
Low Plasma Choline, High Trimethylamine Oxide, and Altered Phosphatidylcholine Subspecies Are Prevalent in Cystic Fibrosis Patients with Pancreatic Insufficiency

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
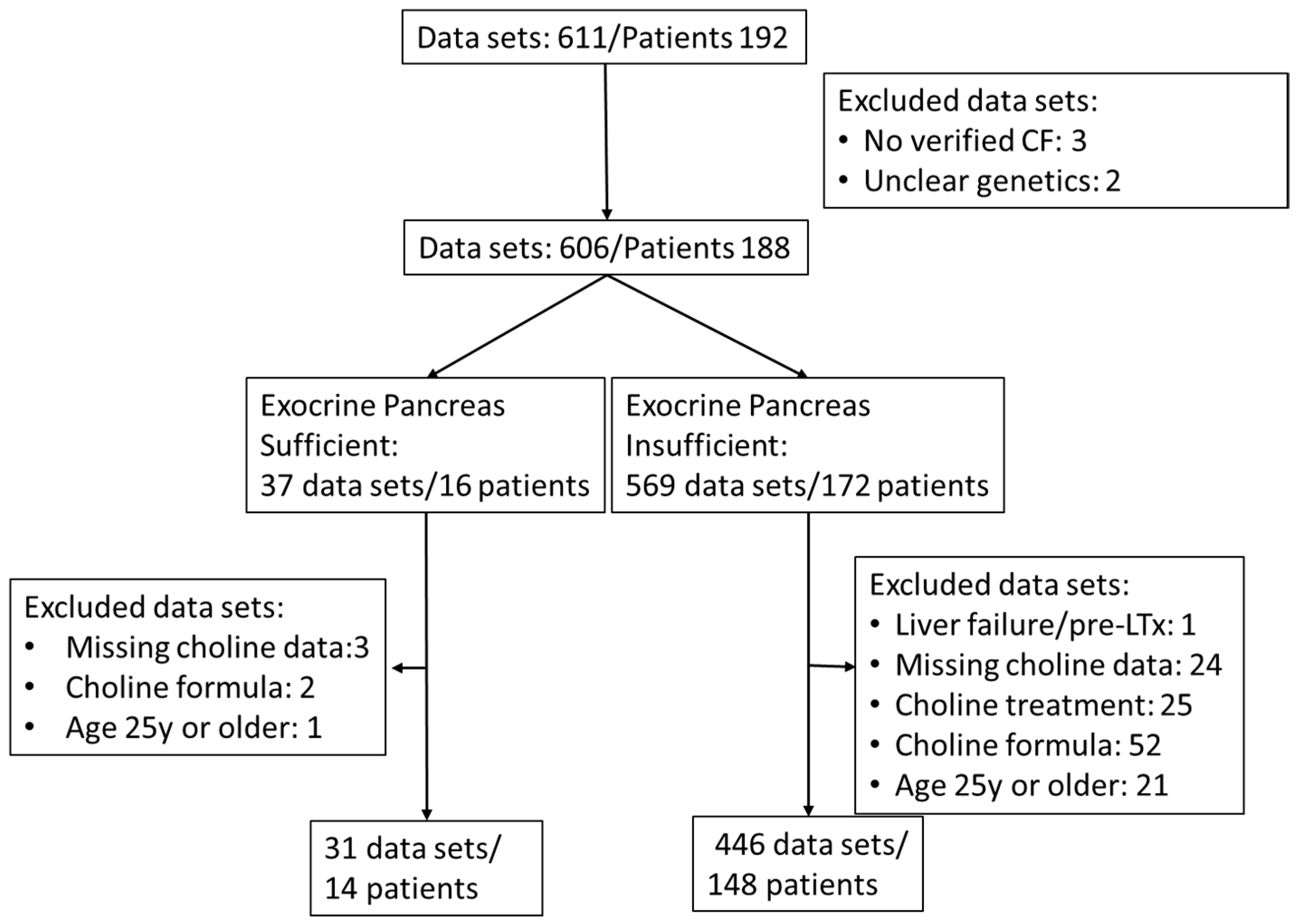
2.1. Study Population
2.2. Exclusion of Additional Choline Intake
2.3. Sample Parameters and Numbers
2.4. Plasma Collection
2.5. Mass Spectrometry
2.6. Clinical Parameters
2.7. Statistics
3. Results
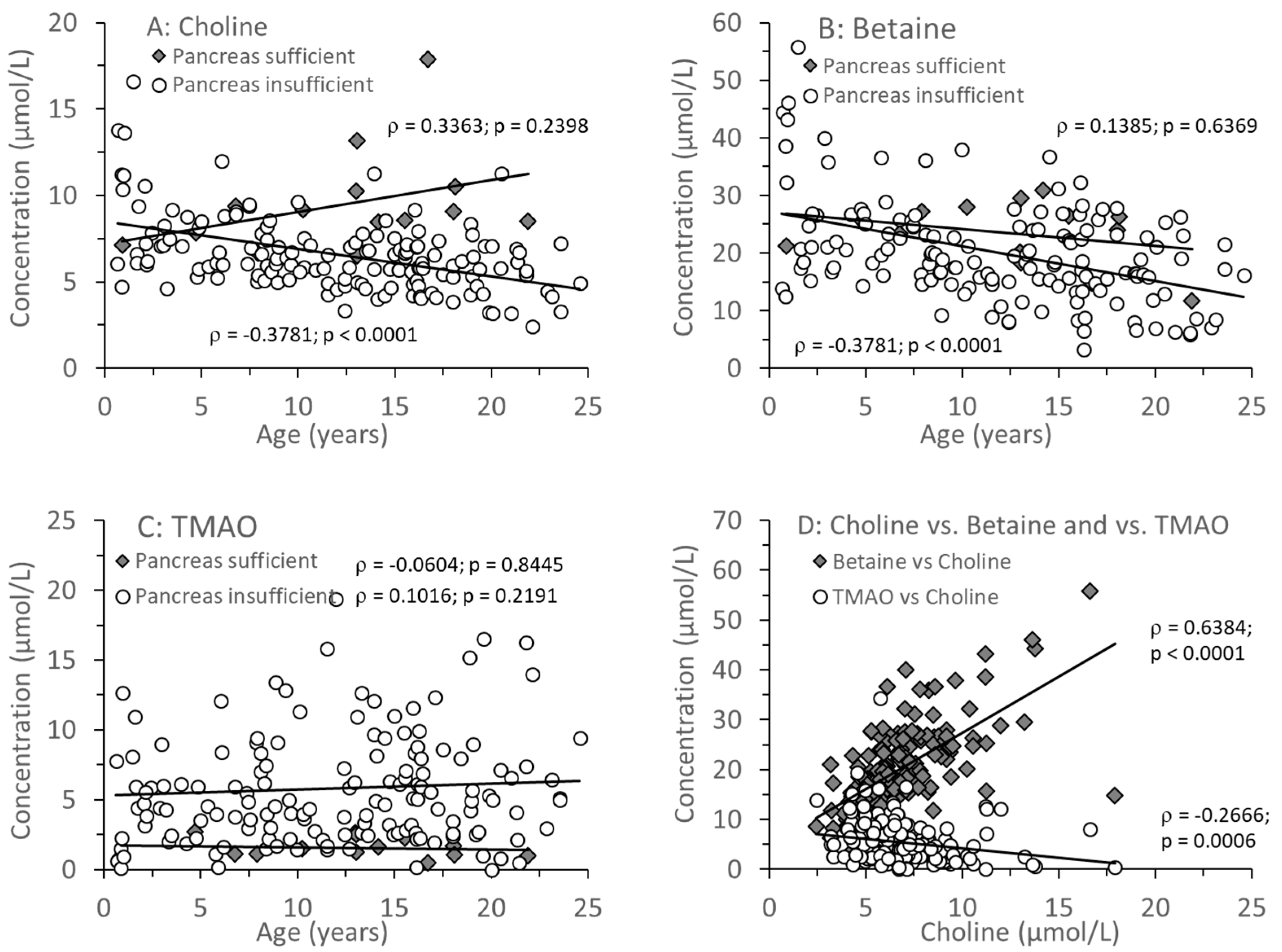
3.1. Parameters of Choline Homeostasis in the Whole Study Group
3.2. Age-Related Changes of Water-Soluble Choline Compounds in Plasma
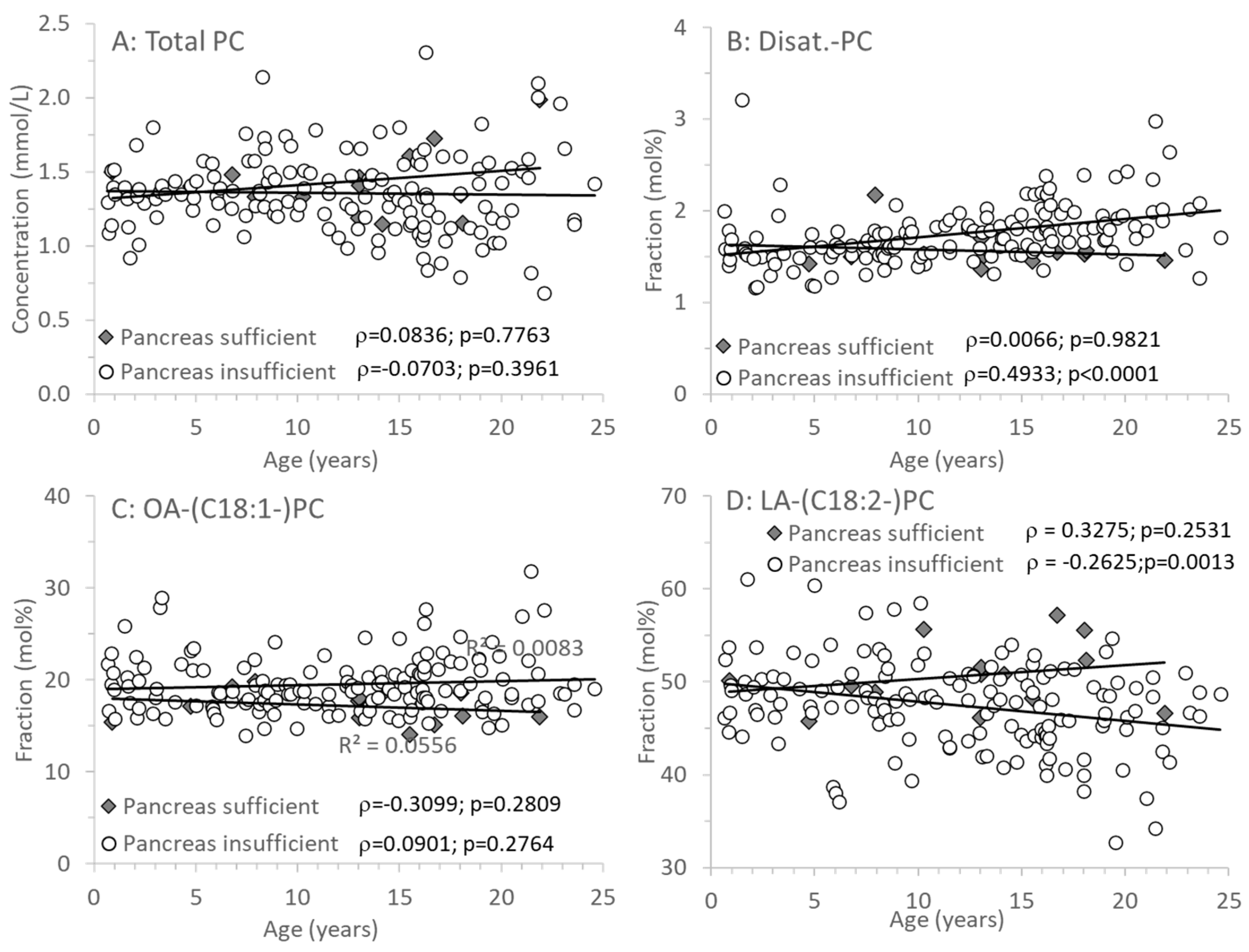
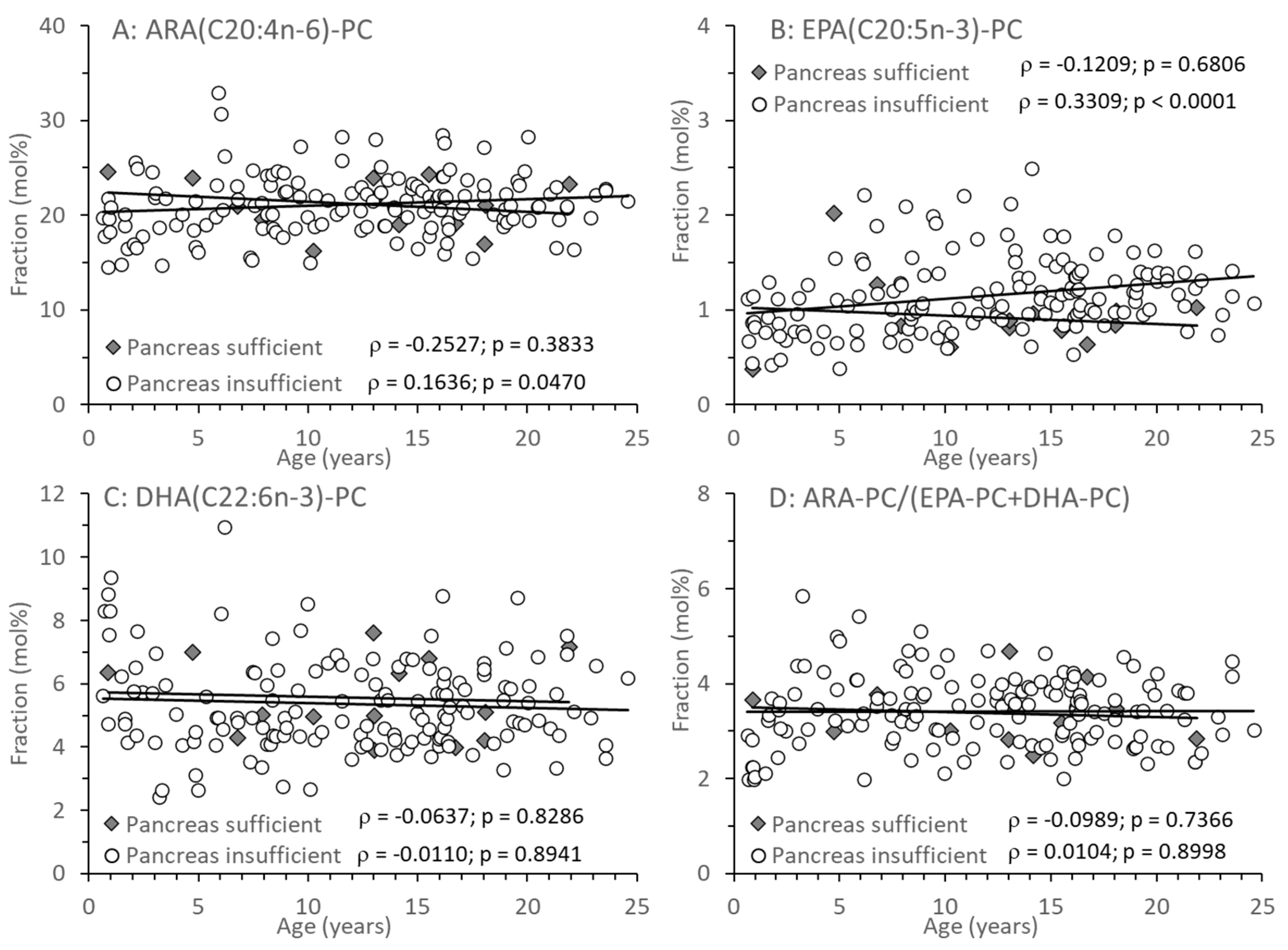
3.3. Age-Related Changes of PC and Its Sub-Groups
4. Discussion
4.1. Plasma Choline and Betaine Levels in Relation to Exocrine Pancreas Function
4.2. The Impact of Small Intestinal Bacterial Colonization and PEMT Genetics
4.3. Effects of Age on Plasma Choline and TMAO Levels
4.4. Plasma Phospholipids and Long-Chain Poly-Unsaturated Fatty Acids (LC-PUFA) in CF
4.5. Perspectives of Clinical Choline Analysis and Application
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kosorok, M.R.; Wei, W.H.; Farrell, P.M. The incidence of cystic fibrosis. Stat. Med. 1996, 15, 449–462. [Google Scholar] [CrossRef]
- Korten, I.; Kieninger, E.; Yammine, S.; Cangiano, G.; Nyilas, S.; Anagnostopoulou, P.; Singer, F.; Kuehni, C.E.; Regamey, N.; Frey, U.; et al. Respiratory rate in infants with cystic fibrosis throughout the first year of life and association with lung clearance index measured shortly after birth. J. Cyst. Fibros. 2018, 18, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Debray, D.; Kelly, D.; Houwen, R.; Strandvik, B.; Colombo, C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J. Cyst. Fibros. 2011, 10 (Suppl. 2), 29–36. [Google Scholar] [CrossRef] [PubMed]
- Gaskin, K.; Gurwitz, D.; Durie, P.; Corey, M.; Levison, H.; Forstner, G. Improved respiratory prognosis in patients with cystic fibrosis with normal fat absorption. J. Pediatr. 1982, 100, 857–862. [Google Scholar] [CrossRef]
- Chen, A.H.; Innis, S.M.; Davidson, A.G.; James, S.J. Phosphatidylcholine and lysophosphatidylcholine excretion is increased in children with cystic fibrosis and is associated with plasma homocysteine, S-adenosylhomocysteine, and S-adenosylmethionine. Am. J. Clin. Nutr. 2005, 81, 686–691. [Google Scholar] [CrossRef]
- Grothe, J.; Riethmüller, J.; Tschürtz, S.M.; Raith, M.; Pynn, C.J.; Stoll, D.; Bernhard, W. Plasma Phosphatidylcholine Alterations in Cystic Fibrosis Patients: Impaired Metabolism and Correlation with Lung Function and Inflammation. Cell Physiol. Biochem. 2015, 35, 1437–1453. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Patient Registry. Annual Data Report. Bethesda, MD, USA. 2010. Available online: http://www.cff.org/UploadedFiles/LivingWithCF/CareCenterNetwork/PatientRegistry/2010-Patient-Registry-Report.pdf (accessed on 22 March 2024).
- Colombo, C.; Battezzati, P.M.; Crosignani, A.; Morabito, A.; Costantini, D.; Padoan, R.; Giunta, A. Liver disease in cystic fi-brosis: A prospective study on incidence, risk factors, and outcome. Hepatology 2002, 36, 1374–1382. [Google Scholar] [CrossRef]
- Buchman, A.L.; Dubin, M.; Jenden, D.; Moukarzel, A.; Roch, M.H.; Rice, K.; Gornbein, J.; Ament, M.E.; Eckhert, C.D. Lecithin increases plasma free choline and decreases hepatic 22 steatosis in long-term total parenteral nutrition patients. Gastroenterology 1992, 102, 1363–1370. [Google Scholar] [CrossRef]
- Scheele, G.A.; Fukuoka, S.I.; Kern, H.F.; Freedman, S.D. Pancreatic dysfunction in cystic fibrosis occurs as a result of impairments in luminal pH, apical trafficking of zymogen granule membranes, and solubilization of secretory enzymes. Pancreas 1996, 12, 1–9. [Google Scholar]
- Bernhard, W. Choline in cystic fibrosis: Relations to pancreas insufficiency, enterohepatic cycle, PEMT and intestinal microbiota. Eur. J. Nutr. 2020, 60, 1737–1759. [Google Scholar] [CrossRef]
- Nilsson, Å.; Duan, R.-D. Pancreatic and mucosal enzymes in choline phospholipid digestion. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G425–G445. [Google Scholar] [CrossRef] [PubMed]
- Day, C.R.; Kempson, S.A. Betaine chemistry, roles, and potential use in liver disease. Biochim. Biophys. Acta 2016, 1860, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M.; Davidson, A.G.; Melynk, S.; James, S.J. Choline-related supplements improve abnormal plasma methio-nine-homocysteine metabolites and glutathione status in children with cystic fibrosis. Am. J. Clin. Nutr. 2007, 85, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Shunova, A.; Machann, J.; Grimmel, M.; Haack, T.B.; Utz, P.; Graepler-Mainka, U. Resolution of severe hepatosteatosis in a cystic fibrosis patient with multifactorial choline deficiency: A case report. Nutrition 2021, 89, 111348. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Lange, R.; Graepler-Mainka, U.; Engel, C.; Machann, J.; Hund, V.; Shunova, A.; Hector, A.; Riethmüller, J. Choline Supplementation in Cystic Fibrosis—The Metabolic and Clinical Impact. Nutrients 2019, 11, 656. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and Its Panel on Folate, Other B Vitamins, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press: Washington, DC, USA, 1998. Available online: https://www.ncbi.nlm.nih.gov/books/NBK114310/pdf/Bookshelf_NBK114310.pdf (accessed on 6 March 2020).
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary reference values for choline EFSA panel on dietetic products, nutrition and allergies (NDA). EFSA J. 2016, 14, 4484. [Google Scholar] [CrossRef]
- Bailey, R.L.; Pac, S.G.; Fulgoni, V.L., III; Reidy, K.C.; Catalano, P.M. Estimation of Total Usual Dietary Intakes of Pregnant Women in the United States. JAMA Netw. Open 2019, 2, e195967. [Google Scholar] [CrossRef]
- The LipidWeb. Plasma Lipoproteins. 2020. Available online: https://lipidmaps.org/resources/lipidweb/lipidweb_html/lipids/simple/lipoprot/index.htm (accessed on 8 November 2024).
- Li, Z.; Agellon, L.B.; Vance, D.E. Choline redistribution during adaptation to choline deprivation. J. Biol. Chem. 2007, 282, 10283–10289. [Google Scholar] [CrossRef]
- da Costa, K.; Corbin, K.D.; Niculescu, M.D.; Galanko, J.A.; Zeisel, S.H. Identification of new genetic polymorphisms that alter the dietary requirement for choline and vary in their distribution across ethnic and racial groups. FASEB J. 2014, 28, 2970–2978. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Kunze, R.; Koch, V.; Heni, M.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Choline concentrations are lower in postnatal plasma of preterm infants than in cord plasma. Eur. J. Nutr. 2014, 54, 733–741. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Koch, V.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Developmental changes in polyunsaturated fetal plasma phospholipids and feto-maternal plasma phospholipid ratios and their association with bronchopulmonary dysplasia. Eur. J. Nutr. 2015, 55, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am. J. Physiol. Physiol. 1998, 275, C323–C326. [Google Scholar] [CrossRef]
- Kogan, I.; Ramjeesingh, M.; Li, C.; Kidd, J.F.; Wang, Y.; Leslie, E.M.; Cole, S.P.; Bear, C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003, 22, 1981–1989. [Google Scholar] [CrossRef]
- Ratjen, F. The future of cystic fibrosis: A global perspective. Pediatr. Pulmonol. 2024. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Baris, E.; Simsek, O.; Arici, M.A.; Tosun, M. Choline and citicoline ameliorate oxidative stress in acute kidney injury in rats. Bratisl. Lek. Listy. 2023, 124, 47–52. [Google Scholar] [CrossRef]
- Buchman, A.L.; Ament, M.E.; Sohel, M.; Dubin, M.; Jenden, D.J.; Roch, M.; Pownall, H.; Farley, W.; Awal, M.; Ahn, C. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: Proof of a human choline requirement: A placebo-controlled trial. JPEN J. Parenter. Enteral Nutr. 2001, 25, 260–268. [Google Scholar] [CrossRef]
- Buchman, A. The addition of choline to parenteral nutrition. Gastroenterology 2009, 137 (Suppl. 5), S119–S128. [Google Scholar] [CrossRef]
- Narkewicz, M.R. Integrating Clinical Ultrasound into Screening for Cystic Fibrosis Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 394–395. [Google Scholar] [CrossRef]
- Buchman, A.L.; Sohel, M.; Moukarzel, A.; Bryant, D.; Schanler, R.; Awal, M.; Burns, P.; Dorman, K.; Belfort, M.; Jenden, D.J.; et al. Plasma choline in normal newborns, infants, toddlers, and in very-low-birth-weight neonates requiring total parenteral nutrition. Nutrition 2001, 17, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Yeung, C.; Peter, R.; Ibarra, C.; Gasser, R.; Itagaki, K.; Philpot, R.; Rettie, A. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: Selective catalysis by FMO3. Biochem. Pharmacol. 1998, 56, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- al-Waiz, M.; Mikov, M.; Mitchell, S.C.; Smith, R.L. The exogenous origin of trimethylamine in the mouse. Metabolism 1992, 41, 135–136. [Google Scholar] [CrossRef]
- Bernhard, W.; Böckmann, K.; Maas, C.; Mathes, M.; Hövelmann, J.; Shunova, A.; Hund, V.; Schleicher, E.; Poets, C.F.; Franz, A.R. Combined choline and DHA supplementation: A randomized controlled trial. Eur. J. Nutr. 2019, 59, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Kumpf, V.J. Parenteral nutrition-associated liver disease in adult and pediatric patients. Nutr. Clin. Pract. 2006, 21, 279–290. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Shunova, A.; Lorenz, S.; Böckmann, K.; Minarski, M.; Poets, C.F.; Franz, A.R. Choline Kinetics in Neonatal Liver, Brain and Lung—Lessons from a Rodent Model for Neonatal Care. Nutrients 2022, 14, 720. [Google Scholar] [CrossRef]
- Bernhard, W.; Maas, C.; Shunova, A.; Mathes, M.; Böckmann, K.; Bleeker, C.; Vek, J.; Poets, C.F.; Schleicher, E.; Franz, A.R. Transport of long-chain polyunsaturated fatty acids in preterm infant plasma is dominated by phosphatidylcholine. Eur. J. Nutr. 2017, 57, 2105–2112. [Google Scholar] [CrossRef]
- Shrestha, N.; Rout-Pitt, N.; McCarron, A.; Jackson, C.A.; Bulmer, A.C.; McAinch, A.J.; Donnelley, M.; Parsons, D.W.; Hryciw, D.H. Changes in Essential Fatty Acids and Ileal Genes Associated with Metabolizing Enzymes and Fatty Acid Transporters in Rodent Models of Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 7194. [Google Scholar] [CrossRef]
- Wilschanski, M.; Munck, A.; Carrion, E.; Cipolli, M.; Collins, S.; Colombo, C.; Declercq, D.; Hatziagorou, E.; Hulst, J.; Kalnins, D.; et al. ESPEN-ESPGHAN-ECFS guideline on nutrition care for cystic fibrosis. Clin. Nutr. 2023, 43, 413–445. [Google Scholar] [CrossRef]
- Böckmann, K.A.; Franz, A.R.; Minarski, M.; Shunova, A.; Maiwald, C.A.; Schwarz, J.; Gross, M.; Poets, C.F.; Bernhard, W. Differential metabolism of choline supplements in adult volunteers. Eur. J. Nutr. 2021, 61, 219–230. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion parameters | Genetically verified cystic fibrosis with or without exocrine pancreas insufficiency Age 0 to <25 years |
| Exclusion parameters | Acute liver failure Pre-transplantation status Choline supplementation via prescribed choline or dietary supplements 25 years or older No data on choline parameters |
| Parameter | Exocrine Pancreatic Sufficiency | Exocrine Pancreatic Insufficiency |
|---|---|---|
| Number of patients (and data sets) | 14 (31) | 148 (446) |
| F508del compound heterozygous F508del homozygous Other | 4 0 10 | 78 53 17 |
| Median age at CF ∂iagnosis (y) | 1.04 (0.92–5.34) | 0.27 (0.10–1.67) p = 0.0009 |
| Age at measurements (y) | 13.03 (8.50–16.42) [0.89–21.90] | 12.66 (7.22–16.36) [0.64–24.58] p = 0.8208 |
| Sex (male/female) | 8/6 | 68/80; p = 0.4258 |
| Body weight (kg) | 44.9 (28.8–56.8) | 38.1 (22.2–54.6); p = 0.3634 |
| Body length (cm) | 156.0 (131.2–161.5) (71–181) | 150.9 (117.0–165.0) (61–188) p = 0.7324 |
| (BMI (kg/m2) | 18.2 (17.2–21.4) | 17.2 (15.8–20.1) p = 0.2199 |
| BMI Percentile (%) | 57.5 (26.8–73.3) | 40.0 (18.6–64.8) p = 0.3051 |
| Coagulation (Quick) [70–120] | 95 (88–103) | 89 (79–100) p = 0.0808 |
| Thrombocytes (103/µL) | 281 (256–298) | 304 (260–355) p = 0.2566 |
| CRP (mg/dL) ESR (mm/h) | 0.02 (0.01–0.04) ** 6 (4–8) ** | 0.02 (0.01–0.08) **; p = 0.7577 8 (5–14) **; p = 0.1572 |
| Cholesterol (mg/dL) [130–190] | 137(126–152) | 128 (105–143) p = 0.3272 |
| Triglycerides (mg/dL) [<200] | 68 (55–74) | 83 (62–117) p = 0.1218 |
| Albumin (g/dL) [3.0–5.0] | 4.3 (4.1–4.4) | 4.0 (3.8–4.2) p = 0.0016 |
| AST [<39 U/L] | 19 (16–32) | 25 (19–36) p = 0.0879 |
| ALT [<39 U/L] | 17 (13–20) | 25 (20–34) p = 0.0021 |
| AP [130–400] | 225 (134–267) | 225(150–279) p = 0.7037 |
| gGT [<30 U/L] | 12 (10–13) | 12 (10–20) p = 0.2901 |
| Lipid-soluble vitamins A (µmol/L) [1.1–2.7] | 1.50 (1.15–1.70) | 1.40 (1.20–1.60) p = 0.6669 |
| E (µmol/L) [10–40] | 24.3 (20.7–24.6) | 20.4 (15.7–24.8) p = 0.0518 |
| D (nmol/L) [50–175] | 54.0 (46.6–59.0) | 51.0 (35.0–63.8) p = 0.5444 |
| Lung function parameters ppFEV1 (%) * FVC (%) * FEF 25 (%) * FEF25–75 (%) * | 97 (88–101) 103 (87–107) 104 (75–125) 93 (74–117) | 96 (83–104) p = 0.9754 100 (93–107) p = 0.8155 80 (57–106) p = 0.0450 84 (64–100) p = 0.0843 |
| Parameter | EPS | EPI | p-Level |
|---|---|---|---|
| A: Choline and water-soluble derivatives | |||
| Choline (µmol/L) | 8.8 (8.0–10.0) | 6.1 (5.2–7.4) | <0.0001 |
| Betaine (µmol/L) | 24.9 (21.4–27.1) | 18.6 (14.8–24.6) | 0.0287 |
| Choline + Betaine | 33.3 (30.9–35.6) | 25.3 (20.5–31.9) | 0.0020 |
| TMAO (µmol/L) | 1.4 (1.1–2.1) | 4.9 (2.6–8.0) | <0.0001 |
| Betaine/Choline | 2.64 (2.36–3.31) | 3.00 (2.473.66) | 0.0772 |
| TMAO/Choline | 0.18 (0.13–0.20) | 0.71 (0.44–1.35) | <0.0001 |
| B: Phospholipids | |||
| Phosphatidylcholine (PC) (mmol/L) | 1.41 (1.33–1.50) | 1.35 (1.19–1.51) | 0.2927 |
| Lyso-PC (% of PC) | 2.57 (1.90–3.41) | 2.56 (2.01–3.09) | 0.7318 |
| SPH (% of PC) | 25.2 (21.1–29.3) | 23.2 (20.6–26.2) | 0.2344 |
| Ceramides (% of PC) | 0.27 (0.17–0.31) | 0.24 (0.19–0.30) | 0.6938 |
| C: PC sub-groups | |||
| Disaturated PC | 1.53 (1.47–1.57) | 1.70 (1.54–1.88) | 0.0056 |
| Oleyl (C18:1)-PC | 17.4 (15.9–18.6) | 18.9 (17.4–21.1) | 0.0031 |
| Linoleoyl (C18:2)-PC | 50.5 (48.4–52.1) | 47.9 (44.5–50.5) | 0.0121 |
| Arachidonoyl (C20:4)-PC | 21.3 (19.2–23.8) | 20.9 (18.9–23.0) | 0.7543 |
| Eicosapentaenoyl (C20:5)-PC | 0.86 (0.79–0.98) | 1.11 (0.88–1.37) | 0.0119 |
| Docosahexaenoyl: C22:6-PC | 5.06 (4.46–6.69) | 5.02 (4.34–6.29) | 0.4949 |
| Other PC * | 2.55 (2.37–2.96) | 2.43 (2.88–4.22) | 0.0005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernhard, W.; Shunova, A.; Boriga, J.; Graepler-Mainka, U.; Hilberath, J. Low Plasma Choline, High Trimethylamine Oxide, and Altered Phosphatidylcholine Subspecies Are Prevalent in Cystic Fibrosis Patients with Pancreatic Insufficiency. Nutrients 2025, 17, 868. https://doi.org/10.3390/nu17050868
Bernhard W, Shunova A, Boriga J, Graepler-Mainka U, Hilberath J. Low Plasma Choline, High Trimethylamine Oxide, and Altered Phosphatidylcholine Subspecies Are Prevalent in Cystic Fibrosis Patients with Pancreatic Insufficiency. Nutrients. 2025; 17(5):868. https://doi.org/10.3390/nu17050868
Chicago/Turabian StyleBernhard, Wolfgang, Anna Shunova, Julia Boriga, Ute Graepler-Mainka, and Johannes Hilberath. 2025. "Low Plasma Choline, High Trimethylamine Oxide, and Altered Phosphatidylcholine Subspecies Are Prevalent in Cystic Fibrosis Patients with Pancreatic Insufficiency" Nutrients 17, no. 5: 868. https://doi.org/10.3390/nu17050868
APA StyleBernhard, W., Shunova, A., Boriga, J., Graepler-Mainka, U., & Hilberath, J. (2025). Low Plasma Choline, High Trimethylamine Oxide, and Altered Phosphatidylcholine Subspecies Are Prevalent in Cystic Fibrosis Patients with Pancreatic Insufficiency. Nutrients, 17(5), 868. https://doi.org/10.3390/nu17050868