Google Scholar]
- Adalsteinsson, B.T.; Gudnason, H.; Aspelund, T.; Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Smith, A.V.; Gudnason, V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One 2012, 7, e46705. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar]
- Romagnolo, D.F.; Zempleni, J.; Selmin, O.I. Nuclear Receptors and Epigenetic Regulation: Opportunities for Nutritional Targeting and Disease Prevention. Adv. Nutr. 2014, 5, 373–385. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pudenz, M.; Roth, K.; Gerhauser, C. Impact of Soy Isoflavones on the Epigenome in Cancer Prevention. Nutrients 2014, 6, 4218-4272. https://doi.org/10.3390/nu6104218
Pudenz M, Roth K, Gerhauser C. Impact of Soy Isoflavones on the Epigenome in Cancer Prevention. Nutrients. 2014; 6(10):4218-4272. https://doi.org/10.3390/nu6104218
Chicago/Turabian StylePudenz, Maria, Kevin Roth, and Clarissa Gerhauser. 2014. "Impact of Soy Isoflavones on the Epigenome in Cancer Prevention" Nutrients 6, no. 10: 4218-4272. https://doi.org/10.3390/nu6104218
APA StylePudenz, M., Roth, K., & Gerhauser, C. (2014). Impact of Soy Isoflavones on the Epigenome in Cancer Prevention. Nutrients, 6(10), 4218-4272. https://doi.org/10.3390/nu6104218