
The Therapeutic Potential of Butyrate and Lauric Acid in Modulating Glial and Neuronal Activity in Alzheimer’s Disease
 ,
,  ,
, 
Abstract
1. Introduction
2. Alzheimer’s Disease
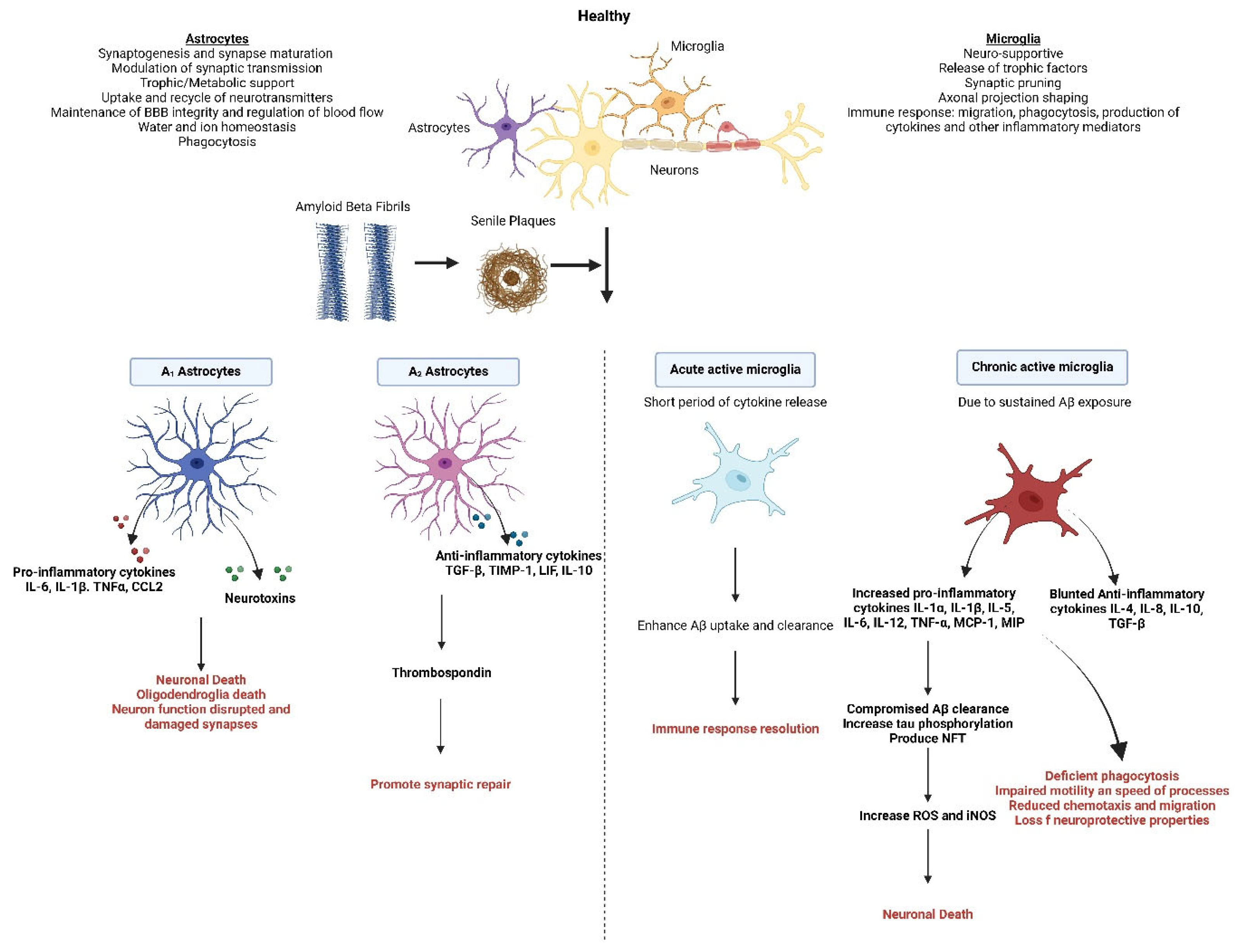
3. Role of Microglia in Alzheimer’s Disease
3.1. Microglia Response to Aβ, Tau, Oxidative Stress, and Neuronal Damage
3.2. Age-Related Changes in Microglia
4. Role of Astrocytes in Alzheimer’s Disease
4.1. Astrocytes’ Response to Aβ, Tau, and Neuronal Damage
4.2. Age Related Changes in Astrocytes
5. Neuronal Activity in Alzheimer’s Disease
Neurons’ Response to Aβ, Tau, and Oxidative Stress
6. Microbiota–Gut–Brain Axis Connection
7. Role of Gut Microbiota in Alzheimer’s Disease Pathology
8. Short Chain Fatty Acids
8.1. Butyrate
8.1.1. Origin, Structure, and Mechanism of Action
8.1.2. Effects on Microglia, Astrocytes, and Neurons
8.1.3. Therapeutic Potential, Challenges, and Limitations of Butyrate Delivery to the CNS

{kind=link}
{kind=link}
| Treatment | Model | Main Finding | Author | |
|---|---|---|---|---|
| Aggregation of AB | Levels of butyrate amongst 89 older people with cognitive performance from normal to impaired | Human | Butyrate was associated with decreased brain amyloid deposition. | [174] |
| Addition of NaB (2 mM) to cell culture. | Mouse neuroblastoma cells (N2a) | Supressed expression of APP and promoted effect of Neprilysin (NEP) | [163] | |
| Addition of butyrate to Aβ40 and Aβ42 monomers at 0:1, 1:1, and 4:1 SCFA: Aβ molar ratio. | In vitro | Butyrate inhibited the self-assembly of Aβ40 and Aβ42 monomers into Aβ fibrils. | [173] | |
| Pro-inflammatory mediators | Butyrate was added to cell cultures at 0.2, 2, and 20 mmol/L | Human monocytes | Inhibited IL-10 production in LPS stimulated human monocytes and MCP1 in both LPS and non-LPS stimulated human monocytes. | [191] |
| Butyrate was added to cell cultures at 0.2, 2, and 20 mmol/L | Human peripheral blood mononuclear cells (PBMC) | inhibited TNF-α and IFN-y secretion in human PBMC | [191] | |
| Oral treatment of 100 mM of sodium butyrate for 3 weeks. | Specific pathogen free (SPF) C57BL/6 mice | NaB promotes the expansion of Foxp3+ regulatory T cells | [192] | |
| Cell lines treated with Butyrate on its own and in combination with other SCFAs | Human THP-1 monocytic cell line | Reduced secretion of MCP-1, compared to cells treated with control and significantly reduced the secretion of IL-1β in human THP-1 monocytic cell line | [193] | |
| Mice were administered streptomycin (5 g/L) containing water and inoculated with 1 × 109 CFUs of Enterotoxigenic Escherichia coli (ETEC). Mice received sodium Butyrate (5 g/L) via water one day before streptomycin and throughout experiment. | Male GPCR109A+/+ and GPCR109A−/− mice | sodium butyrate reversed the increased expression of proinflammatory cytokines IL-1β, IL-6, and TNF-α in GPCR109A+/+ mice but showed no reversal effect in GPCR109A−/− mice | [194] | |
| Reactive Oxygen Species | Addition of NaB (2 mM) to cell culture. | Mouse neuroblastoma cells (N2a) | NaB Inhibited the production of Aβ induced ROS | [163] |
| Histone acetylation | Cell lines were incubated in presence of 10 mmol/L sodium butyrate | Human breast cancer MCF—7 cell line | Sodium butyrate inhibits histone deacetylase | [195] |
| Nab was dissolved in 0.01 M phosphate buffer saline (PSB) and administered daily at a final concentration of 1.2 g/kg of body weight. | APPPS1—21 double transgenic mice that co-express KM670/671NL mutated amyloid protein precursor (ABPP) and L166P mutated presenilin 1 (PS1) | Enhanced associative memory and elevated hippocampal acetylation at H3K14, H4K5, and H4K12 sites. | [166] | |
| Nab was added to chow pellets and was administered at either 5 mh/kg/day, or 15 mg/kg/day for 12 weeks. | Male transgenic mice expressing 5 familial AD mutations [APP: K67ON/M671 L (Swedish) + 1716 V (Florida) + V7171 (London) and PS1: M146 L + L286V], crossed with APOE ε3 | Improved associative memory and cognitive functioning. A 40% decrease in brain Aβ levels. | [23] | |
| Endothelial and colonic epithelial integrity | Nab was administered intraperitoneally at 200 mg/kg body weight. | Male C57BL/6 mice | Increased expression of tight junction (TJ) proteins occluding and ZO-1. | [23] |
| Mice were administered streptomycin (5 g/L) containing water and inoculated with 1 × 109 CFUs of Enterotoxigenic Escherichia coli (ETEC). Mice received sodium Butyrate (5 g/L) via water one day before streptomycin and throughout experiment. | Male GPCR109A+/+ and GPCR109A−/− mice | Sodium butyrate reversed the decreased expression of TJ proteins Cldn1, Cldn2, Cldn3, Ocln, and Zo-1 caused by ETEC in GPCR109A+/+ mice, but not in GPCR109A−/− mice. | [194] |
9. Medium Chain Fatty Acids
9.1. Lauric Acid
9.1.1. Origin, Structure, and Mechanism of Action
9.1.2. Effects on Microglia, Astrocytes, and Neurons
9.1.3. Therapeutic Potential, Challenges, and Limitations of Lauric Acid Delivery to the CNS
| Treatment | Model | Main Findings | Authors |
|---|---|---|---|
| Medium chain triglycerides (including Lauric acid) | Healthy adults | MCTs, including Lauric acid, improve cognitive function in healthy adults, suggesting benefits for brain health and potential support against cognitive decline. | [224] |
| Ketone bodies (including those from Lauric acid) | Transgenic Mouse | Ketone bodies from Lauric acid may contribute to neuroprotection and cognitive enhancement, influenced by dietary fats. | [225] |
| Ketogenic diet (including Lauric acid) | Mild cognitive impairment | Ketogenic diets, enriched with Lauric acid, improve memory in mild cognitive impairment, potentially impacting Alzheimer’s disease progression. | [226] |
| Ketogenic diet (including medium-chain triglycerides like Lauric acid) | Mild to moderate Alzheimer’s disease | Ketogenic diets, including Lauric acid, modestly improve cognitive function in Alzheimer’s patients, suggesting therapeutic potential. | [227] |
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A1 | Pro-inflammatory reactive astrocyte phenotype |
| A2 | Anti-inflammatory reactive astrocyte phenotype |
| Aβ | Amyloid-β |
| ACTH | Adrenocorticotropic Hormone |
| AD | Alzheimer’s Disease |
| ANS | Autonomic Nervous System |
| APP | Amyloid Precursor Protein |
| BBB | Blood–Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| cAMP | Cyclic Adenosine Monophosphate |
| CNS | Central Nervous System |
| COX-2 | Cyclooxygenase-2 |
| CRF | Corticotropin-Releasing Factor |
| CX3CL1 | Chemokine (C-X3-C motif) ligand 1 |
| CX3CR1 | Chemokine (C-X3-C motif) receptor 1 |
| DAMPs | Danger-Associated Molecular Patterns |
| ENS | Enteric Nervous System |
| FDA | Food and Drug Administration |
| FMT | Fecal Microbiota Transplantation |
| GABA | Gamma-Aminobutyric Acid |
| GDNF | Glial Cell Line-Derived Neurotrophic Factor |
| GPCR | G Protein-Coupled Receptor |
| HDAC | Histone Deacetylase |
| HPA | Hypothalamus-Pituitary-Adrenal (Axis) |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| IP3 | Inositol Triphosphate |
| LDH | Lactate Dehydrogenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinase |
| MCFA | Medium-Chain Fatty Acid |
| MCI | Mild Cognitive Impairment |
| MCT | Monocarboxylate Transporter |
| MHC | Major Histocompatibility Complex |
| MIP | Macrophage Inflammatory Protein |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NaB | Sodium Butyrate |
| NF-κB | Nuclear Factor Kappa-light-chain-enhancer of Activated B cells |
| NGF | Nerve Growth Factor |
| NFT | Neurofibrillary Tangles |
| NO | Nitric Oxide |
| NOS | Nitric Oxide Synthase |
| NVU | Neurovascular Unit |
| PET | Positron Emission Tomography |
| PGC-1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha |
| PRRs | Pattern Recognition Receptors |
| PSEN1/2 | Presenilin 1 and 2 |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| SCFA | Short-Chain Fatty Acid |
| TGF-β | Transforming Growth Factor Beta |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor Alpha |
| ZO-1 | Zonula Occludens-1 |
References
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Yaari, R.; Fleisher, A.S.; Tariot, P.N. Updates to Diagnostic Guidelines for Alzheimers Disease. Prim. Care Companion CNS Disord. 2011, 13, 01262. [Google Scholar]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Valles, S.L.; Singh, S.K.; Campos-Campos, J.; Colmena, C.; Campo-Palacio, I.; Alvarez-Gamez, K.; Caballero, O.; Jorda, A. Functions of Astrocytes under Normal Conditions and after a Brain Disease. Int. J. Mol. Sci. 2023, 24, 8434. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Wong-Guerra, M.; Calfio, C.; Maccioni, R.B.; Rojo, L.E. Revisiting the neuroinflammation hypothesis in Alzheimer’s disease: A focus on the druggability of current targets. Front. Pharmacol. 2023, 14, 1161850. [Google Scholar] [CrossRef]
- Edison, P. Astroglial activation: Current concepts and future directions. Alzheimer’s Dement. 2024, 20, 3034–3053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Geng, R.; Tu, Q. Gut microbial involvement in Alzheimer’s disease pathogenesis. Aging 2021, 13, 13359–13371. [Google Scholar] [CrossRef]
- Terzioglu, G.; Young-Pearse, T.L. Microglial function, INPP5D/SHIP1 signaling, and NLRP3 inflammasome activation: Implications for Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 89. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Lecanemab Approved for Treatment of Early Alzheimer’s Disease. 2024. Available online: https://www.alz.org/alzheimers-dementia/treatments/lecanemab-leqembi (accessed on 12 February 2024).
- Liu, J.; Li, T.; Zhong, G.; Pan, Y.; Gao, M.; Su, S.; Liang, Y.; Ma, C.; Liu, Y.; Wang, Q.; et al. Exploring the therapeutic potential of natural compounds for Alzheimer’s disease: Mechanisms of action and pharmacological properties. Biomed. Pharmacother. 2023, 166, 115406. [Google Scholar] [CrossRef]
- Shao, S.; Ye, X.; Su, W.; Wang, Y. Curcumin alleviates Alzheimer’s disease by inhibiting inflammatory response, oxidative stress and activating the AMPK pathway. J. Chem. Neuroanat. 2023, 134, 102363. [Google Scholar] [CrossRef]
- Caruso, G.; Godos, J.; Privitera, A.; Lanza, G.; Castellano, S.; Chillemi, A.; Bruni, O.; Ferri, R.; Caraci, F.; Grosso, G. Phenolic Acids and Prevention of Cognitive Decline: Polyphenols with a Neuroprotective Role in Cognitive Disorders and Alzheimer’s Disease. Nutrients 2022, 14, 819. [Google Scholar] [CrossRef]
- Gustafson, D.R.; Bäckman, K.; Scarmeas, N.; Stern, Y.; Manly, J.J.; Mayeux, R.; Gu, Y. Dietary fatty acids and risk of Alzheimer’s disease and related dementias: Observations from the Washington Heights-Hamilton Heights-Inwood Columbia Aging Project (WHICAP). Alzheimer’s Dement. 2020, 16, 1638–1649. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fernando, W.M.A.D.B.; Martins, I.J.; Morici, M.; Bharadwaj, P.; Rainey-Smith, S.R.; Lim, W.L.F.; Martins, R.N. Sodium Butyrate Reduces Brain Amyloid-β Levels and Improves Cognitive Memory Performance in an Alzheimer’s Disease Transgenic Mouse Model at an Early Disease Stage. J. Alzheimer’s Dis. 2020, 74, 91–99. [Google Scholar] [CrossRef]
- Fernando, W.M.A.D.B.; Rainey-Smith, S.R.; Martins, I.J.; Martins, R.N. In vitro study to assess the potential of short chain fatty acids (scfa) as therapeutic agents for alzheimer’s disease. Alzheimer’s Dement. 2014, 10, P626. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Mori, T.; Goto, K.; Kawahara, I.; Fujiwara-Tani, R.; Kishi, S.; Sasaki, T.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Intake of medium-chain fatty acids induces myocardial oxidative stress and atrophy. Lipids Health Dis. 2018, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Fernando, M.; Eslick, S.; Asih, P.R.; Shadfar, S.; Bandara, E.M.S.; Hillebrandt, H.; Meghwar, S.; Shahriari, M.; Chatterjee, P.; et al. Ketone bodies mediate alterations in brain energy metabolism and biomarkers of Alzheimer’s disease. Front. Neurosci. 2023, 17, 1297984. [Google Scholar] [CrossRef] [PubMed]
- Khatua, P.; Jana, A.K.; Hansmann, U.H.E. Effect of Lauric Acid on the Stability of Aβ42 Oligomers. ACS Omega 2021, 6, 5795–5804. [Google Scholar] [CrossRef]
- Jabir, N.R.; Khan, F.R.; Tabrez, S. Cholinesterase targeting by polyphenols: A therapeutic approach for the treatment of Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 753–762. [Google Scholar] [CrossRef]
- Dementia Australia. Alzheimer’s Disease. 2020. Available online: https://www.dementia.org.au/about-dementia/types-of-dementia/alzheimers-disease (accessed on 15 December 2024).
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-onset Alzheimer Disease and Its Variants. Contin. Lifelong Learn. Neurol. 2019, 25, 34–51. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef]
- Davis, M.; O`Connell, T.; Johnson, S.; Cline, S.; Merikle, E.; Martenyi, F.; Simpson, K. Estimating Alzheimer’s Disease Progression Rates from Normal Cognition Through Mild Cognitive Impairment and Stages of Dementia. Curr. Alzheimer Res. 2018, 15, 777–788. [Google Scholar] [CrossRef]
- Albert, M.S.; Dekosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso, A.d.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Et Biophys. Acta-Mol. Basis Dis. 2005, 1739, 198. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, J.; Xu, Z.; Yan, H.; Tang, B.; Liu, C.; Chen, C.; Meng, Q. Microglia-containing human brain organoids for the study of brain development and pathology. Mol. Psychiatry 2023, 28, 96–107. [Google Scholar] [CrossRef]
- Zhou, M.; Cornell, J.; Salinas, S.; Huang, H.-Y. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705. [Google Scholar] [CrossRef]
- Fakhoury, M. Immune-mediated processes in neurodegeneration: Where do we stand? J. Neurol. 2016, 263, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; De Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Regulation of Inducible Nitric Oxide Synthase Gene in Glial Cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef]
- Sierra, A.; Navascués, J.; Cuadros, M.A.; Calvente, R.; Martín-Oliva, D.; Ferrer-Martín, R.M.; Martín-Estebané, M.; Carrasco, M.-C.; Marín-Teva, J.L. Expression of Inducible Nitric Oxide Synthase (iNOS) in Microglia of the Developing Quail Retina. PLoS ONE 2014, 9, e106048. [Google Scholar] [CrossRef]
- Park, J.; Lee, C.; Kim, Y.T. Effects of Natural Product-Derived Compounds on Inflammatory Pain via Regulation of Microglial Activation. Pharmaceuticals 2023, 16, 941. [Google Scholar] [CrossRef]
- Bartels, A.; Leenders, K. Cyclooxygenase and Neuroinflammation in Parkinsons Disease Neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62–68. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int. J. Mol. Sci. 2023, 24, 6321. [Google Scholar] [CrossRef] [PubMed]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef] [PubMed]
- Novoa, C.; Salazar, P.; Cisternas, P.; Gherardelli, C.; Vera-Salazar, R.; Zolezzi, J.M.; Inestrosa, N.C. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol. Res. 2022, 55, 39. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Wang, H.; Yang, K.; Luan, J.; Wang, S. TREM2: Potential therapeutic targeting of microglia for Alzheimer’s disease. Biomed. Pharmacother. 2023, 165, 115218. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.-N.H.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, C. Microglia activation linking amyloid-β drive tau spatial propagation in Alzheimer’s disease. Front. Neurosci. 2022, 16, 951128. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of Microglial Cells in Alzheimer’s Disease Tau Propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Laurent, C.; Dorothée, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Légeron, F.-P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of Inflammatory Mediators and Microglial Activation in Mice Transgenic for Mutant Human P301S Tau Protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Lindsay, C.B.; Zolezzi, J.M.; Rivera, D.S.; Cisternas, P.; Bozinovic, F.; Inestrosa, N.C. Andrographolide Reduces Neuroinflammation and Oxidative Stress in Aged Octodon degus. Mol. Neurobiol. 2020, 57, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Kovac, A.; Zilka, N.; Kazmerova, Z.; Cente, M.; Zilkova, M.; Novak, M. Misfolded Truncated Protein τ Induces Innate Immune Response via MAPK Pathway. J. Immunol. 2011, 187, 2732–2739. [Google Scholar] [CrossRef]
- Jiang, S.; Maphis, N.M.; Binder, J.; Chisholm, D.; Weston, L.; Duran, W.; Peterson, C.; Zimmerman, A.; Mandell, M.A.; Jett, S.D.; et al. Proteopathic tau primes and activates interleukin-1β via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep. 2021, 36, 109720. [Google Scholar] [CrossRef]
- Antignano, I.; Liu, Y.; Offermann, N.; Capasso, M. Aging microglia. Cell. Mol. Life Sci. 2023, 80, 126. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef]
- Greenwood, E.K.; Brown, D.R. Senescent Microglia: The Key to the Ageing Brain? Int. J. Mol. Sci. 2021, 22, 4402. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Hernández, N.P.; Peña-Ortega, F. Fractalkine/CX3CR1-Dependent Modulation of Synaptic and Network Plasticity in Health and Disease. Neural Plast. 2023, 2023, 4637073. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef] [PubMed]
- Man, J.H.; Breur, M.; van Gelder, C.A.; Marcon, G.; Maderna, E.; Giaccone, G.; Altelaar, M.; van der Knaap, M.S.; Bugiani, M. Region-specific and age-related differences in astrocytes in the human brain. Neurobiol. Aging 2024, 140, 102–115. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.W.; Kim, K.-T. Region-Specific Characteristics of Astrocytes and Microglia: A Possible Involvement in Aging and Diseases. Cells 2022, 11, 1902. [Google Scholar] [CrossRef]
- Yan, Y.Q.; Ma, C.G.; Ding, Z.B.; Song, L.J.; Wang, Q.; Kumar, G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1702–1710. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative Regulators of Neuroinflammation in Stroke and Other Neurological Diseases. Neurotherapeutics 2016, 13, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.L.; Ousman, S.S. Astrocytes and Aging. Front. Aging Neurosci. 2018, 10, 337. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.-I.; Kim, S.U. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Escalada, P.; Ezkurdia, A.; Ramírez, M.J.; Solas, M. Essential Role of Astrocytes in Learning and Memory. Int. J. Mol. Sci. 2024, 25, 1899. [Google Scholar] [CrossRef]
- Liang, Y.; Raven, F.; Ward, J.F.; Zhen, S.; Zhang, S.; Sun, H.; Miller, S.J.; Choi, S.H.; Tanzi, R.E.; Zhang, C. Upregulation of Alzheimer’s Disease Amyloid-β Protein Precursor in Astrocytes Both in vitro and in vivo. J. Alzheimer’s Dis. 2020, 76, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Eisel, U.L.M. Microglia-Astrocyte Communication in Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 95, 785–803. [Google Scholar] [CrossRef] [PubMed]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Cuellar-Santoyo, A.O.; Ruiz-Rodríguez, V.M.; Mares-Barbosa, T.B.; Patrón-Soberano, A.; Howe, A.G.; Portales-Pérez, D.P.; Miquelajáuregui Graf, A.; Estrada-Sánchez, A.M. Revealing the contribution of astrocytes to glutamatergic neuronal transmission. Front. Cell. Neurosci. 2023, 16, 1037641. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, X.; Zhang, Y.; Zheng, X.; Cepeda, C.; Wang, Y.; Duan, S.; Tong, X. Interactions of glial cells with neuronal synapses, from astrocytes to microglia and oligodendrocyte lineage cells. Glia 2023, 71, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, M.; Miyamoto, Y.; Ikeshima-Kataoka, H. Astrocytic Neuroimmunological Roles Interacting with Microglial Cells in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 1599. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-β Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef]
- Stanca, S.; Rossetti, M.; Bongioanni, P. Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways. Int. J. Mol. Sci. 2023, 24, 13880. [Google Scholar] [CrossRef]
- Beltran-Lobo, P.; Matthew; Jimenez-Sanchez, M.; Verkhratsky, A.; Beatriz; Noble, W. Astrocyte adaptation in Alzheimer’s disease: A focus on astrocytic P2X7R. Essays Biochem. 2023, 67, 119–130. [Google Scholar] [CrossRef]
- Zattoni, M.; Mearelli, M.; Vanni, S.; Colini Baldeschi, A.; Tran, T.H.; Ferracin, C.; Catania, M.; Moda, F.; Di Fede, G.; Giaccone, G.; et al. Serpin Signatures in Prion and Alzheimer’s Diseases. Mol. Neurobiol. 2022, 59, 3778–3799. [Google Scholar] [CrossRef]
- Bai, X.; Mai, M.; Yao, K.; Zhang, M.; Huang, Y.; Zhang, W.; Guo, X.; Xu, Y.; Zhang, Y.; Qurban, A.; et al. The role of DHCR24 in the pathogenesis of AD: Re-cognition of the relationship between cholesterol and AD pathogenesis. Acta Neuropathol. Commun. 2022, 10, 35. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]
- Cotto, B.; Natarajaseenivasan, K.; Langford, D. Astrocyte activation and altered metabolism in normal aging, age-related CNS diseases, and HAND. J. Neurovirol. 2019, 25, 722–733. [Google Scholar] [CrossRef]
- Parachikova, A.; Agadjanyan, M.G.; Cribbs, D.H.; Blurton-Jones, M.; Perreau, V.; Rogers, J.; Beach, T.G.; Cotman, C.W. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol. Aging 2007, 28, 1821–1833. [Google Scholar] [CrossRef]
- Zhao, Y.F.; Wei, D.N.; Tang, Y. Gut Microbiota Regulate Astrocytic Functions in the Brain: Possible Therapeutic Consequences. Curr. Neuropharmacol. 2021, 19, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Zißler, J.; Rothhammer, V.; Linnerbauer, M. Gut–Brain Interactions and Their Impact on Astrocytes in the Context of Multiple Sclerosis and Beyond. Cells 2024, 13, 497. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.K.; Stice, S.L. Neural differentiation of human embryonic stem cells. J. Cell. Biochem. 2008, 105, 633–640. [Google Scholar] [CrossRef]
- DiCicco-Bloom, E. Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Kiraly, M.; Foss, J.F.; Giordano, T. Neuroinflammation, Its Role in Alzheimer’s Disease and Therapeutic Strategies. J. Prev. Alzheimer’s Dis. 2023, 10, 686–698. [Google Scholar] [CrossRef]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Nasb, M.; Tao, W.; Chen, N. Alzheimer’s Disease Puzzle: Delving into Pathogenesis Hypotheses. Aging Dis. 2024, 15, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-Mcfarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Miller, D.B.; O’Callaghan, J.P. Neuroendocrine aspects of the response to stress. Metabolism 2002, 51 (Suppl. S1), 5–10. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the BrainGut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef]
- Young, V.B.; Schmidt, T.M. Overview of the Gastrointestinal Microbiota; Springer: New York, NY, USA, 2008; pp. 29–40. [Google Scholar]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Appleton Jeremy, J. The Gut-Brain Axis: Influence of Microbiota on Mood and Mental Health. Integr. Med. A Clin. J. 2018, 17, 28–32. [Google Scholar]
- Dash, S.; Syed, Y.A.; Khan, M.R. Understanding the Role of the Gut Microbiome in Brain Development and Its Association with Neurodevelopmental Psychiatric Disorders. Front. Cell Dev. Biol. 2022, 10, 880544. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A.; Pettigrew, M.M.; Gilbert, J.; Faith, J. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. mBio 2019, 10, e00632-19. [Google Scholar] [CrossRef]
- Inserra, A.; Rogers, G.B.; Licinio, J.; Wong, M.-L. The Microbiota-Inflammasome Hypothesis of Major Depression. BioEssays 2018, 40, 1800027. [Google Scholar] [CrossRef]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimers Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Askarova, S.; Umbayev, B.; Masoud, A.R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The Links Between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell. Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef]
- Kandpal, M.; Indari, O.; Baral, B.; Jakhmola, S.; Tiwari, D.; Bhandari, V.; Pandey, R.K.; Bala, K.; Sonawane, A.; Jha, H.C. Dysbiosis of Gut Microbiota from the Perspective of the Gut–Brain Axis: Role in the Provocation of Neurological Disorders. Metabolites 2022, 12, 1064. [Google Scholar] [CrossRef]
- O’Riordan, K.J.; Collins, M.K.; Moloney, G.M.; Knox, E.G.; Aburto, M.R.; Fülling, C.; Morley, S.J.; Clarke, G.; Schellekens, H.; Cryan, J.F. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 2022, 546, 111572. [Google Scholar] [CrossRef]
- Popescu, C.; Munteanu, C.; Anghelescu, A.; Ciobanu, V.; Spînu, A.; Andone, I.; Mandu, M.; Bistriceanu, R.; Băilă, M.; Postoiu, R.-L.; et al. Novelties on Neuroinflammation in Alzheimer’s Disease–Focus on Gut and Oral Microbiota Involvement. Int. J. Mol. Sci. 2024, 25, 11272. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Romanenko, M.; Kholin, V.; Koliada, A.; Vaiserman, A. Nutrition, Gut Microbiota, and Alzheimer’s Disease. Front. Psychiatry 2021, 12, 712673. [Google Scholar] [CrossRef]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernández, E.N.; Putz, P.; Rowland, I.; Swann, J.; Türk, J.; Vermeiren, J.; et al. Systematic review of the effects of the intestinal microbiota on selected nutrients and non-nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef]
- Chen, H.; Meng, L.; Shen, L. Multiple roles of short-chain fatty acids in Alzheimer disease. Nutrition 2022, 93, 111499. [Google Scholar] [CrossRef] [PubMed]
- Bedford, A.; Gong, J. Implications of butyrate and its derivatives for gut health and animal production. Anim. Nutr. 2018, 4, 151. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef]
- Miao, Z.; Hao, H.; Yan, R.; Wang, X.; Wang, B.; Sun, J.; Li, Z.; Zhang, Y.; Sun, B. Individualization of Chinese alcoholic beverages: Feasibility towards a regulation of organic acids. LWT 2022, 172, 114168. [Google Scholar] [CrossRef]
- Schnekenburger, M. Nutritional Epigenetic Regulators in the Field of Cancer. In Epigenetic Cancer Therapy; Academic Press: Cambridge, MA, USA, 2015; p. 393. [Google Scholar]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Segain, J.P. Butyrate inhibits inflammatory responses through NFkappa B inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Säemann, M.D.; Böhmig, G.A.; Österreicher, C.H.; Burtscher, H.; Parolini, O.; Diakos, C.; Stöckl, J.; Hörl, W.H.; Zlabinger, G.J. Anti-inflammatory effects of sodium butyrate on human monocytes: Potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 2000, 14, 2380–2382. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, E.J.; Lee, J.C.; Kim, W.K.; Kim, H.S. Anti-inflammatory effects of short chain fatty acids in IFN-g-stimulated RAW 264.7 murine macrophage cells: Involvement of NF-B and ERK signaling pathways. Int. Immunopharmacol. 2007, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, J.; Liu, Y.; Xiao, N.; Suo, H.; Xie, K.; Yang, C.; Wu, C. Short-Chain Fatty Acids Suppress Lipopolysaccharide-Induced Production of Nitric Oxide and Proinflammatory Cytokines Through Inhibition of NF-κB Pathway in RAW264.7 Cells. Inflammation 2012, 35, 1676–1684. [Google Scholar] [CrossRef]
- Lachmandas, E.; Van Den Heuvel, C.N.A.M.; Damen, M.S.M.A.; Cleophas, M.C.P.; Netea, M.G.; Van Crevel, R. Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short-Chain Fatty Acids. J. Diabetes Res. 2016, 2016, 6014631. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Sina, C.; Gavrilova, O.; Förster, M.; Till, A.; Derer, S.; Hildebrand, F.; Raabe, B.; Chalaris, A.; Scheller, J.; Rehmann, A.; et al. G Protein-Coupled Receptor 43 Is Essential for Neutrophil Recruitment during Intestinal Inflammation. J. Immunol. 2009, 183, 7514–7522. [Google Scholar] [CrossRef]
- Fu, S.-P.; Wang, J.-F.; Xue, W.-J.; Liu, H.-M.; Liu, B.-R.; Zeng, Y.-L.; Li, S.-N.; Huang, B.-X.; Lv, Q.-K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.-N.; Vitetta, L. Effects of Intestinal Microbial–Elaborated Butyrate on Oncogenic Signaling Pathways. Nutrients 2019, 11, 1026. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Lulla, A.; Debroy, K.; Shikany, J.M.; Yaffe, K.; Meirelles, O.; Launer, L.J. Association of the Gut Microbiota with Cognitive Function in Midlife. JAMA Netw. Open 2022, 5, e2143941. [Google Scholar] [CrossRef]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated with Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Yang, B.; Chen, K.; Kong, Y.; Fang, N.; Gong, T.; Wang, F.; Ling, Z.; Liu, J. Effect of Clostridium butyricum against Microglia-Mediated Neuroinflammation in Alzheimer’s Disease via Regulating Gut Microbiota and Metabolites Butyrate. Mol. Nutr. Food Res. 2020, 64, 1900636. [Google Scholar] [CrossRef]
- Wei, H.; Yu, C.; Zhang, C.; Ren, Y.; Guo, L.; Wang, T.; Chen, F.; Li, Y.; Zhang, X.; Wang, H.; et al. Butyrate ameliorates chronic alcoholic central nervous damage by suppressing microglia-mediated neuroinflammation and modulating the microbiome-gut-brain axis. Biomed. Pharmacother. 2023, 160, 114308. [Google Scholar] [CrossRef]
- Go, J.; Chang, D.-H.; Ryu, Y.-K.; Park, H.-Y.; Lee, I.-B.; Noh, J.-R.; Hwang, D.Y.; Kim, B.-C.; Kim, K.-S.; Lee, C.-H. Human gut microbiota Agathobaculum butyriciproducens improves cognitive impairment in LPS-induced and APP/PS1 mouse models of Alzheimer’s disease. Nutr. Res. 2021, 86, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Saw, G.; Krishna, K.; Gupta, N.; Soong, T.W.; Mallilankaraman, K.; Sajikumar, S.; Dheen, S.T. Epigenetic regulation of microglial phosphatidylinositol 3-kinase pathway involved in long-term potentiation and synaptic plasticity in rats. Glia 2020, 68, 656–669. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, D.; Weng, F.; Jin, Y.; He, L. Sodium butyrate ameliorates the cognitive impairment of Alzheimer’s disease by regulating the metabolism of astrocytes. Psychopharmacology 2022, 239, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Liu, M.; Gong, Y.; Wang, Z.; Jin, X.; Xie, G.; Zhu, M.; Zhang, X.; Luo, S.; Qu, Q.; et al. Sodium butyrate exerts antioxidant stress effects and attenuates Aβ25-35-induced cytotoxicity in PC12 cells. Arch. Biochem. Biophys. 2022, 731, 109448. [Google Scholar] [CrossRef]
- Williams, H.C.; Farmer, B.C.; Piron, M.A.; Walsh, A.E.; Bruntz, R.C.; Gentry, M.S.; Sun, R.C.; Johnson, L.A. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol. Dis. 2020, 136, 104742. [Google Scholar] [CrossRef]
- Sun, J.; Yuan, B.; Wu, Y.; Gong, Y.; Guo, W.; Fu, S.; Luan, Y.; Wang, W. Sodium Butyrate Protects N2a Cells against Aβ Toxicity In Vitro. Mediat. Inflamm. 2020, 2020, 7605160. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Jia, Y.; Yang, S.; Zhao, N.; Hu, Y.; Hong, J.; Gao, S.; Zhao, R. Sodium butyrate protects against high-fat diet-induced oxidative stress in rat liver by promoting expression of nuclear factor E2-related factor 2. Br. J. Nutr. 2019, 122, 400–410. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium Butyrate Improves Memory Function in an Alzheimer’s Disease Mouse Model When Administered at an Advanced Stage of Disease Progression. J. Alzheimer’s Dis. 2011, 26, 187–197. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.J.; Son, Y.; Lee, M.; Moon, C.; Kim, S.H.; Shin, I.S.; Yang, M.; Bae, S. Sodium butyrate prevents radiation-induced cognitive impairment by restoring pCREB/BDNF expression. Neural Regen. Res. 2019, 14, 1530. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of Novel Therapeutics Targeting the Blood–Brain Barrier: From Barrier to Carrier. Adv. Sci. 2021, 8, 2101090. [Google Scholar] [CrossRef]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 695479. [Google Scholar] [CrossRef]
- Bauer, K.C.; Rees, T.; Finlay, B.B. The Gut Microbiota–Brain Axis Expands Neurologic Function: A Nervous Rapport. BioEssays 2019, 41, 1800268. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Recharla, N.; Geesala, R.; Shi, X.-Z. Gut Microbial Metabolite Butyrate and Its Therapeutic Role in Inflammatory Bowel Disease: A Literature Review. Nutrients 2023, 15, 2275. [Google Scholar] [CrossRef]
- Mirzaei, R.; Bouzari, B.; Hosseini-Fard, S.R.; Mazaheri, M.; Ahmadyousefi, Y.; Abdi, M.; Jalalifar, S.; Karimitabar, Z.; Teimoori, A.; Keyvani, H.; et al. Role of microbiota-derived short-chain fatty acids in nervous system disorders. Biomed. Pharmacother. 2021, 139, 111661. [Google Scholar] [CrossRef] [PubMed]
- Giardina, C.; Boulares, H.; Inan, M.S. NSAIDs and butyrate sensitize a human colorectal cancer cell line to TNF-α and Fas ligation: The role of reactive oxygen species. Biochim. Et Biophys. Acta-Mol. Cell Res. 1999, 1448, 425–438. [Google Scholar] [CrossRef]
- Kabir, M.T.; Rahman, H.; Shah, M.; Jamiruddin, M.R.; Basak, D.; Al-Harrasi, A.; Bhatia, S.; Ashraf, G.M.; Najda, A.; El-Kott, A.F.; et al. Therapeutic promise of carotenoids as antioxidants and anti-inflammatory agents in neurodegenerative disorders. Biomed. Pharmacother. 2022, 146, 112610. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-B.; Zhang, Y.-C.; Huang, H.-H.; Lin, J. Prospects for clinical applications of butyrate-producing bacteria. World J. Clin. Pediatr. 2021, 10, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef]
- Geng, Z.-H.; Zhu, Y.; Li, Q.-L.; Zhao, C.; Zhou, P.-H. Enteric Nervous System: The Bridge Between the Gut Microbiota and Neurological Disorders. Front. Aging Neurosci. 2022, 14, 810483. [Google Scholar] [CrossRef]
- Wouw, M.V.D.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: Modulator of Host Metabolism and Appetite. J. Nutr. 2017, 147, 727–745. [Google Scholar] [CrossRef]
- Gagliano, H.; Delgado-Morales, R.; Sanz-Garcia, A.; Armario, A. High doses of the histone deacetylase inhibitor sodium butyrate trigger a stress-like response. Neuropharmacology 2014, 79, 75–82. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef]
- Fu, J.; Zheng, Y.; Gao, Y.; Xu, W. Dietary Fiber Intake and Gut Microbiota in Human Health. Microorganisms 2022, 10, 2507. [Google Scholar] [CrossRef]
- Zhu, X.; Li, B.; Lou, P.; Dai, T.; Chen, Y.; Zhuge, A.; Yuan, Y.; Li, L. The Relationship Between the Gut Microbiome and Neurodegenerative Diseases. Neurosci. Bull. 2021, 37, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Homer, B.; Judd, J.; Mohammadi Dehcheshmeh, M.; Ebrahimie, E.; Trott, D.J. Gut Microbiota and Behavioural Issues in Production, Performance, and Companion Animals: A Systematic Review. Animals 2023, 13, 1458. [Google Scholar] [CrossRef] [PubMed]
- Menafra, D.; Proganò, M.; Tecce, N.; Pivonello, R.; Colao, A. Diet and gut microbiome: Impact of each factor and mutual interactions on prevention and treatment of type 1, type 2, and gestational diabetes mellitus. Hum. Nutr. Metab. 2024, 38, 200286. [Google Scholar] [CrossRef]
- Qu, L.; Li, Y.; Liu, F.; Fang, Y.; He, J.; Ma, J.; Xu, T.; Wang, L.; Lei, P.; Dong, H.; et al. Microbiota-Gut-Brain Axis Dysregulation in Alzheimer’s Disease: Multi-Pathway Effects and Therapeutic Potential. Aging Dis. 2023, 15, 1108–1131. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, G.; Guarnaccia, A.; Fancello, G.; Agrillo, C.; Iannarelli, F.; Sanguinetti, M.; Masucci, L. Fecal Microbiota Transplantation and Other Gut Microbiota Manipulation Strategies. Microorganisms 2022, 10, 2424. [Google Scholar] [CrossRef]
- Cox, M.A.; Jackson, J.; Stanton, M.; Rojas-Triana, A.; Bober, L.; Laverty, M.; Yang, X.; Zhu, F.; Liu, J.; Wang, S.; et al. Short-chain fatty acids act as antiinflammatory mediatorsby regulating prostaglandin E2 and cytokines. World J. Gastroenterol. 2009, 15, 5549. [Google Scholar] [CrossRef]
- Kespohl, M.; Vachharajani, N.; Luu, M.; Harb, H.; Pautz, S.; Wolff, S.; Sillner, N.; Walker, A.; Schmitt-Kopplin, P.; Boettger, T.; et al. The Microbial Metabolite Butyrate Induces Expression of Th1-Associated Factors in CD4+ T Cells. Front. Immunol. 2017, 8, 1036. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Gates, E.J.; Ranger, A.L.; Klegeris, A. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol. Cell. Neurosci. 2020, 105, 103493. [Google Scholar] [CrossRef]
- Gong, Y.; Jin, X.; Yuan, B.; Lv, Y.; Yan, G.; Liu, M.; Xie, C.; Liu, J.; Tang, Y.; Gao, H.; et al. G Protein-Coupled Receptor 109A Maintains the Intestinal Integrity and Protects Against ETEC Mucosal Infection by Promoting IgA Secretion. Front. Immunol. 2021, 11, 583652. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of Histone Deacetylase Activity by Butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Jadhav, H.B.; Annapure, U.S. Triglycerides of medium-chain fatty acids: A concise review. J. Food Sci. Technol. 2023, 60, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Zweemer, A.J.M.; Toraskar, J.; Heitman, L.H.; Ijzerman, A.P. Bias in chemokine receptor signalling. Trends Immunol. 2014, 35, 243. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Takaishi, S.; Nagasaki, M.; Onozawa, Y.; Iino, I.; Maeda, H.; Komai, T.; Oda, T. Medium-chain Fatty Acid-sensing Receptor, GPR84, Is a Proinflammatory Receptor. J. Biol. Chem. 2013, 288, 10684–10691. [Google Scholar] [CrossRef]
- Han, J.; Hamilton, J.A.; Kirkland, J.L.; Corkey, B.E.; Guo, W. Medium-Chain Oil Reduces Fat Mass and Down-regulates Expression of Adipogenic Genes in Rats. Obes. Res. 2003, 11, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, L.; Fu, J.; Yu, P.; Gong, D.; Zeng, C.; Zeng, Z. Effects of Long-Chain and Medium-Chain Fatty Acids on Apoptosis and Oxidative Stress in Human Liver Cells with Steatosis. J. Food Sci. 2016, 81, H794–H800. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Roopashree, P.; Shetty, S.S.; Kumari, N.S. Effect of medium chain fatty acid in human health and disease. J. Funct. Foods 2021, 87, 104724. [Google Scholar] [CrossRef]
- Nafar, F.; Mearow, K.M. Coconut Oil Attenuates the Effects of Amyloid-b on Cortical Neurons in vitro. J. Alzheimer’s Dis. 2014, 39, 233. [Google Scholar] [CrossRef]
- Mustafa, A. Anti-inflammatory activity of lauric acid, thiocolchicoside and thiocolchicoside-lauric acid formulation. Bioinformation 2023, 19, 1075–1080. [Google Scholar] [CrossRef]
- Dayrit, F.M. The Properties of Lauric Acid and Their Significance in Coconut Oil. J. Am. Oil Chem. Soc. 2015, 92, 1–15. [Google Scholar] [CrossRef]
- Saraswathi, V.; Kumar, N.; Gopal, T.; Bhatt, S.; Ai, W.; Ma, C.; Talmon, G.A.; Desouza, C. Lauric Acid versus Palmitic Acid: Effects on Adipose Tissue Inflammation, Insulin Resistance, and Non-Alcoholic Fatty Liver Disease in Obesity. Biology 2020, 9, 346. [Google Scholar] [CrossRef]
- Nakajima, S.; Kunugi, H. Lauric acid promotes neuronal maturation mediated by astrocytes in primary cortical cultures. Heliyon 2020, 6, e03892. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Moriyama, M.; Kawabe, K.; Satoh, H.; Takano, K.; Azuma, Y.-T.; Nakamura, Y. Lauric Acid Alleviates Neuroinflammatory Responses by Activated Microglia: Involvement of the GPR40-Dependent Pathway. Neurochem. Res. 2018, 43, 1723–1735. [Google Scholar] [CrossRef]
- Nonaka, Y.; Takagi, T.; Inai, M.; Nishimura, S.; Urashima, S.; Honda, K.; Aoyama, T.; Terada, S. Lauric Acid Stimulates Ketone Body Production in the KT-5 Astrocyte Cell Line. J. Oleo Sci. 2016, 65, 693–699. [Google Scholar] [CrossRef]
- Preeti; Sambhakar, S.; Saharan, R.; Narwal, S.; Malik, R.; Gahlot, V.; Khalid, A.; Najmi, A.; Zoghebi, K.; Halawi, M.A.; et al. Exploring LIPIDs for their potential to improves bioavailability of lipophilic drugs candidates: A review. Saudi Pharm. J. 2023, 31, 101870. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Aljuraiban, G.S. Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis. Cells 2021, 10, 3388. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.L. The role of fatty acid composition and positional distribution in fat absorption in infants. J. Pediatr. 1994, 125, S62–S68. [Google Scholar] [CrossRef]
- Feltrin, K.L.; Little, T.J.; Meyer, J.H.; Horowitz, M.; Rades, T.; Wishart, J.; Feinle-Bisset, C. Effects of lauric acid on upper gut motility, plasma cholecystokinin and peptide YY, and energy intake are load, but not concentration, dependent in humans. J. Physiol. 2007, 581, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ameena, M.; Arumugham, M.; Ramalingam, K.; Shanmugam, R. Biomedical Applications of Lauric Acid: A Narrative Review. Cureus 2024, 16, e62770. [Google Scholar] [CrossRef]
- Matsue, M.; Mori, Y.; Nagase, S.; Sugiyama, Y.; Hirano, R.; Ogai, K.; Ogura, K.; Kurihara, S.; Okamoto, S. Measuring the Antimicrobial Activity of Lauric Acid against Various Bacteria in Human Gut Microbiota Using a New Method. Cell Transplant. 2019, 28, 1528–1541. [Google Scholar] [CrossRef]
- Gibbons, H.M.; Dragunow, M. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 2006, 1084, 1–15. [Google Scholar] [CrossRef]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef]
- Sanjay; Shin, J.-H.; Park, M.; Lee, H.-J. Cyanidin-3-O-Glucoside Regulates the M1/M2 Polarization of Microglia via PPARγ and Aβ42 Phagocytosis Through TREM2 in an Alzheimer’s Disease Model. Mol. Neurobiol. 2022, 59, 5135–5148. [Google Scholar] [CrossRef]
- Moresco, E.M.Y.; Lavine, D.; Beutler, B. Toll-like receptors. Curr. Biol. 2011, 21, R488–R493. [Google Scholar] [CrossRef] [PubMed]
- Kisioglu, B.; Onal, E.; Karabulut, D.; Onbasilar, I.; Akyol, A. Neuroprotective Roles of Lauric Acid and Resveratrol: Shared Benefits in Neuroinflammation and Anxiety, Distinct Effects on Memory Enhancement. Food Sci. Nutr. 2024, 12, 9735–9748. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zabihi, M.; Li, Q.; Li, X.; Kim, B.J.; Ubogu, E.E.; Raja, S.N.; Wesselmann, U.; Zhao, C. Drug Permeability: From the Blood–Brain Barrier to the Peripheral Nerve Barriers. Adv. Ther. 2023, 6, 2200150. [Google Scholar] [CrossRef]
- Tan, J.Y.B.; Yoon, B.K.; Cho, N.-J.; Lovrić, J.; Jug, M.; Jackman, J.A. Lipid Nanoparticle Technology for Delivering Biologically Active Fatty Acids and Monoglycerides. Int. J. Mol. Sci. 2021, 22, 9664. [Google Scholar] [CrossRef] [PubMed]
- Ameena, M.; Arumugham, M.; Ramalingam, K.; Shanmugam, R. Evaluation of the Anti-inflammatory, Antimicrobial, Antioxidant, and Cytotoxic Effects of Chitosan Thiocolchicoside-Lauric Acid Nanogel. Cureus 2023, 15, e46003. [Google Scholar] [CrossRef]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef]
- Van Der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 28–36. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senarath, R.M.U.S.; Oikari, L.E.; Bharadwaj, P.; Jayasena, V.; Martins, R.N.; Fernando, W.M.A.D.B. The Therapeutic Potential of Butyrate and Lauric Acid in Modulating Glial and Neuronal Activity in Alzheimer’s Disease. Nutrients 2025, 17, 2286. https://doi.org/10.3390/nu17142286
Senarath RMUS, Oikari LE, Bharadwaj P, Jayasena V, Martins RN, Fernando WMADB. The Therapeutic Potential of Butyrate and Lauric Acid in Modulating Glial and Neuronal Activity in Alzheimer’s Disease. Nutrients. 2025; 17(14):2286. https://doi.org/10.3390/nu17142286
Chicago/Turabian StyleSenarath, Rathnayaka Mudiyanselage Uththara Sachinthanie, Lotta E. Oikari, Prashant Bharadwaj, Vijay Jayasena, Ralph N. Martins, and Wanakulasuriya Mary Ann Dipika Binosha Fernando. 2025. "The Therapeutic Potential of Butyrate and Lauric Acid in Modulating Glial and Neuronal Activity in Alzheimer’s Disease" Nutrients 17, no. 14: 2286. https://doi.org/10.3390/nu17142286
APA StyleSenarath, R. M. U. S., Oikari, L. E., Bharadwaj, P., Jayasena, V., Martins, R. N., & Fernando, W. M. A. D. B. (2025). The Therapeutic Potential of Butyrate and Lauric Acid in Modulating Glial and Neuronal Activity in Alzheimer’s Disease. Nutrients, 17(14), 2286. https://doi.org/10.3390/nu17142286