
A Data-Driven Approach to Enhance the Prediction of Bacteria–Metabolite Interactions in the Human Gut Microbiome Using Enzyme Encodings and Metabolite Structural Embeddings


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical–Microbe Interactions
2.2. Curation of Metabolite Classes
2.3. Functional Annotation of Protein Sequences Using DeepECTransformer
2.4. Random Forest-Based Prediction of Enzyme Substrates and Products
2.5. Benchmarking Random Forest Models Against kNN
2.6. Analysis of Microbe–Metabolite Interactions
2.7. Curation of Negative Set
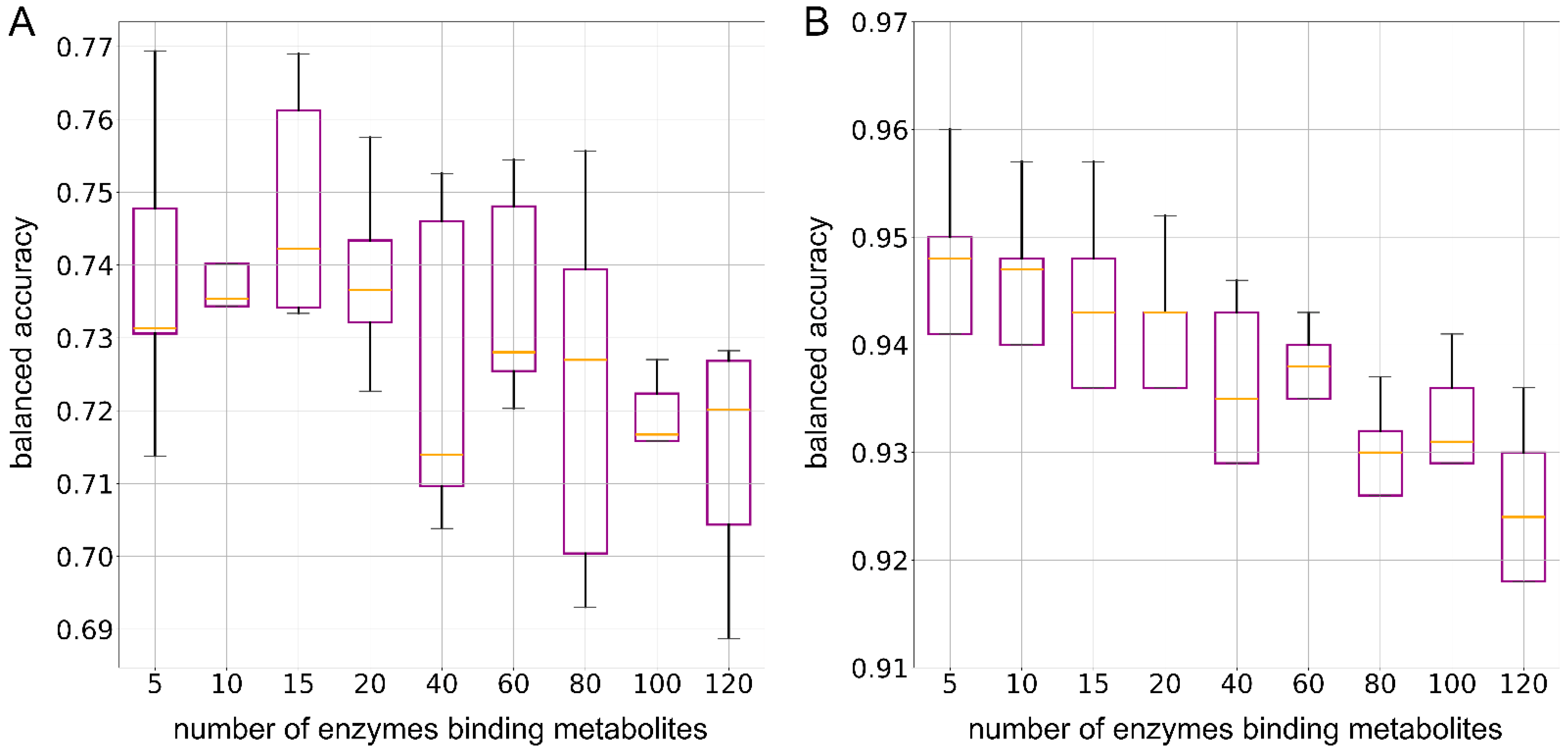
2.8. Minimum Number of Enzymes for Classification Models
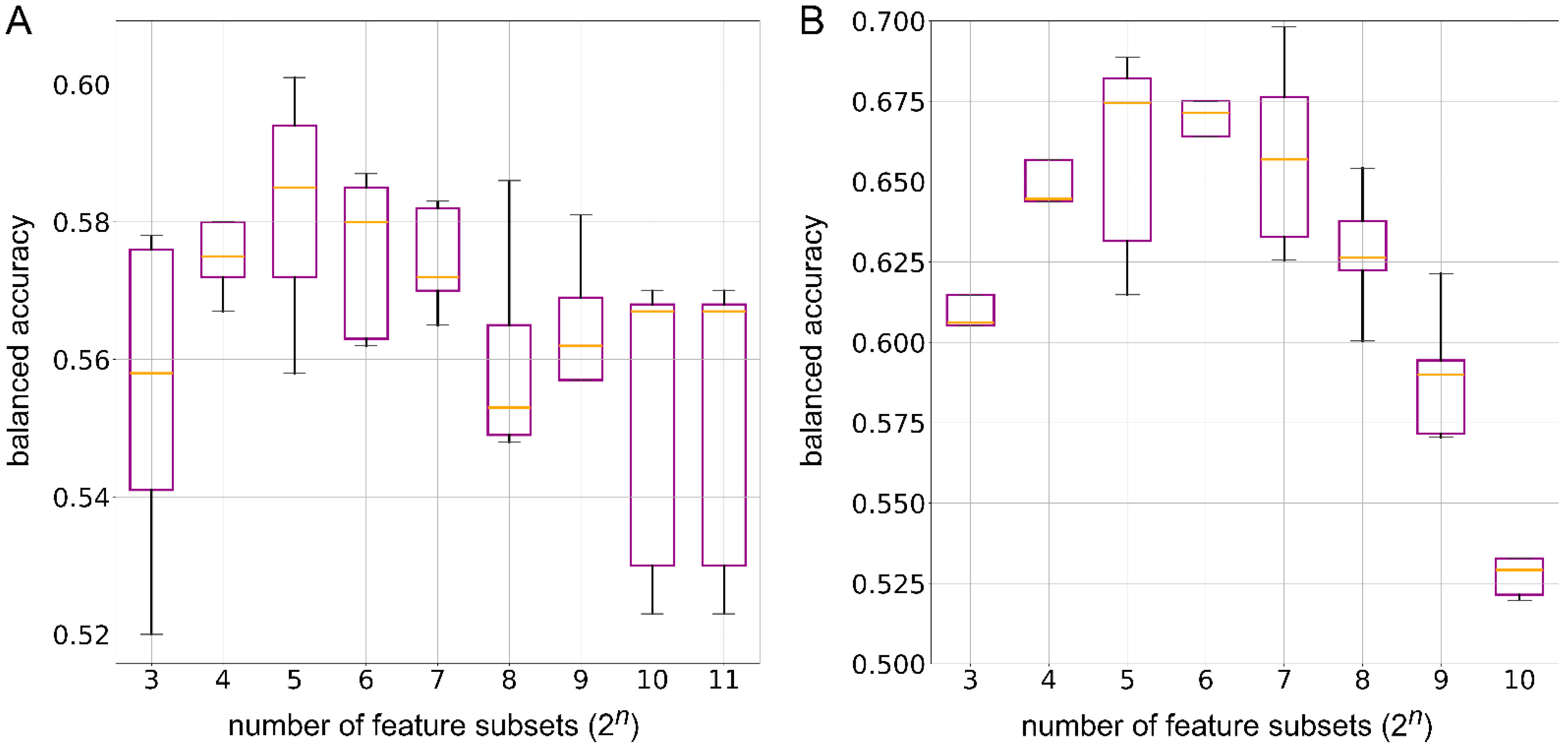
2.9. Dimensionality Reduction Using Kernel PCA
2.10. Preparation of Unseen Data
3. Results
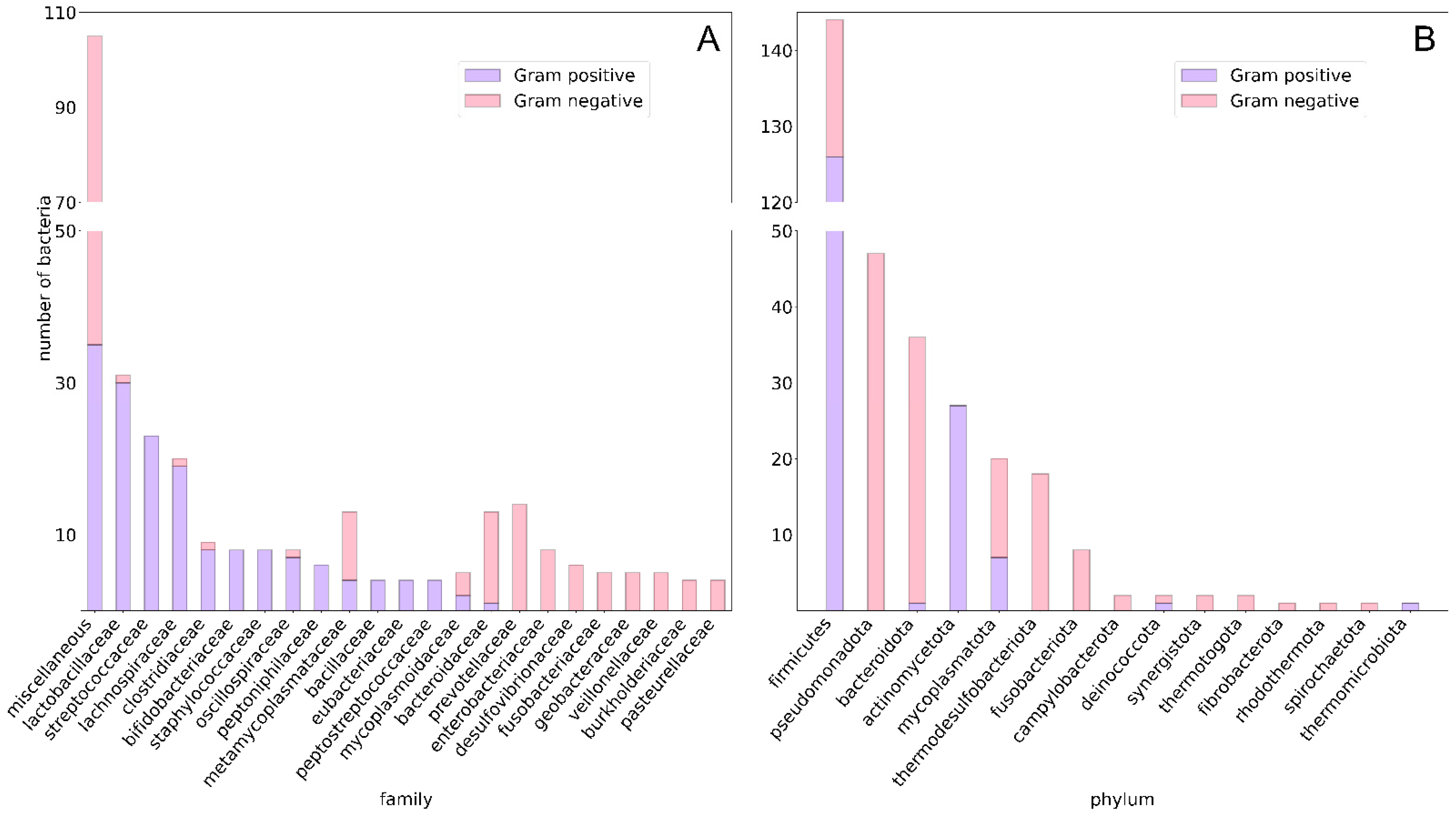
3.1. Data Collection, Curation and Analysis
3.2. Accuracy of Functional Annotation with DeepECTransformer
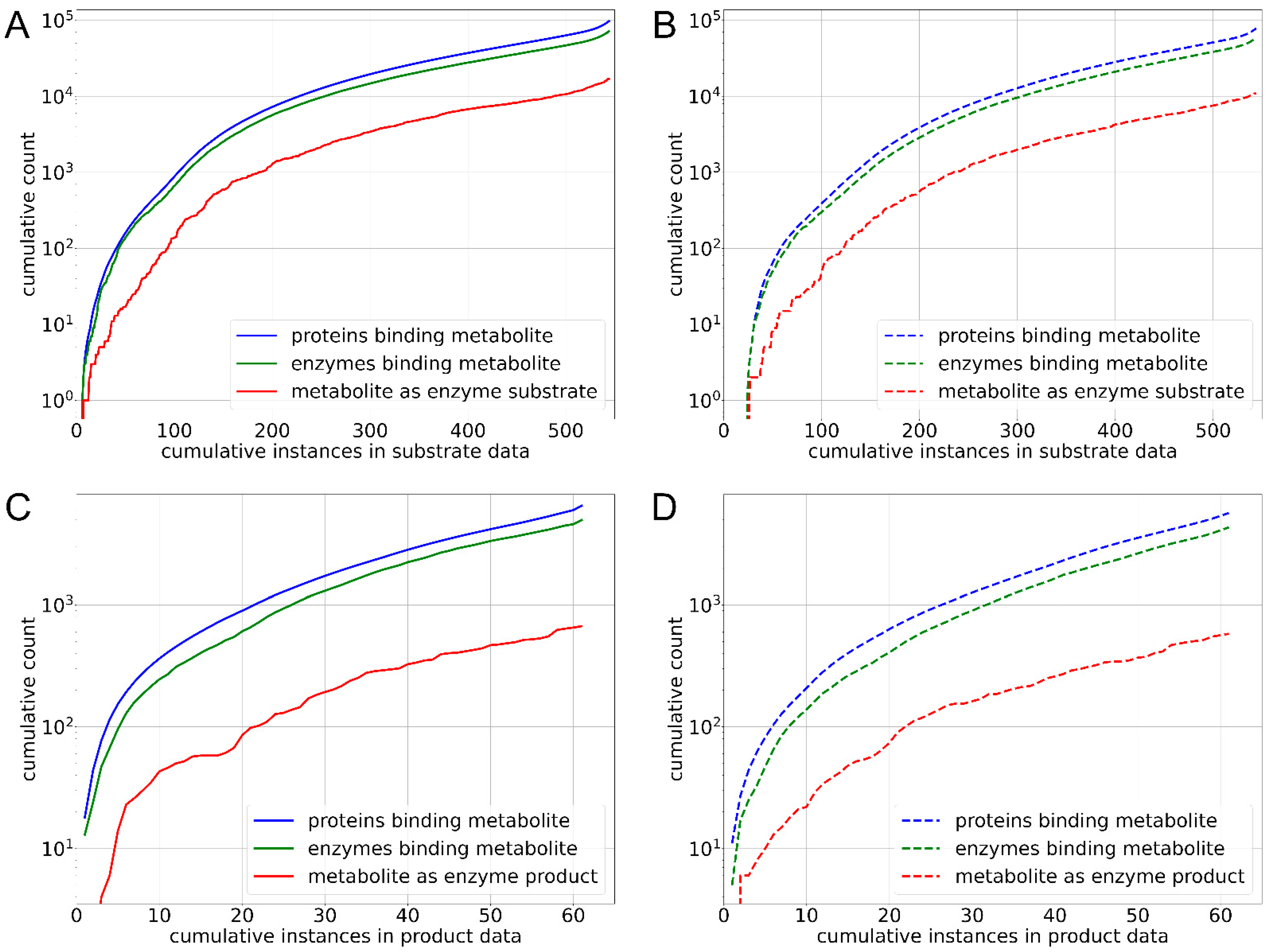
3.3. Feasibility of EC Number Encodings and Chemical Embeddings
3.4. Curation of Negative Set and RF-Based Prediction of Metabolism
3.5. Kernel Principal Component Analysis
3.6. Validation Against Unseen Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.M.P. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Hanning, I.; Diaz-Sanchez, S. The functionality of the gastrointestinal microbiome in non-human animals. Microbiome 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.J.; Goodman, A.L. Cross-feeding in the gut microbiome: Ecology and mechanisms. Cell Host Microbe 2023, 31, 485–499. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Wang, T.; Dubinkina, V.; Maslov, S. Ecology-guided prediction of cross-feeding interactions in the human gut microbiome. Nat. Commun. 2021, 12, 1335. [Google Scholar] [CrossRef]
- Sung, J.; Kim, S.; Cabatbat, J.J.T.; Jang, S.; Jin, Y.-S.; Jung, G.Y.; Chia, N.; Kim, P.-J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 2017, 8, 15393. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Drasar, B.S.; Renwick, A.G.; Williams, R.T. The role of the gut flora in the metabolism of cyclamate. Biochem. J. 1972, 129, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003. [Google Scholar] [CrossRef]
- Yim, Y.J.; Seo, J.; Kang, S.I.; Ahn, J.H.; Hur, H.G. Reductive dechlorination of methoxychlor and DDT by human intestinal bacterium Eubacterium limosum under an-aerobic conditions. Arch. Environ. Contam. Toxicol. 2008, 54, 406–411. [Google Scholar] [CrossRef]
- Joly, C.; Gay-Quéheillard, J.; Léké, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 2013, 20, 2726–2734. [Google Scholar] [CrossRef]
- Renwick, A.G.; Williams, R.T. Gut bacteria and the metabolism of cyclamate in the rat. Biochem. J. 1969, 114, 78P. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, J.; Shah, H.A.; Feng, J. A novel hybrid framework for metabolic pathways prediction based on the graph attention network. BMC Bioinform. 2022, 23, 329. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Zhou, M.; Wang, Y.; Wang, X.; Zhou, X.; Song, Q. DrugFormer: Graph-Enhanced Language Model to Predict Drug Sensitivity. Adv. Sci. 2024, 11, e2405861. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, B.; Ji, H.; Li, W. RT-Transformer: Retention time prediction for metabolite annotation to assist in metabolite identification. Bioinformatics 2024, 40, btae084. [Google Scholar] [CrossRef] [PubMed]
- Bakir-Gungor, B.; Hacılar, H.; Jabeer, A.; Nalbantoglu, O.U.; Aran, O.; Yousef, M. Inflammatory bowel disease biomarkers of human gut microbiota selected via different feature selection methods. PeerJ 2022, 10, e13205. [Google Scholar] [CrossRef] [PubMed]
- Thombre, I.; Perepu, P.K.; Sudhakar, S.K. Application of data engineering approaches to address challenges in microbiome data for optimal medical decision-making. arXiv 2023, arXiv:2307.00033. [Google Scholar]
- Fang, L.; Wang, Y.; Ye, C. Integration of multiview microbiome data for deciphering microbiome-metabolome-disease pathways. arXiv 2024, arXiv:2402.08222. [Google Scholar]
- Schölkopf, B.; Smola, A.; Müller, K.-R. Kernel principal component analysis. In International Conference on Artificial Neural Networks; Springer: New York, NY, USA, 1997. [Google Scholar]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of us-er-uploaded gene/measurement sets. Nucleic. Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Stothard, P.; Van Domselaar, G.; Shrivastava, S.; Guo, A.; O’Neill, B.; Cruz, J.; Ellison, M.; Wishart, D.S. BacMap: An interactive picture atlas of annotated bacterial genomes. Nucleic Acids Res. 2005, 33, D317–D320. [Google Scholar] [CrossRef]
- Cruz, J.; Liu, Y.; Liang, Y.; Zhou, Y.; Wilson, M.; Dennis, J.J.; Stothard, P.; Van Domselaar, G.; Wishart, D.S. BacMap: An up-to-date electronic atlas of annotated bacterial genomes. Nucleic Acids Res. 2012, 40, D599–D604. [Google Scholar] [CrossRef] [PubMed]
- Reimer, L.C.; Carbasse, J.S.; Koblitz, J.; Ebeling, C.; Podstawka, A.; Overmann, J. BacDive in 2022: The knowledge base for standardized bacterial and archaeal data. Nucleic Acids Res. 2022, 50, D741–D746. [Google Scholar] [CrossRef]
- Söhngen, C.; Bunk, B.; Podstawka, A.; Gleim, D.; Overmann, J. BacDive—The Bacterial Diversity Metadatabase. Nucleic Acids Res. 2014, 42, D592–D599. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2008, 36, D684–D688. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An open source chemical structure curation pipeline using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Kim, J.Y.; Lee, J.A.; Norsigian, C.J.; Palsson, B.O.; Lee, S.Y. Functional annotation of enzyme-encoding genes using deep learning with transformer layers. Nat. Commun. 2023, 14, 7370. [Google Scholar] [CrossRef]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002, 30, 47–49. [Google Scholar] [CrossRef]
- Sun, D.; Cheng, X.; Tian, Y.; Ding, S.; Zhang, D.; Cai, P.; Hu, Q.-N. EnzyMine: A comprehensive database for enzyme function annotation with enzymatic reaction chemical feature. Database 2020, 2023, baaa065. [Google Scholar] [CrossRef]
- Chung, N.C.; Miasojedow, B.; Startek, M.; Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinform. 2019, 20 (Suppl. S15), 644. [Google Scholar] [CrossRef]
- Steck, H.; Ekanadham, C.; Kallus, N. Is cosine-similarity of embeddings really about similarity? In Proceedings of the Companion Proceedings of the ACM on Web Conference, Singapore, 13–17 May 2024. [Google Scholar]
- Fay, M.P.; Proschan, M.A. Wilcoxon-Mann-Whitney or t-test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Stat. Surv. 2010, 4, 1–39. [Google Scholar] [CrossRef]
- McHugh, M.L. The Chi-square test of independence. Biochem. Med. 2013, 23, 143–149. [Google Scholar] [CrossRef]
- Rodionova, I.A.; Scott, D.A.; Grishin, N.V.; Osterman, A.L.; Rodionov, D.A. Tagaturonate-fructuronate epimerase UxaE, a novel enzyme in the hexuronate catabolic network in Thermotoga maritima. Environ. Microbiol. 2012, 14, 2920–2934. [Google Scholar] [CrossRef]
- Zhang, W.; Lang, R. Succinate metabolism: A promising therapeutic target for inflammation, ischemia/reperfusion injury and cancer. Front. Cell Dev. Biol. 2023, 11, 1266973. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Alpha-glucosidase inhibitors. Endocrinol. Metab. Clin. North Am. 1997, 26, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef]
- Hogeveen, M.; Heijer, M.D.; Semmekrot, B.A.; Sporken, J.M.; Ueland, P.M.; Blom, H.J. Umbilical choline and related methylamines betaine and dimethylglycine in relation to birth weight. Pediatr. Res. 2013, 73, 783–787. [Google Scholar] [CrossRef]
- Sharma, M.J.; Sabir, S.; Sharma, S. GABA Receptor; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Parrella, N.-F.; Hill, A.T.; Dipnall, L.M.; Loke, Y.J.; Enticott, P.G.; Ford, T.C. Inhibitory dysfunction and social processing difficulties in autism: A comprehensive narrative review. J. Psychiatr. Res. 2024, 169, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Borsom, E.M.; Lee, K.; Cope, E.K. Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alz-heimer’s Disease. Brain Sci. 2020, 10, 814. [Google Scholar] [CrossRef]
- Dadi, P.; Pauling, C.W.; Shrivastava, A.; Shah, D.D. Synthesis of versatile neuromodulatory molecules by a gut microbial glutamate decarboxylase. bioRxiv, 2024; preprint. [Google Scholar]
- Tiefenbacher, K.F. Technology of Main Ingredients—Sweeteners and Lipids. In Wafer and Waffle; Tiefenbacher, K.F., Ed.; Academic Press: London, UK, 2017; pp. 123–225. [Google Scholar]
- Fultz, R.; Ticer, T.; Ihekweazu, F.D.; Horvath, T.D.; Haidacher, S.J.; Hoch, K.M.; Bajaj, M.; Spinler, J.K.; Haag, A.M.; Buffington, S.A.; et al. Unraveling the Metabolic Requirements of the Gut Commensal Bacteroides ovatus. Front. Microbiol. 2021, 12, 745469. [Google Scholar] [CrossRef]
- Nilsson, U.; Jägerstad, M. Hydrolysis of lactitol, maltitol and Palatinit by human intestinal biopsies. Br. J. Nutr. 1987, 58, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Hauner, H.; Hornef, M.; Clavel, T. Allulose in human diet: The knowns and the unknowns. Br. J. Nutr. 2021, 128, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Jiang, J.; Hang, X.; Zhang, M.; Liu, X.; Li, D.; Yang, H. Diversity of bile salt hydrolase activities in different lactobacilli toward human bile salts. Ann. Microbiol. 2009, 60, 81–88. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Lawal, S.A.; Voisin, A.; Olof, H.; Bording-Jorgensen, M.; Armstrong, H. Diversity of the microbiota communities found in the various regions of the intestinal tract in healthy individuals and inflammatory bowel diseases. Front. Immunol. 2023, 14, 1242242. [Google Scholar] [CrossRef]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human gut microbiota in health and disease: Unveiling the relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef] [PubMed]
- Calvigioni, M.; Panattoni, A.; Biagini, F.; Donati, L.; Mazzantini, D.; Massimino, M.; Daddi, C.; Celandroni, F.; Vozzi, G.; Ghelardi, E. Impact of Bacillus cereus on the Human Gut Microbiota in a 3D In Vitro Model. Microorganisms 2023, 11, 1826. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.M.; Harris, H.M.; Lynch, D.B.; Ross, R.P.; O’Toole, P.W. Lactobacillus ruminis strains cluster according to their mammalian gut source. BMC Microbiol. 2015, 15, 80. [Google Scholar]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; A Lugli, G.; James, K.; Duranti, S.; Turroni, F.; Ferrario, C.; Ossiprandi, M.C.; van Sinderen, D.; et al. Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J. 2017, 11, 2834–2847. [Google Scholar] [CrossRef]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS ONE 2012, 7, e36957. [Google Scholar] [CrossRef]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Armanini, F.; Turroni, F.; James, K.; Ferretti, P.; Gorfer, V.; Ferrario, C.; Milani, C.; et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, G.; Milani, C.; Duranti, S.; Mancabelli, L.; Ranjanoro, T.; Modica, S.; Carnevali, L.; Statello, R.; Bottacini, F.; Turroni, F.; et al. Ability of bifidobacteria to metabolize chitin-glucan and its impact on the gut microbiota. Sci. Rep. 2019, 9, 5755. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Marchesi, J.R.; Foroni, E.; Gueimonde, M.; Shanahan, F.; Margolles, A.; van Sinderen, D.; Ventura, M. Microbiomic analysis of the bifidobacterial population in the human distal gut. ISME J. 2009, 3, 745–751. [Google Scholar] [CrossRef]
- Collado, M.C.; Gueimonde, M.; Sanz, Y.; Salminen, S. Adhesion properties and competitive pathogen exclusion ability of bifidobacteria with acquired acid resistance. J. Food Prot. 2006, 69, 1675–1679. [Google Scholar] [CrossRef]
- Turroni, F.; Duranti, S.; Milani, C.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Bifidobacterium bifidum: A Key Member of the Early Human Gut Microbiota. Microorganisms 2019, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Cionci, N.B.; Baffoni, L.; Gaggìa, F.; Di Gioia, D. Therapeutic Microbiology: The Role of Bifidobacterium breve as Food Supplement for the Prevention/Treatment of Paediatric Diseases. Nutrients 2018, 10, 1723. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Kim, J.; Kim, S.-H.; Ban, O.-H.; Yang, J.; Park, M.-K. Safety Evaluation of Bifidobacterium breve IDCC4401 Isolated from Infant Feces for Use as a Commercial Probiotic. J. Microbiol. Biotechnol. 2021, 31, 949–955. [Google Scholar] [CrossRef]
- Seo, Y.S.; Lee, H.-B.; Kim, Y.; Park, H.-Y. Dietary Carbohydrate Constituents Related to Gut Dysbiosis and Health. Microorganisms 2020, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Mora-Flores, L.P.; Casildo, R.M.-T.; Fuentes-Cabrera, J.; Pérez-Vicente, H.A.; de Anda-Jáuregui, G.; Neri-Torres, E.E. The Role of Carbohydrate Intake on the Gut Microbiome: A Weight of Evidence Systematic Review. Microorganisms 2023, 11, 1728. [Google Scholar] [CrossRef]
- Jarboe, L.R.; Royce, L.A.; Liu, P. Understanding biocatalyst inhibition by carboxylic acids. Front. Microbiol. 2013, 4, 272. [Google Scholar] [CrossRef]
- Quillin, S.J.; Tran, P.; Prindle, A. Potential roles for gamma-aminobutyric acid signaling in bacterial communities. Bioelectricity 2021, 3, 120–125. [Google Scholar] [CrossRef]
- Cooper, G.M.; Adams, K. The Cell: A Molecular Approach; Oxford University Press: Oxford, UK, 2022. [Google Scholar]
- Bruns, A.; Philipp, H.; Cypionka, H.; Brinkhoff, T. Aeromicrobium marinum sp. nov., an abundant pelagic bacterium isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 2003, 53 Pt 6, 1917–1923. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Dissimilatory amino acid metabolism in human colonic bacteria. Anaerobe 1997, 3, 327–337. [Google Scholar] [CrossRef]
- Sauer, J.-D.; Herskovits, A.A.; O’riordan, M.X. Metabolism of the Gram-Positive Bacterial Pathogen Listeria monocytogenes. Microbiol. Spectr. 2019, 7, 864–872. [Google Scholar] [CrossRef]
- Czuprynski, C.J.; Faith, N.G. Sodium bicarbonate enhances the severity of infection in neutropenic mice orally inoculated with Listeria monocytogenes EGD. Clin. Vaccine Immunol. 2002, 9, 477–481. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactant | Classifier | BAC | AUC | PPV | TPR | FPR | F1-Score | MCC |
|---|---|---|---|---|---|---|---|---|
| Substrate | RF | 0.788 | 0.870 | 0.794 | 0.775 | 0.200 | 0.785 | 0.575 |
| 3NN | 0.508 | 0.508 | 0.508 | 0.454 | 0.438 | 0.479 | 0.016 | |
| Product | RF | 0.791 | 0.870 | 0.799 | 0.775 | 0.194 | 0.787 | 0.582 |
| 3NN | 0.491 | 0.491 | 0.489 | 0.479 | 0.496 | 0.484 | −0.017 |
| Metabolite | Microbe | Original Label | Predicted Label | ||
|---|---|---|---|---|---|
| Consumption Model | Production Model | Consensus | |||
| miglitol | Gluconobacter oxydans | production | production (0.64, 0.36) | consumption (0.87, 0.13) | consumption (0.62, 0.38) |
| betaine | Bifidobacterium bifidum | production | production (0.62, 0.38) | production (0.48, 0.52) | production (0.57, 0.43) |
| 4-aminobutyrate | Bacteroides fragilis | production | production (0.62, 0.38) | production (0.08, 0.92) | production (0.77, 0.23) |
| maltitol | Bacteroides ovatus | consumption | unspecified (0.5, 0.5) | consumption (0.80, 0.20) | consumption (0.35, 0.65) |
| D-psicose | Clostridium carboxidivorans | consumption | consumption (0.27, 0.73) | consumption (0.96, 0.04) | consumption (0.16, 0.84) |
| taurochenodeoxycholate | Lactobacillus acidophilus | consumption | consumption (0.06, 0.94) | production (0.26, 0.74) | consumption (0.40, 0.60) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, G.; Brylinski, M. A Data-Driven Approach to Enhance the Prediction of Bacteria–Metabolite Interactions in the Human Gut Microbiome Using Enzyme Encodings and Metabolite Structural Embeddings. Nutrients 2025, 17, 469. https://doi.org/10.3390/nu17030469
Srivastava G, Brylinski M. A Data-Driven Approach to Enhance the Prediction of Bacteria–Metabolite Interactions in the Human Gut Microbiome Using Enzyme Encodings and Metabolite Structural Embeddings. Nutrients. 2025; 17(3):469. https://doi.org/10.3390/nu17030469
Chicago/Turabian StyleSrivastava, Gopal, and Michal Brylinski. 2025. "A Data-Driven Approach to Enhance the Prediction of Bacteria–Metabolite Interactions in the Human Gut Microbiome Using Enzyme Encodings and Metabolite Structural Embeddings" Nutrients 17, no. 3: 469. https://doi.org/10.3390/nu17030469
APA StyleSrivastava, G., & Brylinski, M. (2025). A Data-Driven Approach to Enhance the Prediction of Bacteria–Metabolite Interactions in the Human Gut Microbiome Using Enzyme Encodings and Metabolite Structural Embeddings. Nutrients, 17(3), 469. https://doi.org/10.3390/nu17030469