
Urolithin A Exhibits Antidepressant-like Effects by Modulating the AMPK/CREB/BDNF Pathway

 and
and
Abstract
1. Introduction
2. Materials
2.1. Cell Culture and Treatment
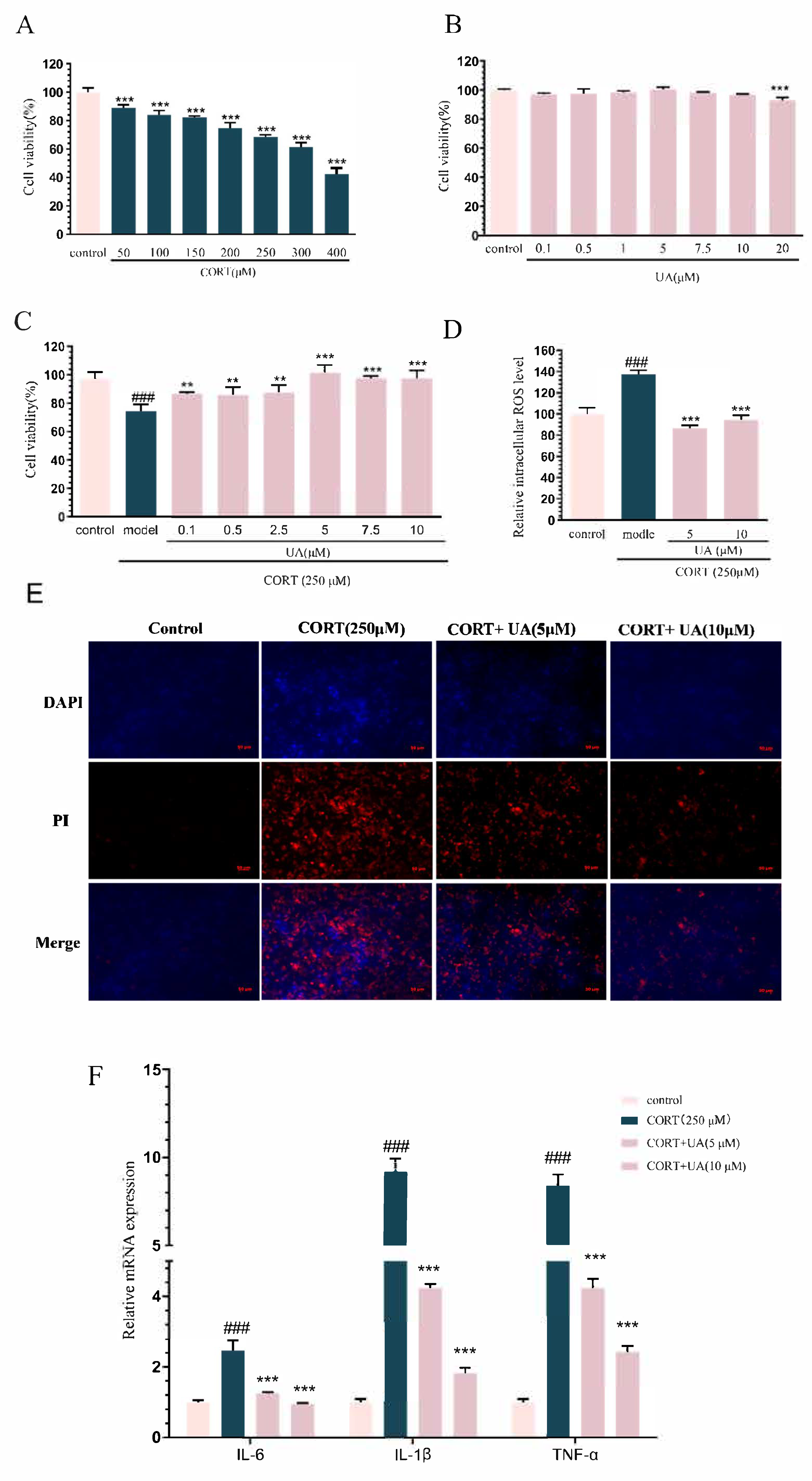
2.2. Cell Viability Assay
2.3. Reactive Oxygen Species Assay
2.4. Determination of Apoptosis
2.5. Animals and Treatments
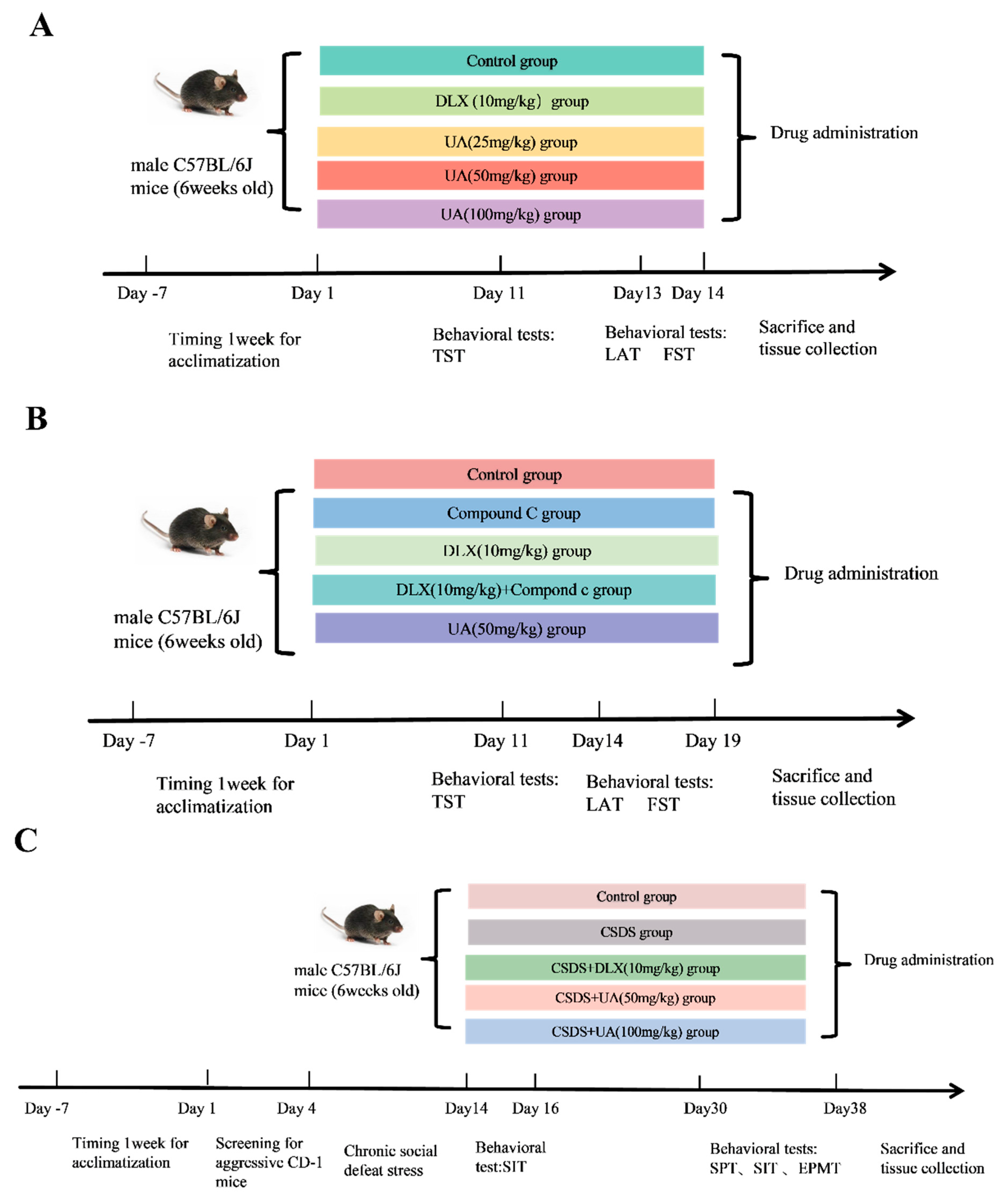
3. Experimental Design
3.1. Forced Swimming Test (FST)
3.2. Tail Suspension Test (TST)
3.3. Locomotor Activity Test (LAT)
3.4. Chronic Social Defeat Stress (CSDS) Model
3.4.1. Screening Process for Aggressor CD-1 Mice
3.4.2. CSDS Procedure
3.5. Sucrose Preference Test (SPT)
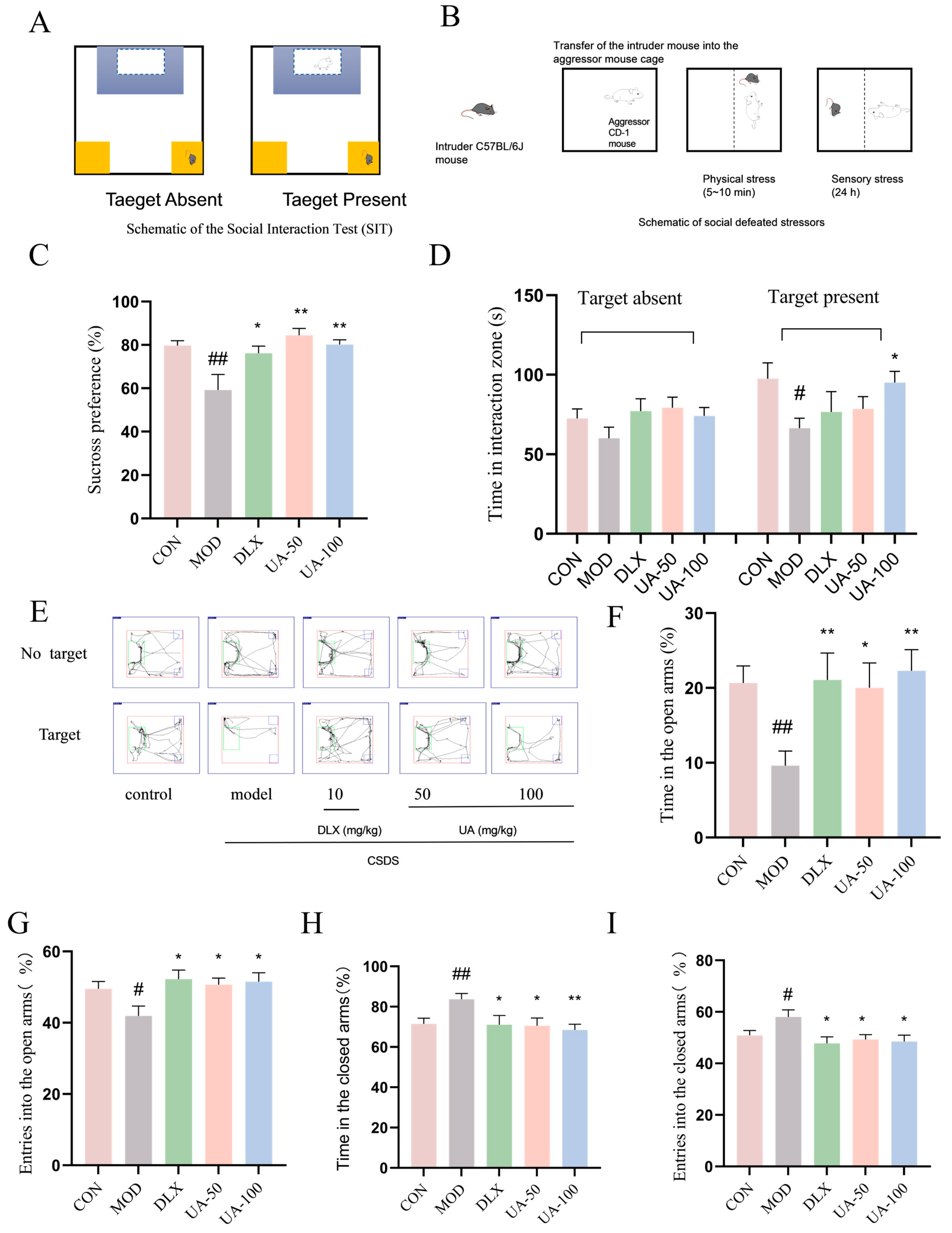
3.6. Social Interaction Test (SIT)
3.7. Elevated Plus Maze Test (EPMT)
3.8. Measurement of Adrenocorticotropic Hormone (ACTH) and Corticosterone (CORT) Levels
3.9. RT-qPCR
3.10. Western Blot Analysis
3.11. Statistical Analysis
4. Results
4.1. UA Inhibited CORT-Induced Damage in PC12 Cells
4.2. UA Upregulated AMPK Activity and CREB-Mediated Neurotrophic Signaling in CORT-Treated PC12 Cells
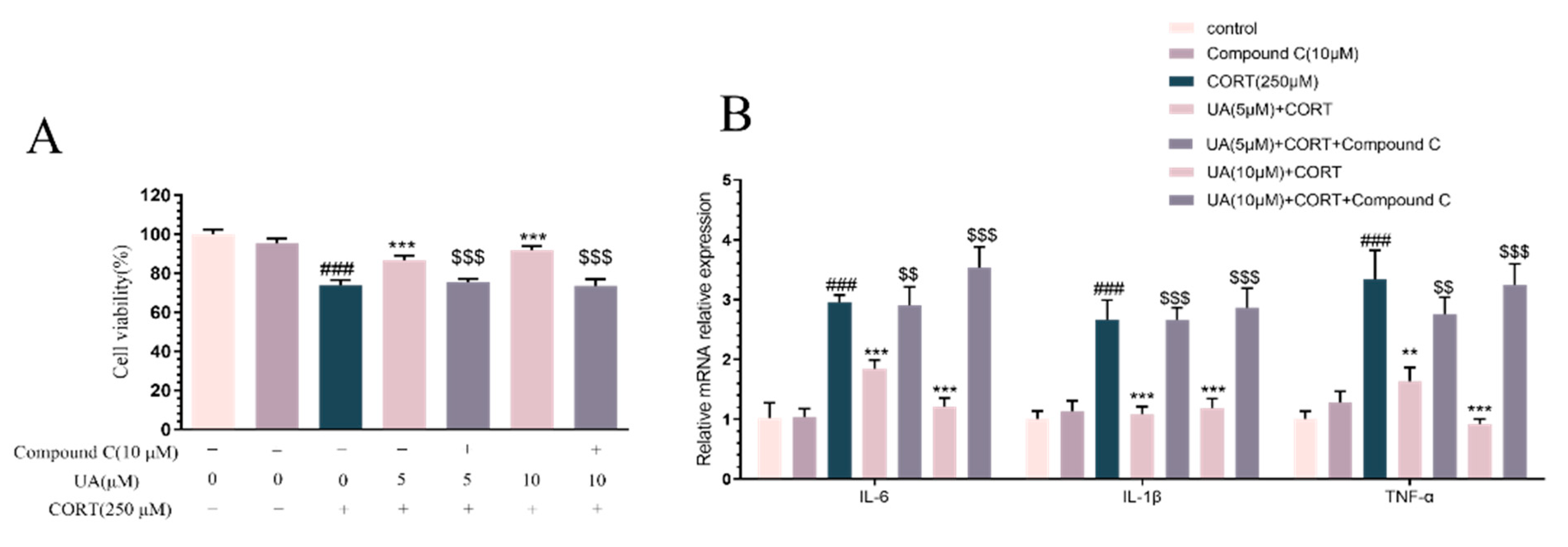
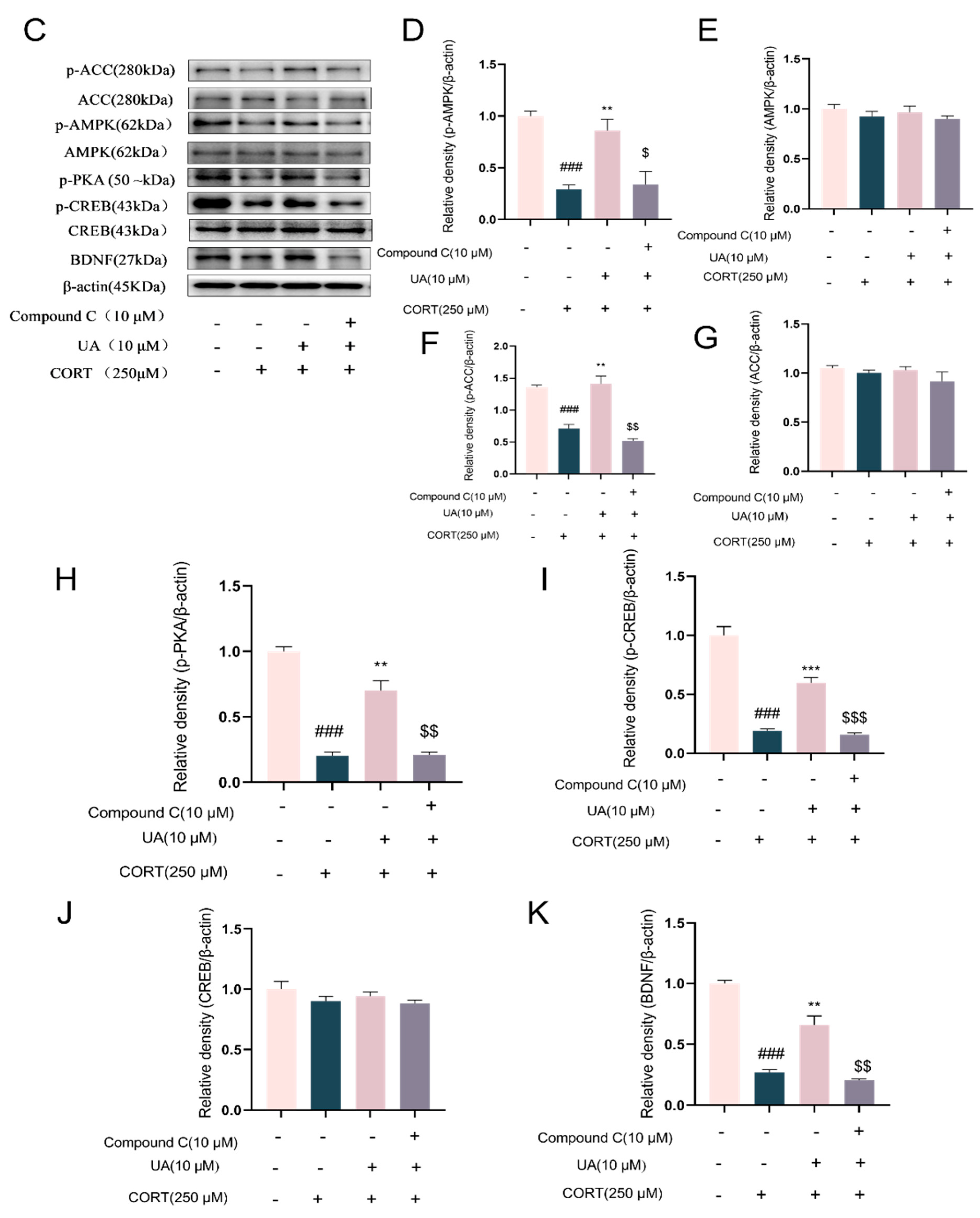
4.3. Blocking AMPK Activity Abolished the Cytoprotective and Neurotrophic Effects of UA in CORT-Treated PC12 Cells
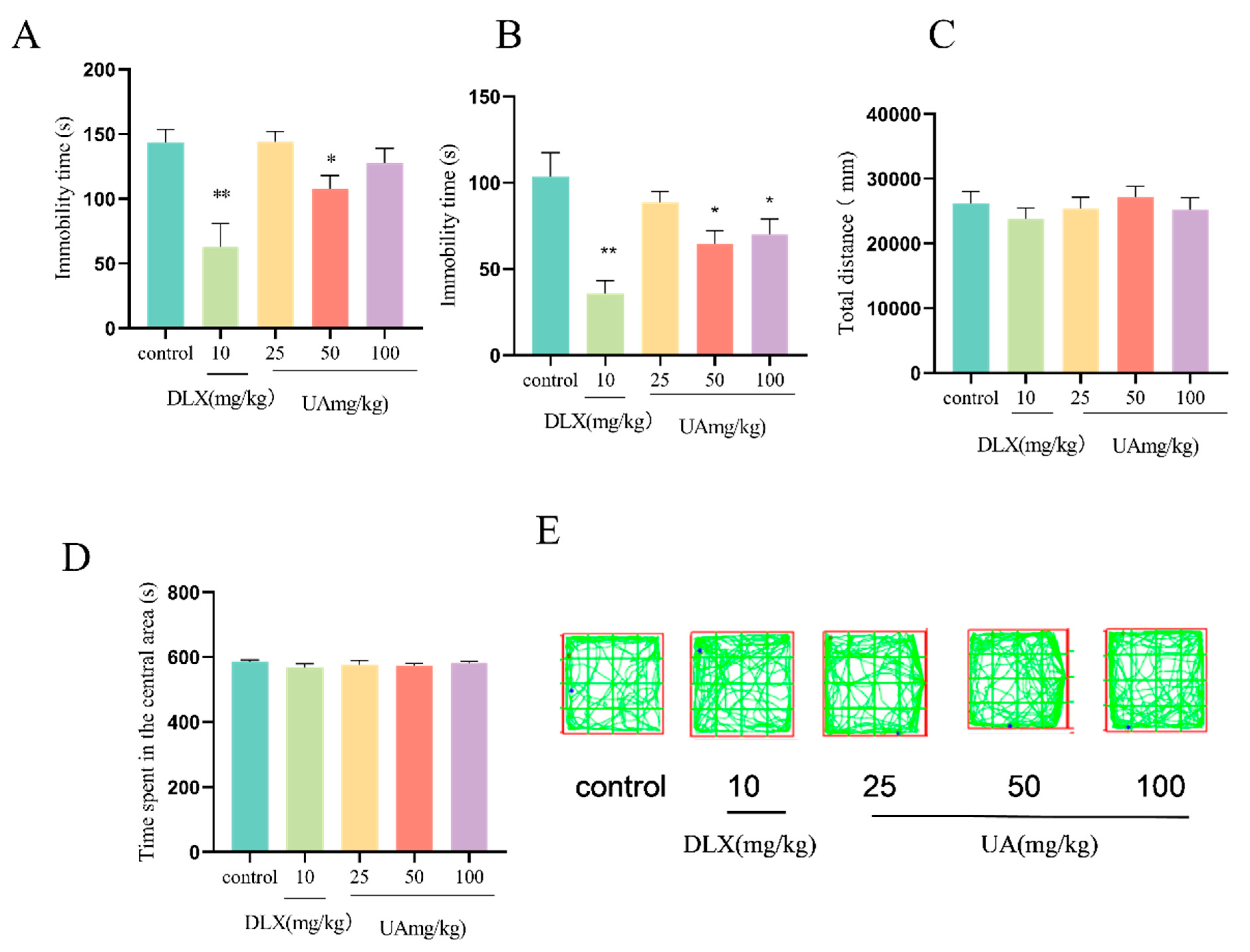
4.4. UA Produced Antidepressant-like Effects in the FST and TST in Mice
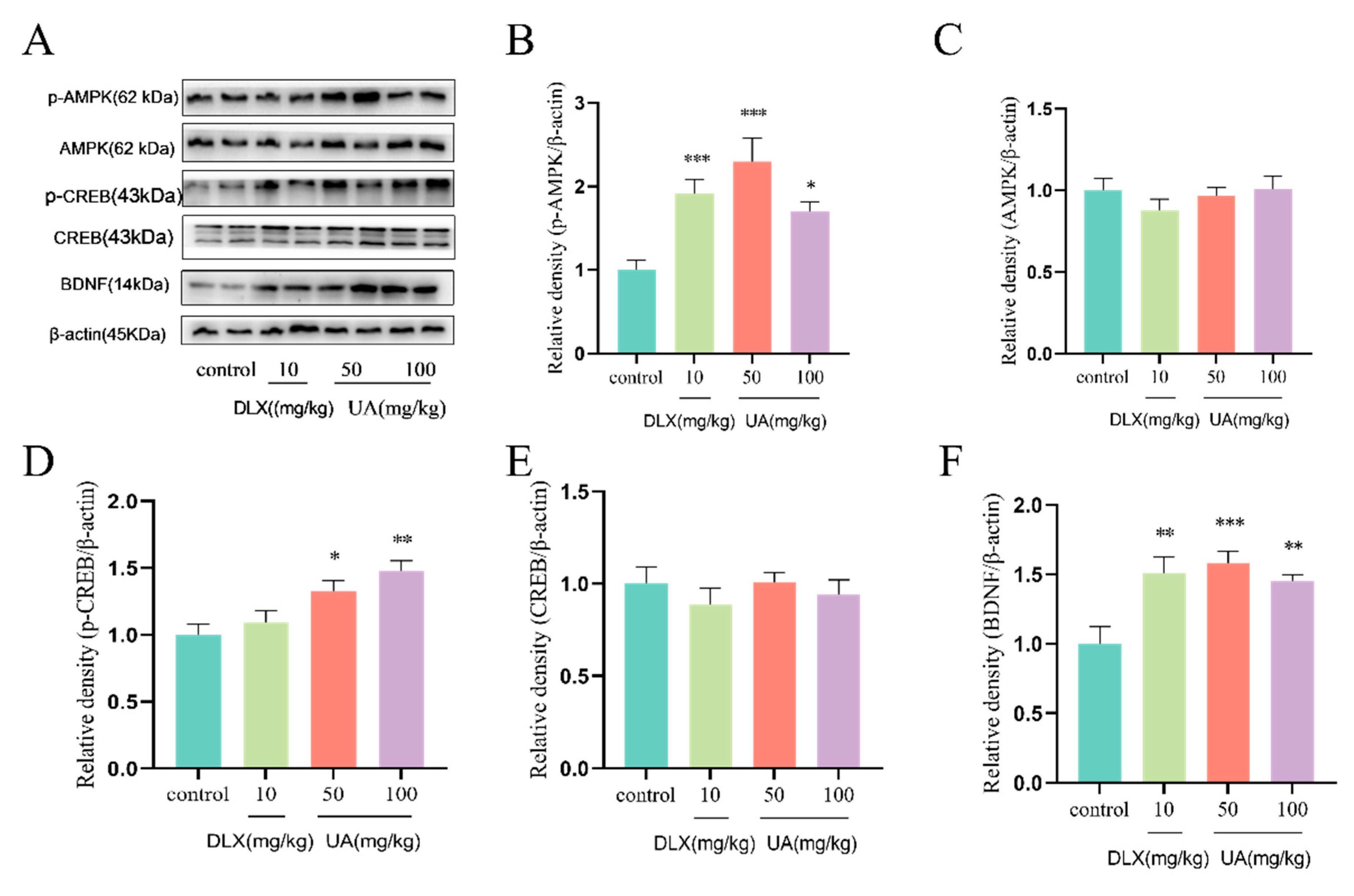
4.5. UA Increased AMPK Activity and CREB-Mediated Neurotrophic Signaling in the Mouse Hippocampus
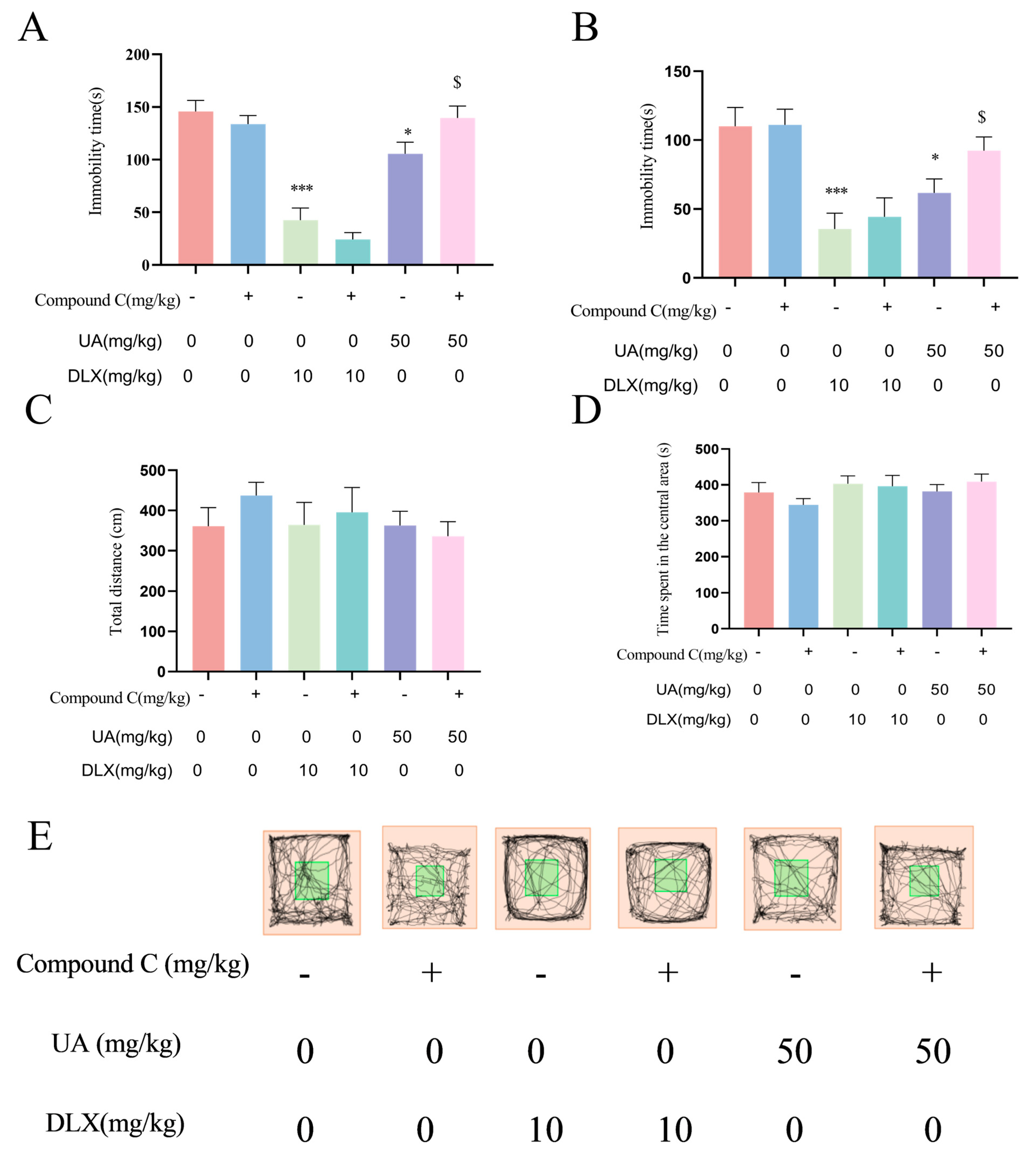
4.6. Activation of AMPK Is Necessary for the Antidepressant Effects of UA in the TST and FST
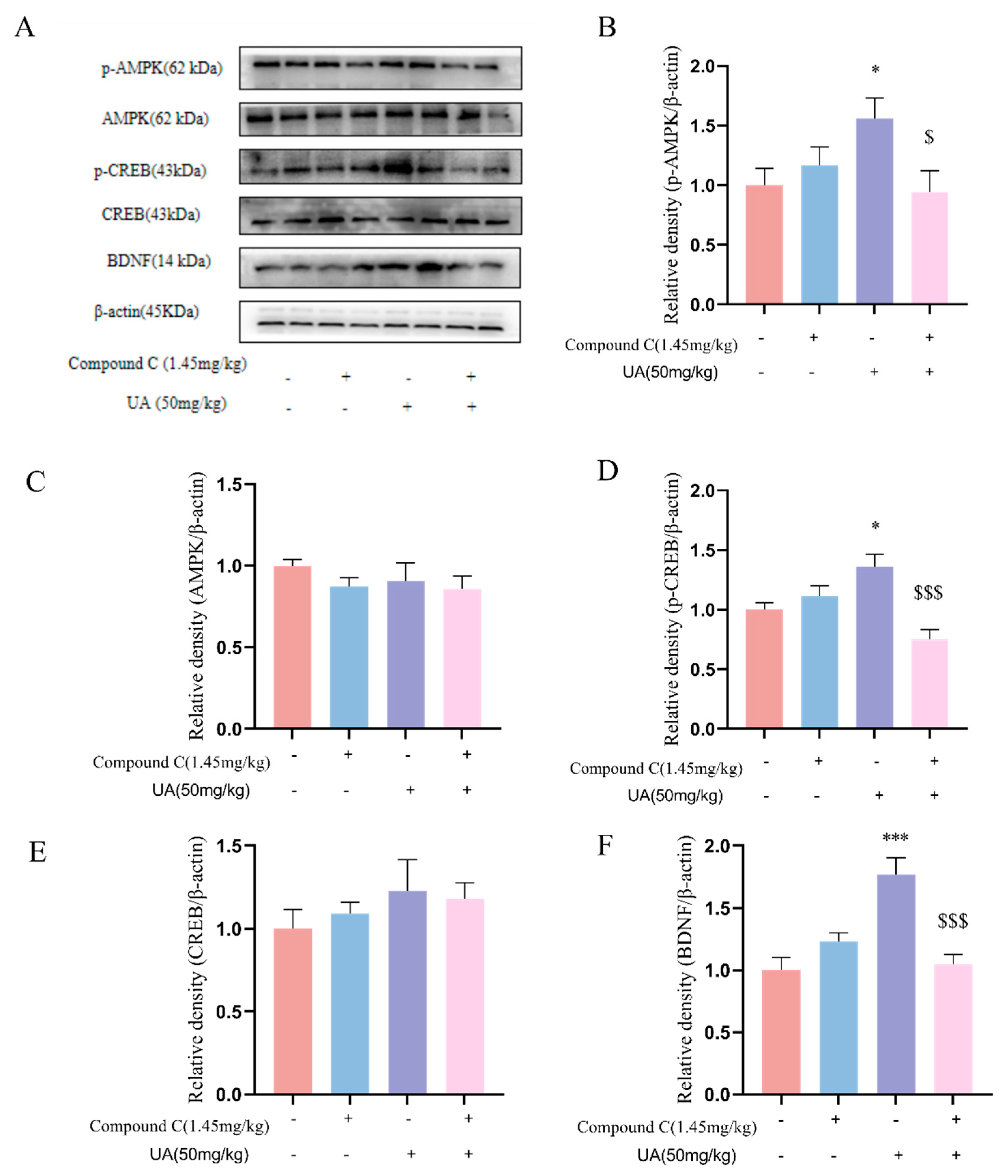
4.7. Activation of AMPK Is Necessary for the Neurotrophic Effects of UA in the Hippocampus of Mice
4.8. UA Alleviated the CSDS-Induced Depression-like and Anxiety-like Behaviors in Mice
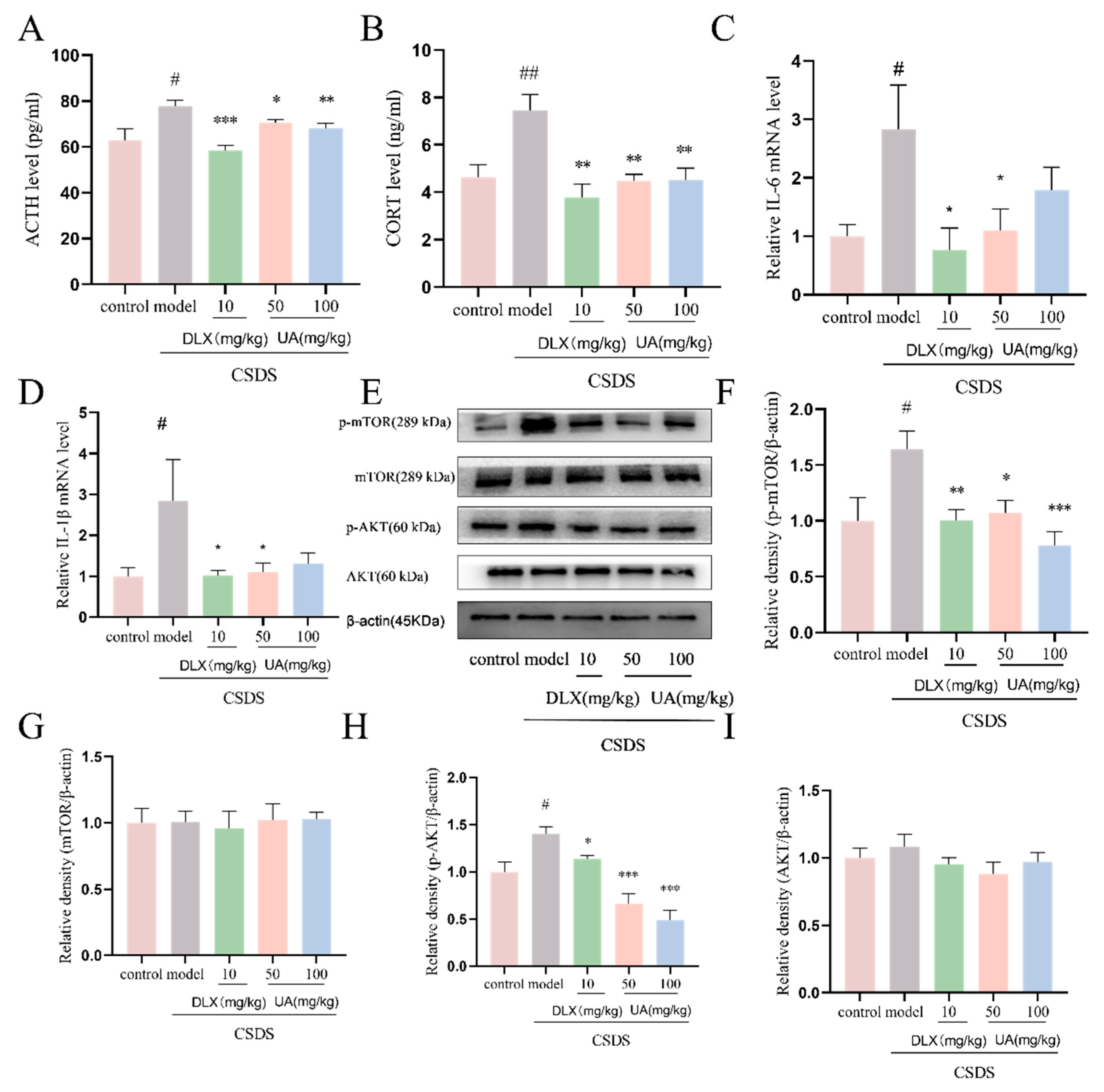
4.9. UA Attenuated the CSDS-Induced Oversecretion of Serum Stress Hormones and Hippocampal Inflammation
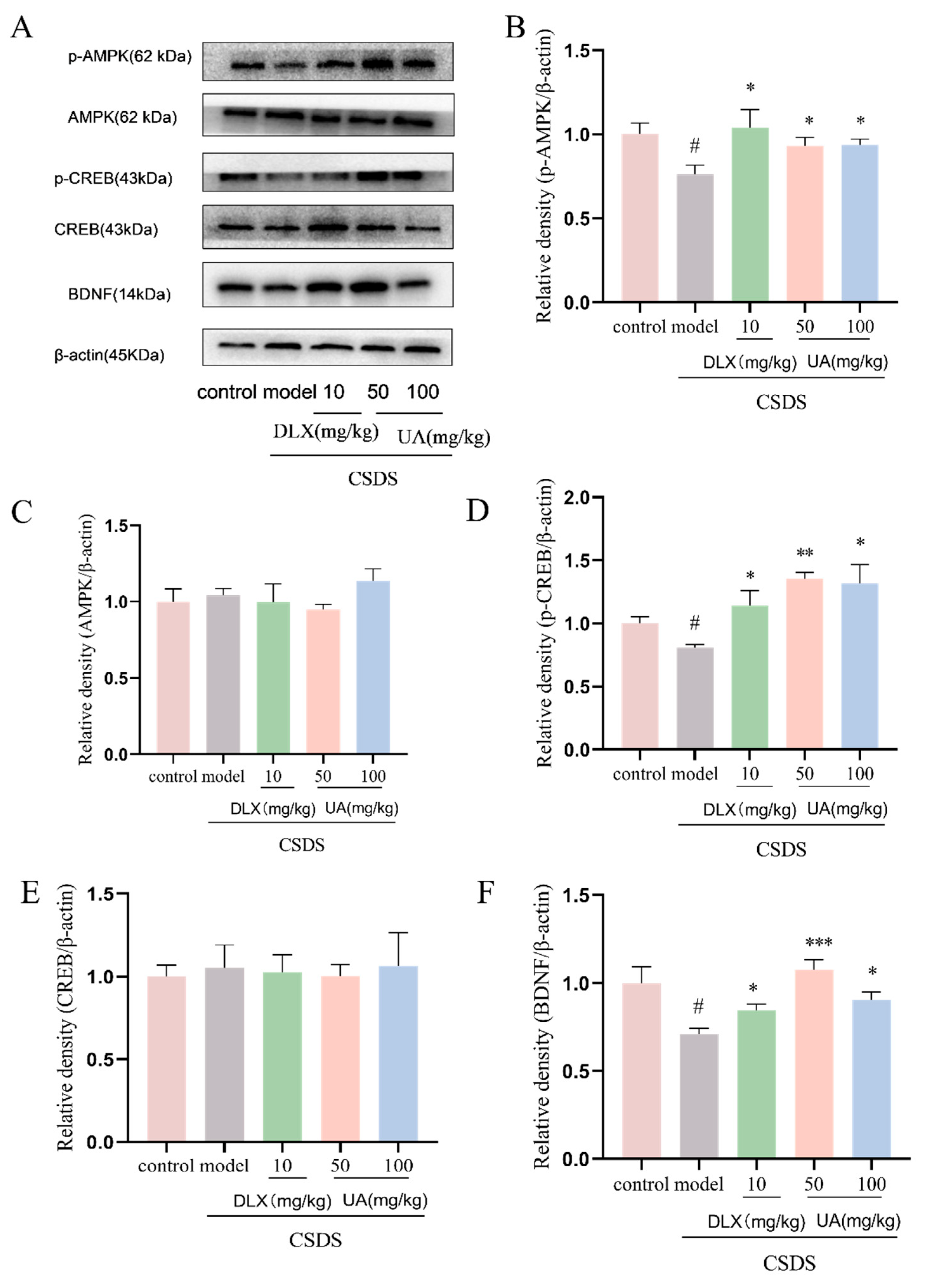
4.10. UA Reversed the CSDS-Induced AMPK/CREB/BDNF Signaling Downregulation in the Hippocampus
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hajiluian, G.; Karegar, S.J.; Shidfar, F.; Aryaeian, N.; Salehi, M.; Lotfi, T.; Farhangnia, P.; Heshmati, J.; Delbandi, A.A. The effects of Ellagic acid supplementation on neurotrophic, inflammation, and oxidative stress factors, and indoleamine 2,3-dioxygenase gene expression in multiple sclerosis patients with mild to moderate depressive symptoms: A randomized, triple-blind, placebo-controlled trial. Phytomedicine 2023, 121, 155094. [Google Scholar] [PubMed]
- Kujawska, M.; Jourdes, M.; Kurpik, M.; Szulc, M.; Szaefer, H.; Chmielarz, P.; Kreiner, G.; Krajka-Kuźniak, V.; Mikołajczak, P.Ł.; Teissedre, P.-L.; et al. Neuroprotective Effects of Pomegranate Juice against Parkinson’s Disease and Presence of Ellagitannins-Derived Metabolite-Urolithin A-In the Brain. Int. J. Mol. Sci. 2019, 21, 202. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, R.; Lepiarz, I.; El-Bakoush, A.; Katola, F.O.; Bhatia, H.; Fiebich, B.L.; Olajide, O.A. Induction of Autophagy and Activation of SIRT-1 Deacetylation Mechanisms Mediate Neuroprotection by the Pomegranate Metabolite Urolithin A in BV2 Microglia and Differentiated 3D Human Neural Progenitor Cells. Mol. Nutr. Food Res. 2019, 63, e1801237. [Google Scholar] [CrossRef]
- Suez, J.; Elinav, E. The path towards microbiome-based metabolite treatment. Nat. Microbiol. 2017, 2, 17075. [Google Scholar] [CrossRef]
- Zheng, Y.; Bonfili, L.; Wei, T.; Eleuteri, A.M. Understanding the Gut-Brain Axis and Its Therapeutic Implications for Neurodegenerative Disorders. Nutrients 2023, 15, 4631. [Google Scholar] [CrossRef]
- Arab, L.; Guo, R.; Elashoff, D. Lower Depression Scores among Walnut Consumers in NHANES. Nutrients 2019, 11, 275. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, J.; Zhu, J.X.; Xu, G.H.; Huang, W.F.; Yi, L.T. Identification, quantification, and antidepressant-like evaluation of anthocyanin-rich extracts from different dietary berries. Food Sci. Nutr. 2024, 12, 6045–6895. [Google Scholar] [CrossRef]
- Estrada-Camarena, E.; López-Rubalcava, C.; Vega-Rivera, N.M.; González-Trujano, M.E. Antidepressant- and Anxiolytic-like Effects of Pomegranate: Is It Acting by Common or Well-Known Mechanisms of Action? Plants 2024, 13, 2205. [Google Scholar] [CrossRef]
- Cásedas, G.; Les, F.; Choya-Foces, C.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 9, 177. [Google Scholar] [CrossRef]
- Chen, P.; Chen, F.; Lei, J.; Wang, G.; Zhou, B. The Gut Microbiota Metabolite Urolithin B Improves Cognitive Deficits by Inhibiting Cyt C-Mediated Apoptosis and Promoting the Survival of Neurons Through the PI3K Pathway in Aging Mice. Front. Pharmacol. 2021, 12, 768097. [Google Scholar] [CrossRef]
- Singh, R.; Chandrashekharappa, S.; Vemula, P.K.; Haribabu, B.; Jala, V.R. Microbial Metabolite Urolithin B Inhibits Recombinant Human Monoamine Oxidase a Enzyme. Metabolites 2020, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Li, M.; Zou, C.; Wang, K.; Zhang, W.; Huang, X.; Wang, Y. Walnut polyphenols and the active metabolite urolithin A improve oxidative damage in SH-SY5Y cells by up-regulating PKA/CREB/BDNF signaling. Food Funct. 2023, 14, 2698–2709. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, J.; Zhang, Y.; Li, V.; Kong, J.; He, J.; Li, X.M. Unpredictable chronic mild stress induces anxiety and depression-like behaviors and inactivates AMP-activated protein kinase in mice. Brain Res. 2014, 1576, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflammation 2019, 16, 62. [Google Scholar] [CrossRef]
- Toney, A.M.; Albusharif, M.; Works, D.; Polenz, L.; Schlange, S.; Chaidez, V.; Ramer-Tait, A.E.; Chung, S. Differential Effects of Whole Red Raspberry Polyphenols and Their Gut Metabolite Urolithin A on Neuroinflammation in BV-2 Microglia. Int. J. Environ. Res. Public Health 2020, 18, 68. [Google Scholar] [CrossRef]
- Lin, J.; Zhuge, J.; Zheng, X.; Wu, Y.; Zhang, Z.; Xu, T.; Meftah, Z.; Xu, H.; Wu, Y.; Tian, N.; et al. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radic. Biol. Med. 2020, 150, 109–119. [Google Scholar] [CrossRef]
- Lee, G.; Park, J.S.; Lee, E.J.; Ahn, J.H.; Kim, H.S. Anti-inflammatory and antioxidant mechanisms of urolithin B in activated microglia. Phytomedicine 2019, 55, 50–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Aisker, G.; Dong, H.; Halemahebai, G.; Zhang, Y.; Tian, L. Urolithin A suppresses glucolipotoxicity-induced ER stress and TXNIP/NLRP3/IL-1β inflammation signal in pancreatic β cells by regulating AMPK and autophagy. Phytomedicine 2021, 93, 153741. [Google Scholar] [CrossRef]
- Han, Q.A.; Su, D.; Shi, C.; Liu, P.; Wang, Y.; Zhu, B.; Xia, X. Urolithin A attenuated ox-LDL-induced cholesterol accumulation in macrophages partly through regulating miR-33a and ERK/AMPK/SREBP1 signaling pathways. Food Funct. 2020, 11, 3432–3440. [Google Scholar] [CrossRef]
- Rodriguez, J.; Pierre, N.; Naslain, D.; Bontemps, F.; Ferreira, D.; Priem, F.; Deldicque, L.; Francaux, M. Urolithin B, a newly identified regulator of skeletal muscle mass. J. Cachexia Sarcopenia Muscle 2017, 8, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Mazrooei, Z.; Dehkordi, H.T.; Shahraki, M.H.; Lorigooini, Z.; Zarean, E.; Amini-Khoei, H. Ellagic acid through attenuation of neuro-inflammatory response exerted antidepressant-like effects in socially isolated mice. Heliyon 2023, 9, e15550. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, W.; You, B.; Tang, W.; Gan, T.; Feng, C.; Li, C.; Yang, R. Serum Metabonomic Study on the Antidepressant-like Effects of Ellagic Acid in a Chronic Unpredictable Mild Stress-Induced Mouse Model. J. Agric. Food Chem. 2020, 68, 9546–9556. [Google Scholar] [CrossRef] [PubMed]
- Odaira, T.; Nakagawasai, O.; Takahashi, K.; Nemoto, W.; Sakuma, W.; Lin, J.R.; Tan-No, K. Mechanisms underpinning AMP-activated protein kinase-related effects on behavior and hippocampal neurogenesis in an animal model of depression. Neuropharmacology 2019, 150, 121–133. [Google Scholar] [CrossRef]
- Bedel, H.A.; Usta, C. Effect of ellagic acid on BDNF/PI3K/AKT-mediated signaling pathways in mouse models of depression. Iran J. Basic Med. Sci. 2025, 28, 493–497. [Google Scholar]
- Luo, J.; Tang, C.; Chen, X.; Ren, Z.; Qu, H.; Chen, R.; Tong, Z. Impacts of Aerobic Exercise on Depression-Like Behaviors in Chronic Unpredictable Mild Stress Mice and Related Factors in the AMPK/PGC-1α Pathway. Int. J. Environ. Res. Public Health 2020, 17, 2042. [Google Scholar] [CrossRef]
- Jin, H.; Xu, G.; Lu, Y.; Niu, C.; Zhang, X.; Kan, T.; Cao, J.; Yang, X.; Cheng, Q.; Zhang, J.; et al. Fluoxetine partially alleviates inflammation in the kidney of socially stressed male C57 BL/6 mice. FEBS Open Bio 2023, 13, 1723–1736. [Google Scholar] [CrossRef]
- Bedel, H.A.; Kencebay Manas, C.; Özbey, G.; Usta, C. The antidepressant-like activity of ellagic acid and its effect on hippocampal brain derived neurotrophic factor levels in mouse depression models. Nat. Prod. Res. 2018, 32, 2932–2935. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, M.; Tang, Q.; Lu, M.; An, X.; Cui, Y.; Zhao, M.; Qian, N.; Shao, J.; Shi, H.; et al. Juvenile chronic social defeat stress reduces prosocial behavior in adult male mice. Pharmacol. Biochem. Behav. 2025, 247, 173941. [Google Scholar] [CrossRef]
- Yao, C.; Jiang, N.; Sun, X.; Zhang, Y.; Pan, R.; He, Q.; Chang, Q.; Liu, X. Effects of inulin-type oligosaccharides (JSO) from Cichorium intybus L. on behavioral deficits induced by chronic restraint stress in mice and associated molecular alterations. Front. Pharmacol. 2024, 15, 1484337. [Google Scholar] [CrossRef]
- García-Villalba, R.; Tomás-Barberán, F.A.; Iglesias-Aguirre, C.E.; Giménez-Bastida, J.A.; González-Sarrías, A.; Selma, M.V.; Espín, J.C. Ellagitannins, urolithins, and neuroprotection: Human evidence and the possible link to the gut microbiota. Mol. Aspects Med. 2023, 89, 101109. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Lu, Q.; Wang, K.; Wang, Y. Urolithins: A Prospective Alternative against Brain Aging. Nutrients 2023, 15, 3884. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, G.; Stevanoska, M.; Beekmann, K.; Sturla, S.J. Physiologically-Based Pharmacokinetic Modeling of the Postbiotic Supplement Urolithin a Predicts its Bioavailability Is Orders of Magnitude Lower than Concentrations that Induce Toxicity, but also Neuroprotective Effects. Mol. Nutr. Food Res. 2023, 67, 2300009. [Google Scholar] [CrossRef] [PubMed]
- Piwowarski, J.P.; Granica, S.; Stefańska, J.; Kiss, A.K. Differences in Metabolism of Ellagitannins by Human Gut Microbiota ex Vivo Cultures. J. Nat. Prod. 2017, 80, 434–441. [Google Scholar] [CrossRef]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 743–758. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence |
|---|---|---|
| IL-6 | forward | 5′-ACATCGACCCGTCCACAGTAT-3′ |
| IL-6 | reverse | 5′-AGTGGTATAGACAGGTCTGTTGG-3′ |
| IL-1β | forward | 5′-GAAATGCCACCTTTTGACAGTG-3′ |
| IL-1β | reverse | 5′-TGGATGCTCTCATCAGGACAG-3′ |
| TNF-α | forward | 5′-CAGGCGGTGCCTATGTCTC-3′ |
| TNF-α | reverse | 5′-CGATCACCCCGAAGTTCAGTAG-3′ |
| GAPDH | forward | 5′-AGCCTCGTCCCGTAGACAAAA-3′ |
| GAPDH | reverse | 5′-TGGCAACAATCTCCACTTTGC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di, Y.; Xue, R.; Li, X.; Jin, Z.; Li, H.; Wu, L.; Zhang, Y.; An, L. Urolithin A Exhibits Antidepressant-like Effects by Modulating the AMPK/CREB/BDNF Pathway. Nutrients 2025, 17, 2294. https://doi.org/10.3390/nu17142294
Di Y, Xue R, Li X, Jin Z, Li H, Wu L, Zhang Y, An L. Urolithin A Exhibits Antidepressant-like Effects by Modulating the AMPK/CREB/BDNF Pathway. Nutrients. 2025; 17(14):2294. https://doi.org/10.3390/nu17142294
Chicago/Turabian StyleDi, Yaqian, Rui Xue, Xia Li, Zijia Jin, Hanying Li, Lanrui Wu, Youzhi Zhang, and Lei An. 2025. "Urolithin A Exhibits Antidepressant-like Effects by Modulating the AMPK/CREB/BDNF Pathway" Nutrients 17, no. 14: 2294. https://doi.org/10.3390/nu17142294
APA StyleDi, Y., Xue, R., Li, X., Jin, Z., Li, H., Wu, L., Zhang, Y., & An, L. (2025). Urolithin A Exhibits Antidepressant-like Effects by Modulating the AMPK/CREB/BDNF Pathway. Nutrients, 17(14), 2294. https://doi.org/10.3390/nu17142294