Consequences of Essential Fatty Acids

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
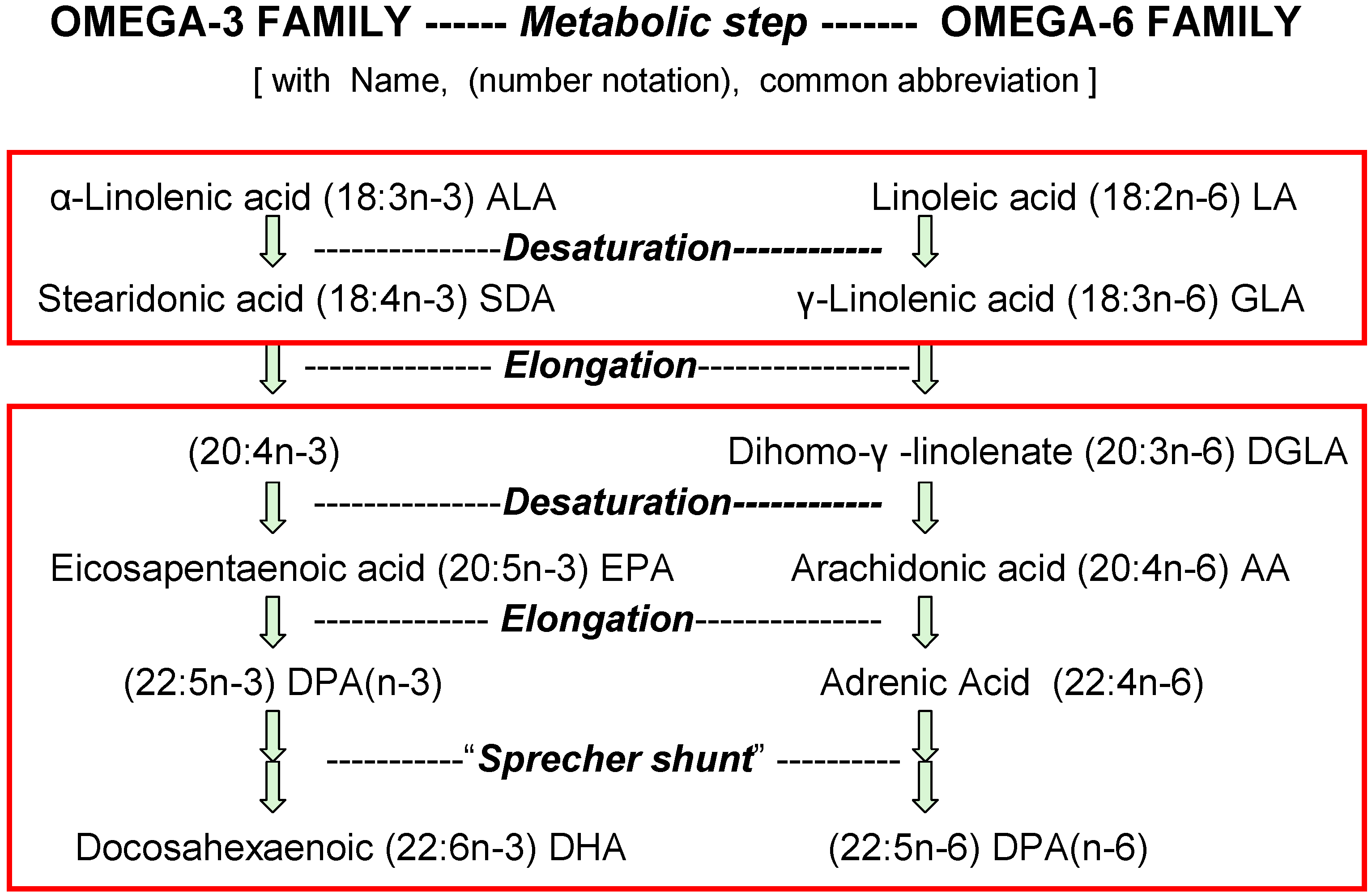
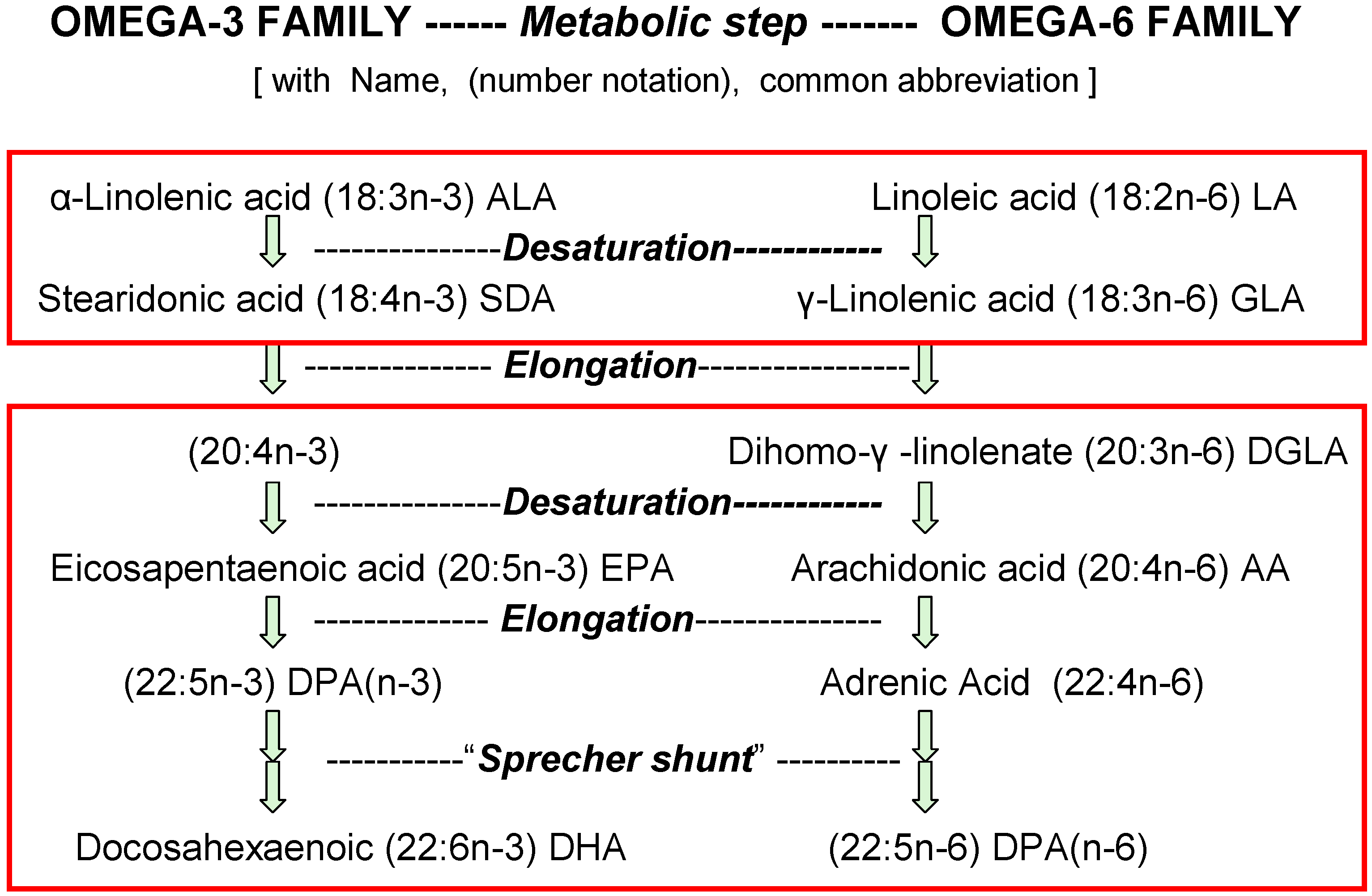
2. Non-Selective Competitions When Converting n-3 and n-6 Nutrients into Tissue Esters
2.1. Remodeling Reactions with Membrane Phospholipids
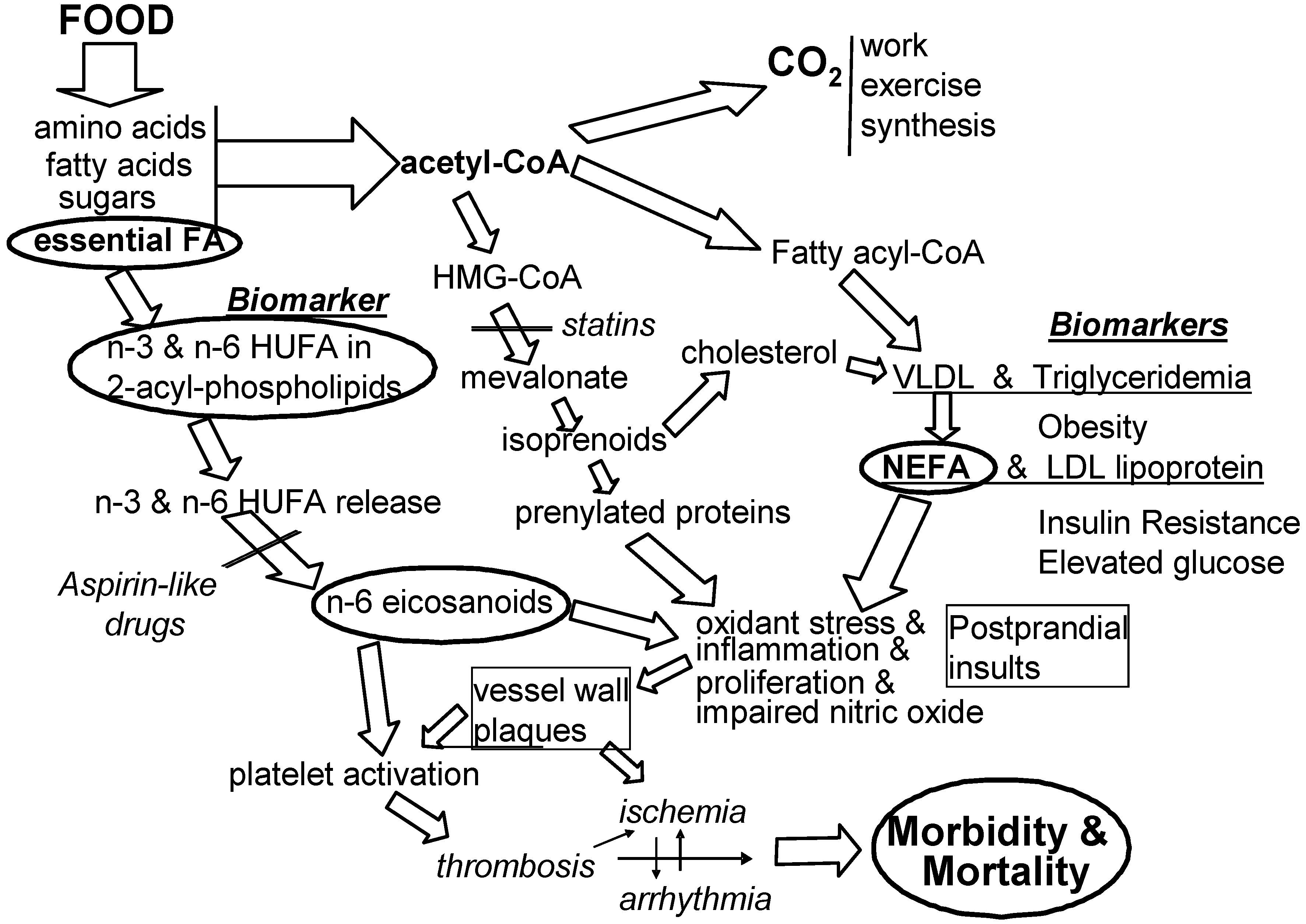
2.2. The External Environment Affects the Internal Environment
2.3. Impact of Dietary 18-Carbon EFA
3. Selective Conversion of HUFA into Transient Signaling Actions
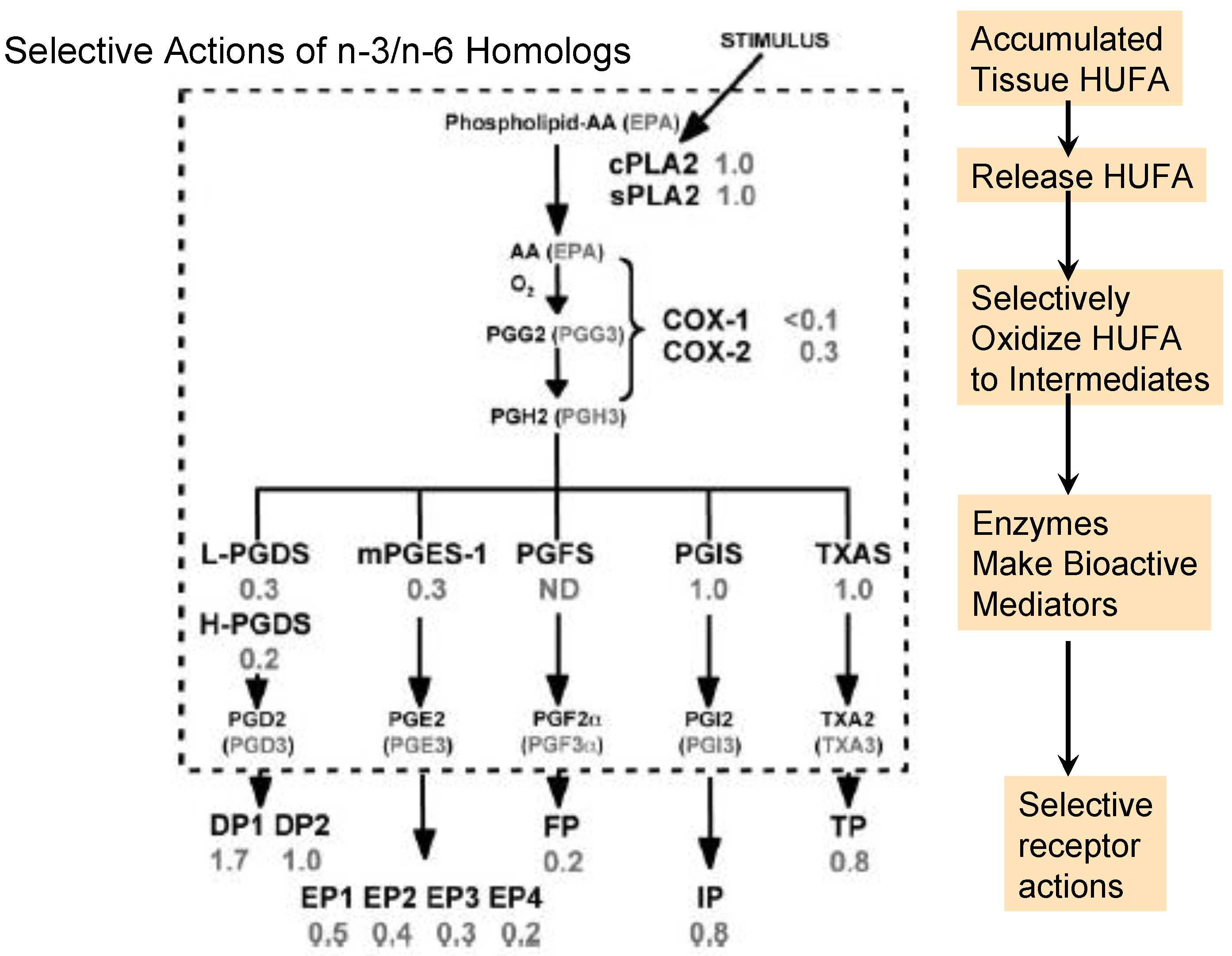
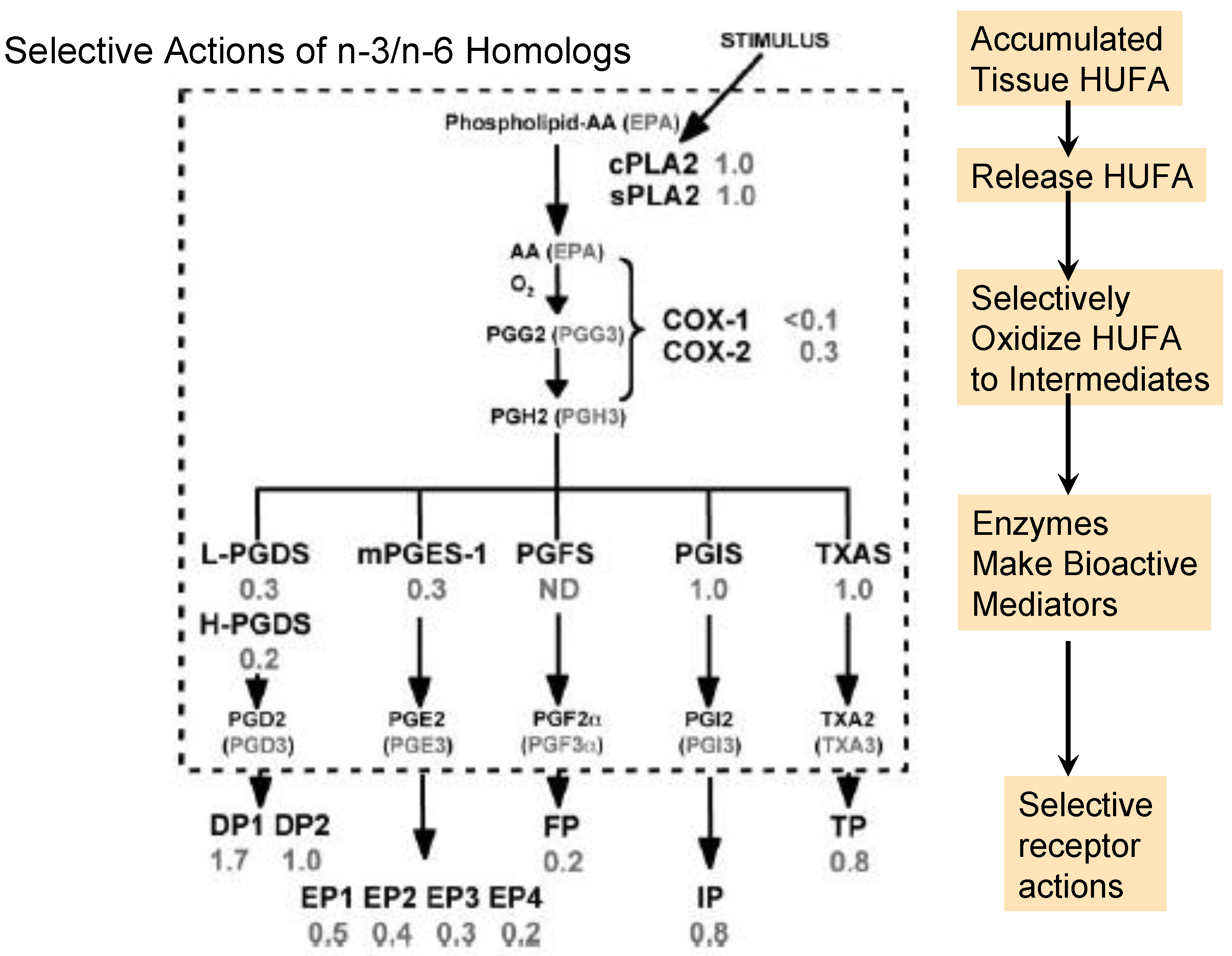
3.1. Stimulated Release of Tissue HUFA
3.2. Oxidation to Intermediate Forms
3.3. Rearrangement of PGH into Active Eicosanoids
3.4. Endocannabinoid Signaling
3.5. Selective Binding and Signaling at Cellular Receptors
3.6. Lipoxygenase and Leukotriene Formation and Action
3.7. Other Active Eicosanoid Signaling Agents
4. Preventing the Consequences of EFA Imbalances
4.1. Omega 3 Fatty Acids in Cardiovascular Health
4.2. Focusing on Mediators in Preventable Disorders
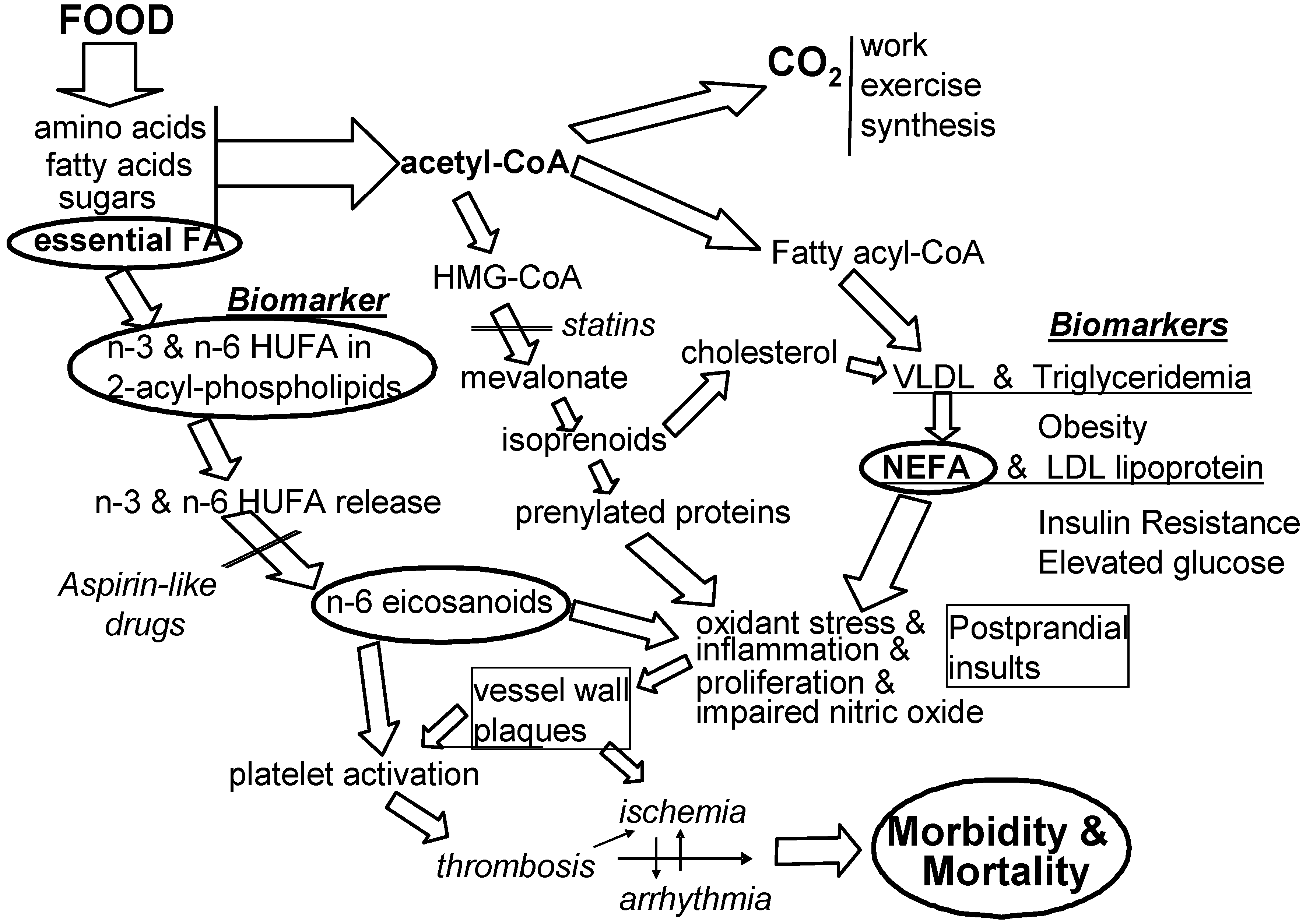
5. Conclusions
Acknowledgments
Apparent Conflict of Interest
References
- Burr, G.O.; Burr, M.M.; Miller, E.S. On the fatty acids essential in nutrition. J. Biol. Chem. 1932, 97, 1–9. [Google Scholar]
- Thomasson, H.J. Essential fatty acids. Nature 1962, 194, 973. [Google Scholar] [CrossRef]
- Hansen, A.E.; Wiese, H.F.; Boelsche, A.N.; Haggard, M.E.; Adam, D.J.D.; Davis, H. Role of linoleic acid in infant nutrition. Clinical and chemical study of 428 infants fed on milk mixtures varying in kind and amount of fat. Pediatrics 1963, 31, 171–192. [Google Scholar]
- Cuthbertson, W.F.J. Essential fatty acid requirements in infancy. Am. J. Clin. Nutr. 1976, 20, 559–568. [Google Scholar]
- Lands, W.E.M. Diets could prevent many diseases. Lipids 2003, 38, 317–321. [Google Scholar] [CrossRef]
- Lands, B. A critique of paradoxes in current advice on dietary lipids. Prog. Lipid Res. 2008, 47, 77–106. [Google Scholar] [CrossRef]
- Lands, W.E.M. Fish, Omega-3 and Human Health, 2nd ed; AOCS Press: Champaign, IL, USA, 2005. [Google Scholar]
- Cockbain, A.J.; Toogood, G.J.; Hull, M.A. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut 2012, 61, 135–149. [Google Scholar] [CrossRef]
- Alvheim, A.R.; Malde, M.K.; Osei-Hyiaman, D.; Lin, Y.H.; Pawlosky, R.J.; Madsen, L.; Kristiansen, K.; Frøyland, L.; Hibbeln, J.R. Dietary linoleic acid elevates endogenous 2-AG and anandamide and induces obesity. Obesity 2012. [Google Scholar] [CrossRef]
- Hibbeln, J.R.; Nieminen, L.R.; Blasbalg, T.L.; Riggs, J.A.; Lands, W.E.M. Healthy intakes of n-3 and n-6 fatty acids: Estimations considering worldwide diversity. Am. J. Clin. Nutr. 2006, 83, 1483–1493. [Google Scholar]
- Freeman, M.P.; Hibbeln, J.R.; Wisner, K.L.; Davis, J.M.; Mischoulon, D.; Peet, M.; Keck, P.E., Jr.; Marangell, L.B.; Richardson, A.J.; Lake, J.; Stoll, A.L. Omega-3 fatty acids: Evidence basis for treatment and future research in psychiatry. J. Clin. Psychiatry 2006, 67, 1954–1967. [Google Scholar]
- Wei, C.; Hua, J.; Bin, C.; Klassen, K. Impact of lipid emulsion containing fish oil on outcomes of surgical patients: Systematic review of randomized controlled trials from Europe and Asia. Nutrition 2010, 26, 474–481. [Google Scholar] [CrossRef]
- Lands, B. Prevent the cause, not just the symptoms. Prostaglandins Other Lipid Mediat. 2011, 96, 90–93. [Google Scholar] [CrossRef]
- Lands, W.E.M. Biosynthesis of Prostaglandins. Ann. Rev. Nutr. 1991, 11, 41–60. [Google Scholar] [CrossRef]
- Wada, M.; DeLong, C.J.; Hong, Y.H.; Rieke, C.J.; Song, I.; Sidhu, R.S.; Yuan, C.; Warnock, M.; Schmaier, A.H.; Yokoyama, C.; et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid derived substrates and products. J. Biol. Chem. 2007, 282, 22254–22266. [Google Scholar]
- Lands, W.E.M. Learning how membrane fatty acids affect cardiovascular integrity. J. Membr. Biol. 2006, 206, 75–83. [Google Scholar]
- Lands, W.E.M.; Inoue, M.; Sugiura, Y.; Okuyama, H. Selective incorporation of polyunsaturated fatty acids into phosphatidylcholine by rat liver microsomes. J. Biol. Chem. 1982, 257, 14968–14972. [Google Scholar]
- Mohrhauer, H.; Holman, R.T. The effect of dose level of essential fatty acids upon fatty acid composition of the rat liver. J. Lipid Res. 1963, 41, 151–159. [Google Scholar]
- Lands, W.E.M. Long-term fat intake and biomarkers. Am. J. Clin. Nutr. 1995, 61, 721–725. [Google Scholar]
- Collins, F.D.; Sinclair, A.J.; Royle, J.P.; Coats, D.A.; Maynard, A.T.; Leonard, R.F. Plasma lipids in human linoleic acid deficiency. Nutr. Metab. 1971, 13, 150–167. [Google Scholar] [CrossRef]
- Wilton, D.C. Phospholipases. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th; Vance, D.E., Vance, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 305–329. [Google Scholar]
- Stark, K.D.; Beblo, S.; Murthy, M.; Buda-Abela, M.; Janisse, J.; Rockett, H.; Whitty, J.E.; Martier, S.S.; Sokol, R.J.; Hannigan, J.H.; Salem, N., Jr. Comparison of bloodstream fatty acid composition from African-American women at gestation, delivery, and postpartum. J. Lipid Res. 2005, 46, 516–525. [Google Scholar]
- Stark, K.D. The percentage of n-3 highly unsaturated fatty acids in total HUFA as a biomarker for omega-3 fatty acid status in tissues. Lipids 2008, 43, 45–53. [Google Scholar] [CrossRef]
- Bell, J.G.; Mackinlay, E.E.; Dick, J.R.; Younger, I.; Lands, B.; Gilhooly, T. Using a fingertip whole blood sample for rapid fatty acid measurement: Method validation and correlation with erythrocyte polar lipid compositions in UK subjects. Br. J. Nutr. 2011, 106, 1408–1415. [Google Scholar] [CrossRef]
- Lands, B. Measuring blood fatty acids as a surrogate indicator for coronary heart disease. World Rev. Nutr. Diet. 2009, 100, 22–34. [Google Scholar] [CrossRef]
- Lands, B.; Lamoreaux, E. Using 3–6 differences in essential fatty acids rather than 3/6 ratios gives useful food balance scores. Nutr. Metab. (Lond.) 2012, 9. [Google Scholar] [CrossRef]
- Strandjord, S.E.; Lands, B.; Hibbeln, J.R. Validation of an equation predicting highly unsaturated fatty acid compositions of human blood fractions from dietary intake. Nutr. Metab. (Lond.) 2012, in press. [Google Scholar]
- Kramer, R.; Sharp, J. Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2). FEBS Lett. 1997, 410, 49–53. [Google Scholar] [CrossRef]
- Hanel, A.M.; Schüttel, S.; Gelb, M.H. Processive interfacial catalysis by mammalian 85-kilodalton phospholipase A2 enzymes on product-containing vesicles: Application to the determination of substrate preferences. Biochemistry 1993, 32, 5949–5958. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar]
- Shikano, M.; Masuzawa, Y.; Yazawa, K.; Takayama, K.; Kudo, I.; Inoue, K. Complete discrimination of docosahexaenoate from arachidonate by 85 kDa cytosolic phospholipase A2 during the hydrolysis of diacyl- and alkenylacylglycerophosphoethanolamine. Biochim. Biophys. Acta 1994, 1212, 211–216. [Google Scholar]
- Preuss, I.; Patscheke, H. Regulation of the concentration of free arachidonic acid in homogenates of human platelets. Agents Actions Suppl. 1992, 37, 34–40. [Google Scholar]
- Lands, W.E.M.; Libelt, B.; Morris, A.; Kramer, N.C.; Prewitt, T.E.; Bowen, P.; Schmeisser, D.; Davidson, M.H.; Burns, J.H. Maintenance of lower proportions of n-6 eicosanoid precursors in phospholipids of human plasma in response to added dietary n-3 fatty acids. Biochem. Biophys. Acta 1992, 1180, 147–162. [Google Scholar]
- Diet Balance Spreadsheet. Available online: http://efaeducation.nih.gov/sig/dietbalance.html (accessed on 1 June 2012).
- Lands, W.E.M.; Hamazaki, T.; Yamazaki, K.; Okuyama, H.; Sakai, K.; Goto, Y.; Hubbard, V.S. Changing dietary patterns. Am. J. Clin. Nutr. 1990, 51, 991–993. [Google Scholar]
- Lands, W.E.M. Functional foods in primary prevention or nutraceuticals in secondary prevention? Curr. Top. Nutraceut. Res. 2003, 1, 113–120. [Google Scholar]
- Kato, H.; Tillotwn, J.; Nichaman, M.Z.; Rhoads, G.G.; Hamilton, H.B. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: Serum lipids and diet. Am. J. Epidemiol. 1973, 97, 372–385. [Google Scholar]
- Birtwhistle, R. What ever happened to the Mediterranean diet? QJM 2008, 101, 741–742. [Google Scholar] [CrossRef]
- National Nutrient Database for Standard Reference Release 24. Available online: http://ndb.nal.usda.gov/ndb/foods/list (accessed on 1 June 2012).
- Omega 3–6 Balance Scores. Available online: http://www.fastlearner.org/Omega3-6Balance.htm (accessed on 1 June 2012).
- An “App” for Omega 3–6 Balance Scores. Available online: http://www.fastlearner.org/Omega3-6BalanceApp.htm (accessed on 1 June 2012).
- Sergeant, S.; Hugenschmidt, C.E.; Rudock, M.E.; Ziegler, J.T.; Ivester, P.; Ainsworth, H.C.; Vaidya, D.; Case, L.D.; Langefeld, C.D.; Freedman, B.I.; et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012, 107, 547–555. [Google Scholar] [CrossRef]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.V.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain Omega-3 and Omega-6 fatty acids. Am. J. Human Genet. 2012. [Google Scholar] [CrossRef]
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080. [Google Scholar] [CrossRef]
- Na, H.K.; Park, J.M.; Lee, H.G.; Lee, H.N.; Myung, S.J.; Surh, Y.J. 15-Hydroxyprostaglandin dehydrogenase as a novel molecular target for cancer chemoprevention and therapy. Biochem. Pharmacol. 2011, 82, 1352–1360. [Google Scholar] [CrossRef]
- Folco, G.; Murphy, R.C. Eicosanoid transcellular biosynthesis: From cell-cell interactions to in vivo tissue responses. Pharmacol. Rev. 2006, 58, 375–388. [Google Scholar] [CrossRef]
- Rosenwaks, Z.; Jones, G.S.; Henzl, M.R.; Dubin, N.H.; Ghodgaonkar, R.B.; Hoffman, S. Naproxen sodium, aspirin, and placebo in primary dysmenorrhea. Reduction of pain and blood levels of prostaglandin F2-alpha metabolite. Am. J. Obstet. Gynecol. 1981, 140, 592–598. [Google Scholar]
- Olson, D.M.; Ammann, C. Role of the prostaglandins in labour and prostaglandin receptor inhibitors in the prevention of preterm labour. Front Biosci. 2007, 12, 1329–1343. [Google Scholar] [CrossRef]
- Oudin, M.J.; Hobbs, C.; Doherty, P. DAGL-dependent endocannabinoid signalling: Roles in axonal pathfinding, synaptic plasticity and adult neurogenesis. Eur. J. Neurosci. 2011, 34, 1634–1646. [Google Scholar] [CrossRef]
- Pini, A.; Mannaioni, G.; Pellegrini-Giampietro, D.; Passani, M.B.; Mastroianni, R.; Bani, D.; Masini, E. The role of cannabinoids in inflammatory modulation of allergic respiratory disorders, inflammatory pain and ischemic stroke. Curr. Drug Targets 2012, 13, 984–993. [Google Scholar] [CrossRef]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavon, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoyl glycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Kishimoto, S.; Miyashita, T.; Nakane, S.; Kodaka, T.; Suhara, Y.; Takayama, H.; Waku, K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000, 275, 605–612. [Google Scholar]
- Annuzzi, G.; Piscitelli, F.; di Marino, L.; Patti, L.; Giacco, R.; Costabile, G.; Bozzetto, L.; Riccardi, G.; Verde, R.; Petrosino, S.; et al. Differential alterations of the concentrations of endocannabinoids and related lipids in the subcutaneous adipose tissue of obese diabetic patients. Lipids Health Dis. 2010, 9, 43–51. [Google Scholar] [CrossRef]
- Kunos, G.; Osei-Hyiaman, D.; Liu, J.; Godlewski, G.; Bátkai, S. Endocannabinoids and the control of energy homeostasis. J. Biol. Chem. 2008, 283, 33021–33025. [Google Scholar]
- Furuyashiki, T.; Narumiya, S. Stress responses: The contribution of prostaglandin E(2) and its receptors. Nat. Rev. Endocrinol. 2011, 7, 163–175. [Google Scholar] [CrossRef]
- Yuhki, K.; Kojima, F.; Kashiwagi, H.; Kawabe, J.; Fujino, T.; Narumiya, S.; Ushikubi, F. Roles of prostanoids in the pathogenesis of cardiovascular diseases: Novel insights from knockout mouse studies. Pharmacol. Ther. 2011, 129, 195–205. [Google Scholar] [CrossRef]
- Narumiya, S.; Furuyashiki, T. Fever, inflammation, pain and beyond: Prostanoid receptor research during these 25 years. FASEB J. 2011, 25, 813–818. [Google Scholar] [CrossRef]
- Tanaka, K.; Furuyashiki, T.; Kitaoka, S.; Senzai, Y.; Imoto, Y.; Segi-Nishida, E.; Deguchi, Y.; Breyer, R.M.; Breyer, M.D.; Narumiya, S. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J. Neurosci. 2012, 32, 4319–4329. [Google Scholar] [CrossRef]
- Suzuki, C.; Miyamoto, C.; Furuyashiki, T.; Narumiya, S.; Ohinata, K. Central PGE2 exhibits anxiolytic-like activity via EP1 and EP4 receptors in a manner dependent on serotonin 5-HT1A, dopamine D1 and GABAA receptors. FEBS Lett. 2011, 585, 2357–2362. [Google Scholar] [CrossRef]
- Yokomizo, T. Leukotriene B4 receptors: Novel roles in immunological regulations. Adv. Enzym. Regul. 2011, 51, 59–64. [Google Scholar] [CrossRef]
- Lee, T.H.; Mencia-Huerta, J.M.; Shih, C.; Corey, E.J.; Lewis, R.A.; Austen, K.F. Effects of exogenous arachidonic, eicosapentaenoic, and docosahexaenoic acids on the generation of 5-lipoxygenase pathway products by ionophore-activated human neutrophils. J. Clin. Invest. 1984, 74, 1922–1933. [Google Scholar] [CrossRef]
- Lee, T.H.; Sethi, T.; Crea, A.E.; Peters, W.; Arm, J.P.; Horton, C.E.; Walport, M.J.; Spur, B.W. Characterization of leukotriene B3: Comparison of its biological activities with leukotriene B4 and leukotriene B5 in complement receptor enhancement, lysozyme release and chemotaxis of human neutrophils. Clin. Sci. (Lond.) 1988, 74, 467–475. [Google Scholar]
- Grimminger, F.; Wahn, H.; Mayer, K.; Kiss, L.; Walmrath, D.; Seeger, W. Impact of arachidonic versus eicosapentaenoic acid on exotonin-induced lung vascular leakage: Relation to 4-series versus 5-series leukotriene generation. Am. J. Respir. Crit. Care Med. 1997, 5, 513–519. [Google Scholar]
- Spite, M.; Hellmann, J.; Tang, Y.; Mathis, S.P.; Kosuri, M.; Bhatnagar, A.; Jala, V.R.; Haribabu, B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J. Immunol. 2011, 187, 1942–1949. [Google Scholar] [CrossRef]
- Snelgrove, R.J. Leukotriene A4 hydrolase: An anti-inflammatory role for a proinflammatory enzyme. Thorax 2011, 66, 550–551. [Google Scholar] [CrossRef]
- Serhan, C.N.; Krishnamoorthy, S.; Recchiuti, A.; Chiang, N. Novel anti-inflammatory—Pro-resolving mediators and their receptors. Curr. Top. Med. Chem. 2011, 11, 629–647. [Google Scholar]
- Imig, J.D. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 2012, 92, 101–130. [Google Scholar] [CrossRef]
- Fleming, I. Cytochrome P450-dependent eicosanoid production and crosstalk. Curr. Opin. Lipidol. 2011, 22, 403–409. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention: Morbidity and Mortality Weekly Report. Available online: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6036a4.htm (accessed on 1 June 2012).
- Heidenreich, P.A.; Trogdon, J.G.; Khavjou, O.A.; Butler, J.; Dracup, K.; Ezekowitz, M.D.; Finkelstein, E.A.; Hong, Y.; Johnston, C.; Khera, A.; et al. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 2011, 123, 933–944. [Google Scholar] [CrossRef]
- Robert Wood Johnson Foundation, High and Rising Health Care Costs: Demystifying U.S. Health Care Spending. October 2008. Available online: http://www.rwjf.org/files/research/35368.highrisingcosts.rpt.pdf (accessed on 1 June 2012).
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Lands, B. Planning primary prevention of coronary disease. Curr. Atheroscler. Rep. 2009, 11, 272–280. [Google Scholar] [CrossRef]
- Lee, J.H.; O’Keefe, J.H.; Lavie, C.J.; Marchioli, R.; Harris, W.S. Omega-3 fatty acids for cardioprotection. Mayo Clin. Proc. 2008, 83, 324–332. [Google Scholar] [CrossRef]
- Miles, J.M.; Nelson, R.H. Contribution of triglyceride-rich lipoproteins to plasma free fatty acids. Horm. Metab. Res. 2007, 39, 726–729. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar]
- Clark, C.R.; Coull, B.; Berkman, L.F.; Buring, J.E.; Ridker, P.M. Geographic variation in cardiovascular inflammation among healthy women in the Women’s Health Study. PLoS One 2011, 6, e27468. [Google Scholar]
- Verschuren, W.M.; Jacobs, D.R.; Bloemberg, B.P.; Kromhout, D.; Menotti, A.; Aravanis, C.; Blackburn, H.; Buzina, R.; Dontas, A.S.; Fidanza, F.; et al. Serum total cholesterol and long-term coronary heart disease mortality in different cultures. Twenty-five year follow-up of the seven countries study. JAMA 1995, 274, 131–136. [Google Scholar]
- Ogushi, Y.; Hamazaki, T.; Kirihara, Y. Blood cholesterol as a good marker of health in Japan. World Rev. Nutr. Diet. 2009, 100, 63–70. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Hibbeln, J.R.; Majchrzak, S.F.; Davis, J.M. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2010, 104, 1586–1600. [Google Scholar] [CrossRef]
- Harris, W.S.; Mozaffarian, D.; Rimm, E.; Kris-Etherton, P.; Rudel, L.L.; Appel, L.J.; Engler, M.M.; Engler, M.B.; Sacks, F. Omega-6 fatty acids and risk for cardiovascular disease: A science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 2009, 119, 902–907. [Google Scholar] [CrossRef]
- Czernichow, S.; Thomas, D.; Bruckert, E. n-6 Fatty acids and cardiovascular health: A review of the evidence for dietary intake recommendations. Br. J. Nutr. 2010, 104, 1–9. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lands, B. Consequences of Essential Fatty Acids. Nutrients 2012, 4, 1338-1357. https://doi.org/10.3390/nu4091338
Lands B. Consequences of Essential Fatty Acids. Nutrients. 2012; 4(9):1338-1357. https://doi.org/10.3390/nu4091338
Chicago/Turabian StyleLands, Bill. 2012. "Consequences of Essential Fatty Acids" Nutrients 4, no. 9: 1338-1357. https://doi.org/10.3390/nu4091338
APA StyleLands, B. (2012). Consequences of Essential Fatty Acids. Nutrients, 4(9), 1338-1357. https://doi.org/10.3390/nu4091338