


Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling


Abstract
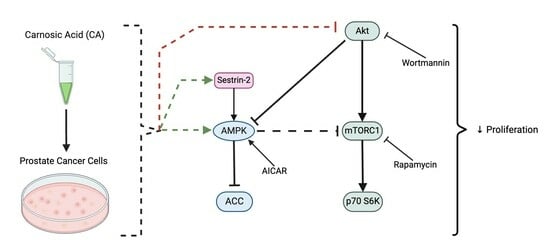
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Treatment
2.3. Clonogenic Survival Assay
2.4. Cell Proliferation Assay
2.5. Immunoblotting
2.6. Statistical Analysis
3. Results
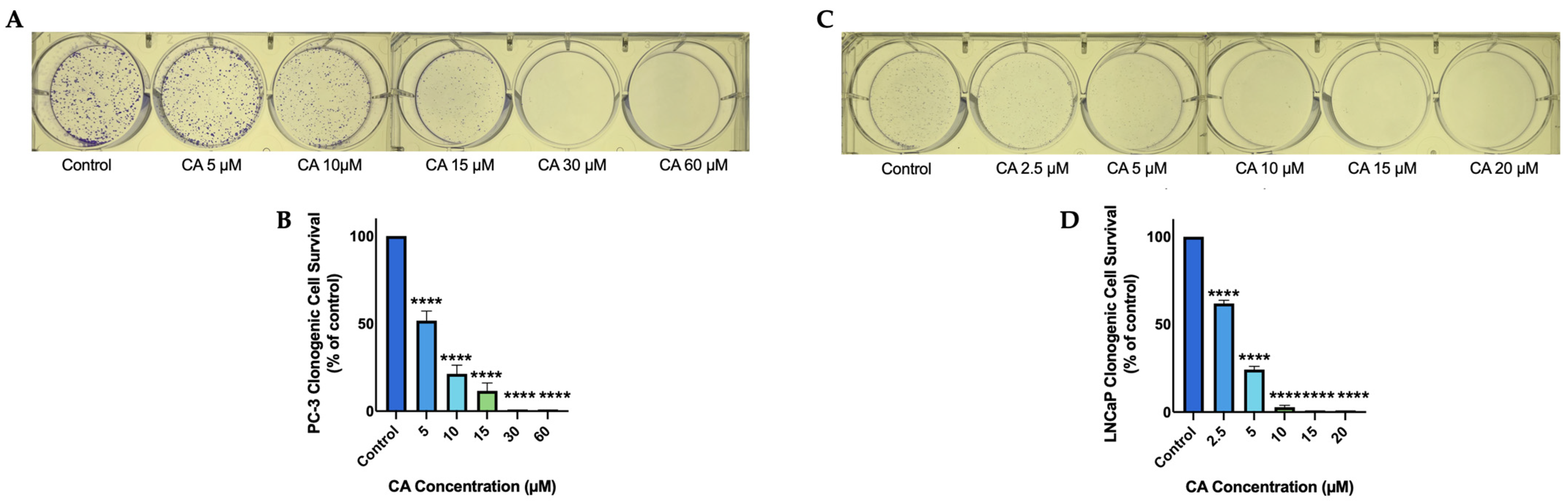
3.1. Carnosic Acid Inhibits Prostate Cancer Cell Survival
3.2. Carnosic Acid Inhibits Prostate Cancer Cell Proliferation
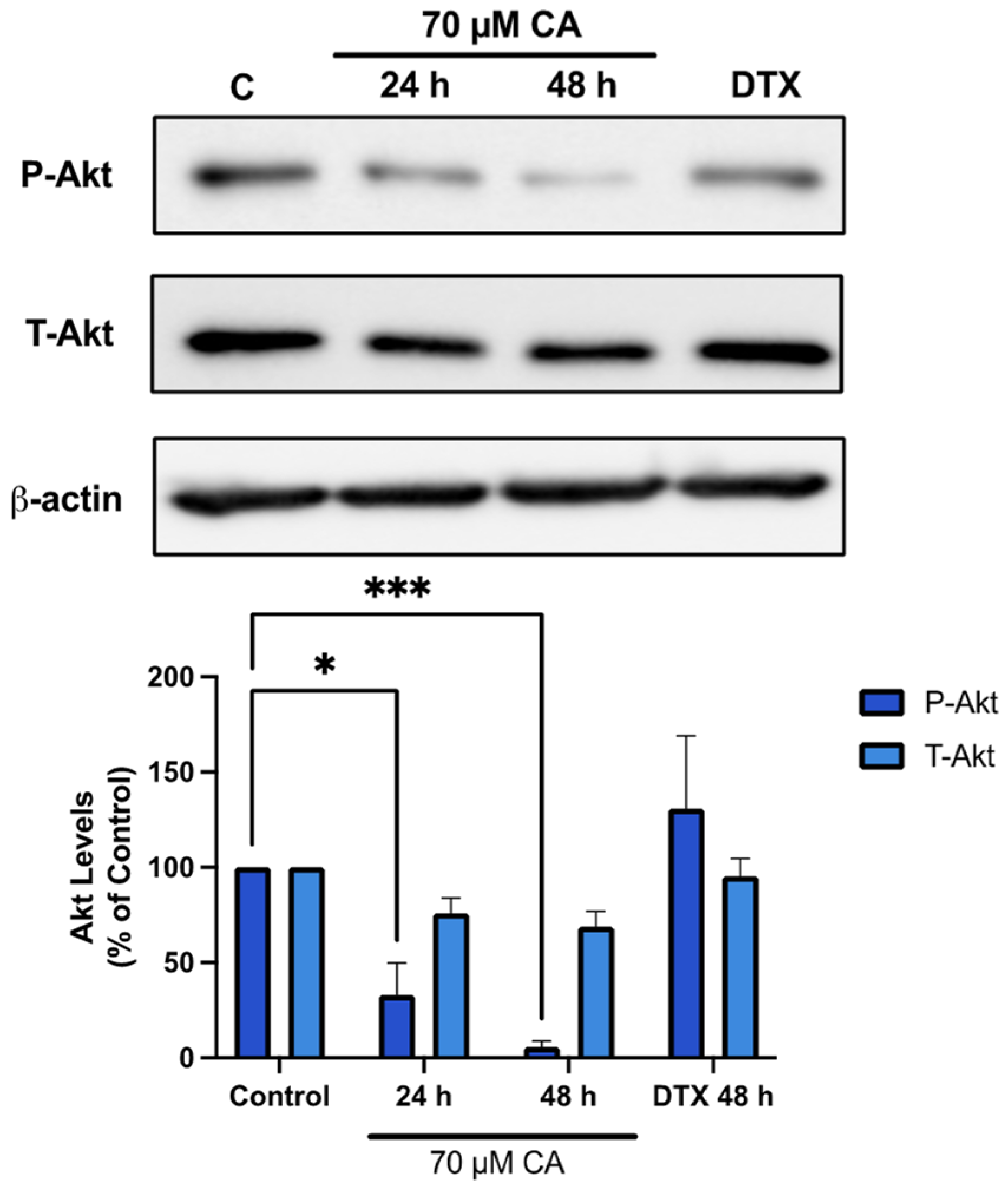
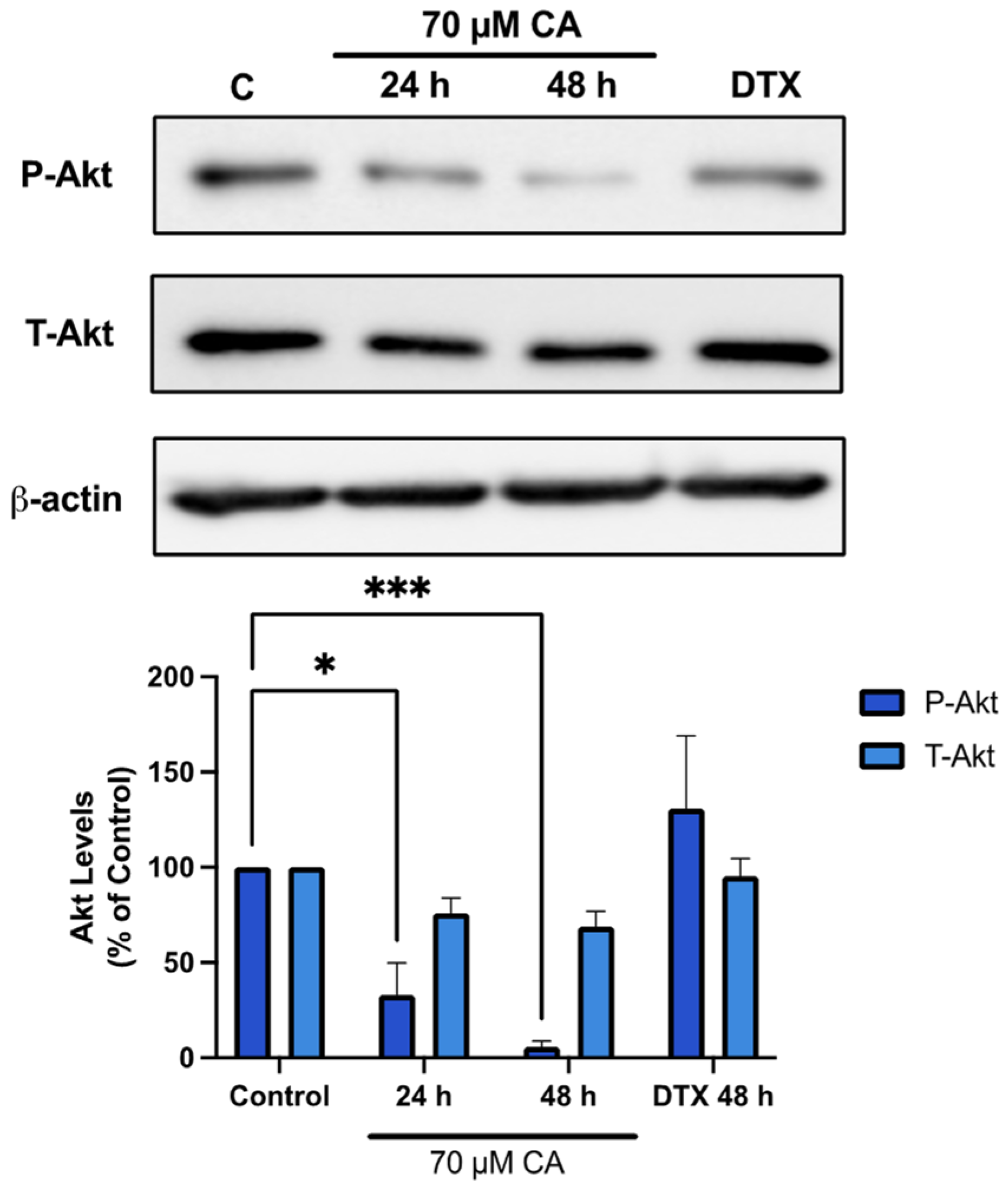
3.3. Carnosic Acid Inhibits Akt Signaling
3.4. Carnosic Acid Inhibits mTORC1-p70 S6K Signaling
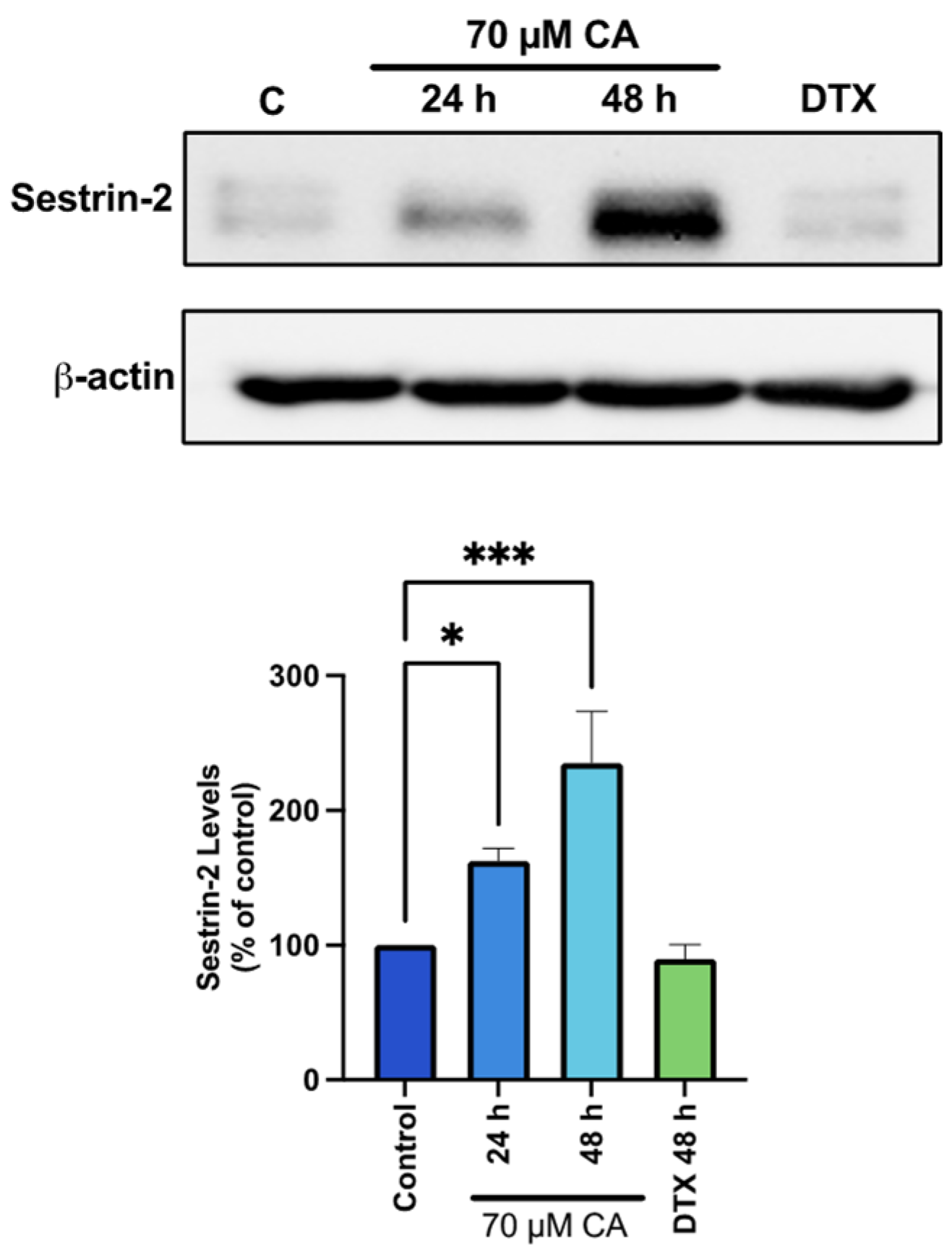
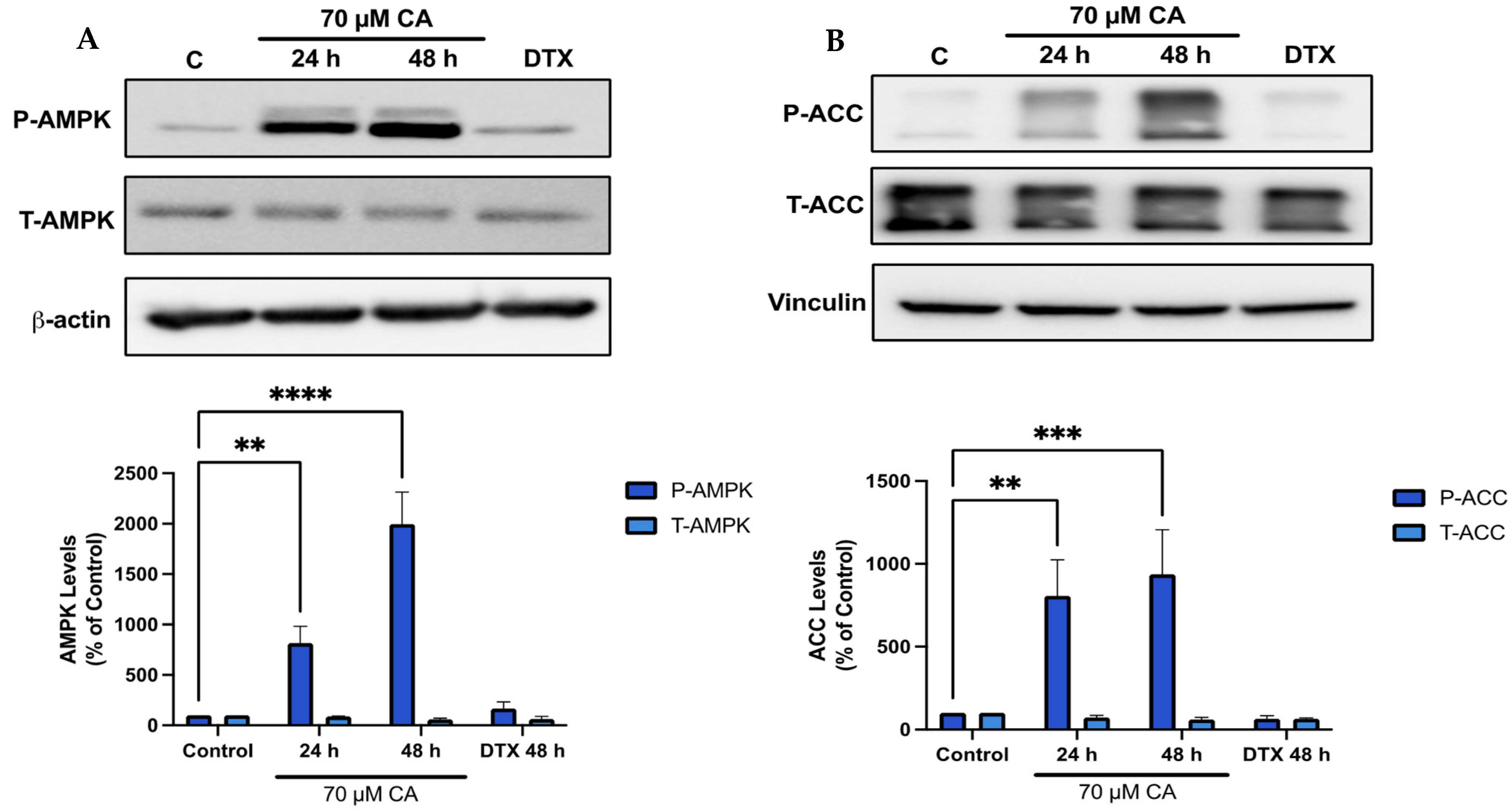
3.5. Carnosic Acid Activates AMPK Signaling
3.6. Carnosic Acid Activates Upstream Regulators of AMPK Signaling
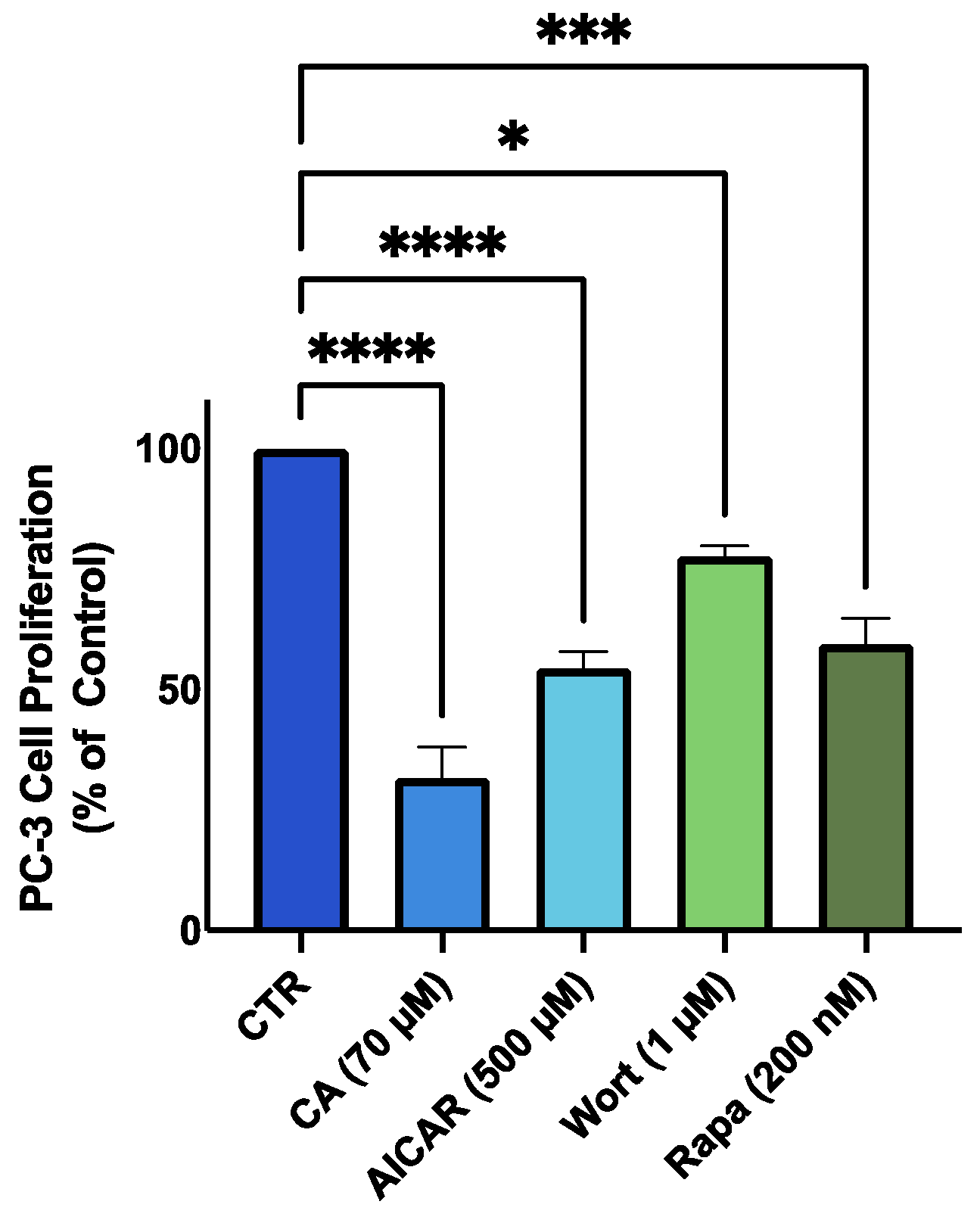
3.7. AMPK Activation; Akt and mTOR Inhibition Mimic the Effects of Carnosic Acid
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D. Epidemiology of Prostate Cancer. Urology 2003, 62, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 Prostate, Breast, and Colon Cancer Risk Loci Show Tissue-Specific Long-Range Interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Eeles, R. Germline Genetic Profiling in Prostate Cancer: Latest Developments and Potential Clinical Applications. Future Sci. OA 2015, 2, FSO87. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR Signaling Network in Cancer. Chin. J. Cancer 2013, 32, 253–265. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt Pathway in Oncology Therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primer 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Saltzman, A.; Yeh, S.; Young, W.; Keller, E.; Lee, H.J.; Wang, C.; Mizokami, A. Androgen Receptor: An Overview. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular Biology of the Androgen Receptor: From Molecular Understanding to the Clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef]
- Modi, P.K.; Faiena, I.; Kim, I.Y. Chapter 3—Androgen Receptor. In Prostate Cancer, 2nd ed.; Mydlo, J.H., Godec, C.J., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 21–28. ISBN 978-0-12-800077-9. [Google Scholar]
- Tyagi, V.; Scordo, M.; Yoon, R.S.; Liporace, F.A.; Greene, L.W. Revisiting the Role of Testosterone: Are We Missing Something? Rev. Urol. 2017, 19, 16–24. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2019, 37, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Huang, J. Phosphatidylinositol 3-Kinase-AKT-Mammalian Target of Rapamycin Pathway Is Essential for Neuroendocrine Differentiation of Prostate Cancer. J. Biol. Chem. 2007, 282, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt Pathway in Prostate Cancer: Challenges and Opportunities (Review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef]
- Roudsari, N.M.; Lashgari, N.-A.; Momtaz, S.; Abaft, S.; Jamali, F.; Safaiepour, P.; Narimisa, K.; Jackson, G.; Bishayee, A.; Rezaei, N.; et al. Inhibitors of the PI3K/Akt/mTOR Pathway in Prostate Cancer Chemoprevention and Intervention. Pharmaceutics 2021, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef]
- Carling, D. AMPK Signalling in Health and Disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic Target for Diabetes and Cancer Prevention. Curr. Pharm. Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New Insights into Activation and Function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. The AMP-Activated Protein Kinase Cascade—A Unifying System for Energy Control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A Common Bicyclic Protein Kinase Cascade Inactivates the Regulatory Enzymes of Fatty Acid and Cholesterol Biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Goldstein, J.L.; Brown, M.S. Replacement of Serine-871 of Hamster 3-Hydroxy-3-Methylglutaryl-CoA Reductase Prevents Phosphorylation by AMP-Activated Kinase and Blocks Inhibition of Sterol Synthesis Induced by ATP Depletion. Proc. Natl. Acad. Sci. USA 1993, 90, 9261–9265. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-Activated Protein Kinase Kinase from Rat Liver and Identification of Threonine 172 as the Major Site at Which It Phosphorylates AMP-Activated Protein Kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 Activates AMPK-Dependent Cytoprotective Autophagy in TRAIL-Treated Epithelial Cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, D.; Dyck, J.R.B.; Li, Y.; Zhang, H.; Morishima, M.; Mann, D.L.; Taffet, G.E.; Baldini, A.; Khoury, D.S.; et al. A Pivotal Role for Endogenous TGF-Beta-Activated Kinase-1 in the LKB1/AMP-Activated Protein Kinase Energy-Sensor Pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 17378–17383. [Google Scholar] [CrossRef]
- Gong, L.; Wang, Z.; Wang, Z.; Zhang, Z. Sestrin2 as a Potential Target for Regulating Metabolic-Related Diseases. Front. Endocrinol. 2021, 12, 751020. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, S.; Jayachandran, I.; Balasubramanyam, M.; Mohan, V.; Venkatesan, B.; Manickam, N. Sestrin2 Regulates Monocyte Activation through AMPK-mTOR Nexus under High-Glucose and Dyslipidemic Conditions. J. Cell. Biochem. 2019, 120, 8201–8213. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2008, 134, 451–460, Erratum in Cell 2009, 136, 378. [Google Scholar] [CrossRef]
- Kennedy, D.O.; Wightman, E.L. Herbal Extracts and Phytochemicals: Plant Secondary Metabolites and the Enhancement of Human Brain Function. Adv. Nutr. 2011, 2, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/Paclitaxel Kills Cancer Cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterom, A.T.; Schrijvers, D. Docetaxel (Taxotere), a Review of Preclinical and Clinical Experience. Part II: Clinical Experience. Anticancer. Drugs 1995, 6, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Abal, M.; Andreu, J.M.; Barasoain, I. Taxanes: Microtubule and Centrosome Targets, and Cell Cycle Dependent Mechanisms of Action. Curr. Cancer Drug Targets 2003, 3, 193–203. [Google Scholar] [CrossRef]
- Pienta, K.J. Preclinical Mechanisms of Action of Docetaxel and Docetaxel Combinations in Prostate Cancer. Semin. Oncol. 2001, 28, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Liu, C.; Sanli, T.; Tsiani, E.; Singh, G.; Bristow, R.G.; Dayes, I.; Lukka, H.; Wright, J.; Tsakiridis, T. Resveratrol Enhances Prostate Cancer Cell Response to Ionizing Radiation. Modulation of the AMPK, Akt and mTOR Pathways. Radiat. Oncol. 2011, 6, 144. [Google Scholar] [CrossRef]
- Moore, J.; Megaly, M.; MacNeil, A.J.; Klentrou, P.; Tsiani, E. Rosemary Extract Reduces Akt/mTOR/p70S6K Activation and Inhibits Proliferation and Survival of A549 Human Lung Cancer Cells. Biomed. Pharmacother. 2016, 83, 725–732. [Google Scholar] [CrossRef]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer Effects of Rosemary (Rosmarinus officinalis L.) Extract and Rosemary Extract Polyphenols. Nutrients 2016, 8, 731. [Google Scholar] [CrossRef] [PubMed]
- Jaglanian, A.; Tsiani, E. Rosemary Extract Inhibits Proliferation, Survival, Akt, and mTOR Signaling in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 810. [Google Scholar] [CrossRef] [PubMed]
- Jaglanian, A.; Termini, D.; Tsiani, E. Rosemary (Rosmarinus officinalis L.) Extract Inhibits Prostate Cancer Cell Proliferation and Survival by Targeting Akt and mTOR. Biomed. Pharmacother. 2020, 131, 110717. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.J.; Moore, J.; Song, J.; Tsiani, E.L. Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK. Life 2021, 12, 52. [Google Scholar] [CrossRef]
- O’Neill, E.J.; Hartogh, D.J.D.; Azizi, K.; Tsiani, E. Anticancer Properties of Carnosol: A Summary of in Vitro and In Vivo Evidence. Antioxidants 2020, 9, 961. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.J.; Sze, N.S.K.; MacPherson, R.E.K.; Tsiani, E. Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/LKB1/AMPK Signalling. Int. J. Mol. Sci. 2024, 25, 1950. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Lines. Plant Foods Hum. Nutr. Dordr. Neth. 2010, 65, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Palit, S.; Ball, W.B.; Das, P.K. Carnosic Acid Modulates Akt/IKK/NF-κB Signaling by PP2A and Induces Intrinsic and Extrinsic Pathway Mediated Apoptosis in Human Prostate Carcinoma PC-3 Cells. Apoptosis 2012, 17, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Petiwala, S.M.; Li, G.; Bosland, M.C.; Lantvit, D.D.; Petukhov, P.A.; Johnson, J.J. Carnosic Acid Promotes Degradation of the Androgen Receptor and Is Regulated by the Unfolded Protein Response Pathway in Vitro and in Vivo. Carcinogenesis 2016, 37, 827–838. [Google Scholar] [CrossRef]
- Ossikbayeva, S.; Khanin, M.; Sharoni, Y.; Trachtenberg, A.; Tuleukhanov, S.; Sensenig, R.; Rom, S.; Danilenko, M.; Orynbayeva, Z. Curcumin and Carnosic Acid Cooperate to Inhibit Proliferation and Alter Mitochondrial Function of Metastatic Prostate Cancer Cells. Antioxidants 2021, 10, 1591. [Google Scholar] [CrossRef]
- Moore, J.; Pickering, G.; Gaudette, N.J.; Tsiani, E. Resveratrol-Fortification of Red Wine Does Not Provide Greater Inhibition of Human Lung Cancer Cell Survival Compared to Non-Fortified Wine. J. Mol. Biochem. 2015, 4, 52–62. [Google Scholar]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR Signalling Controls Tumour Cell Growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Naimi, M.; Vlavcheski, F.; Murphy, B.; Hudlicky, T.; Tsiani, E. Carnosic Acid as a Component of Rosemary Extract Stimulates Skeletal Muscle Cell Glucose Uptake via AMPK Activation. Clin. Exp. Pharmacol. Physiol. 2017, 44, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Den Hartogh, D.J.; Vlavcheski, F.; Giacca, A.; MacPherson, R.E.K.; Tsiani, E. Carnosic Acid Attenuates the Free Fatty Acid-Induced Insulin Resistance in Muscle Cells and Adipocytes. Cells 2022, 11, 167. [Google Scholar] [CrossRef]
- Hardie, D.G. The AMP-Activated Protein Kinase Pathway—New Players Upstream and Downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Khan, A.S.; Frigo, D.E. A Spatiotemporal Hypothesis for the Regulation, Role, and Targeting of AMPK in Prostate Cancer. Nat. Rev. Urol. 2017, 14, 164–180. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, Y.M. Quercetin Regulates Sestrin 2-AMPK-mTOR Signaling Pathway and Induces Apoptosis via Increased Intracellular ROS in HCT116 Colon Cancer Cells. J. Cancer Prev. 2013, 18, 264–270. [Google Scholar] [CrossRef]
- Seo, K.; Ki, S.H.; Park, E.Y.; Shin, S.M. 5-Fluorouracil Inhibits Cell Migration by Induction of Sestrin2 in Colon Cancer Cells. Arch. Pharm. Res. 2017, 40, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.G.; Bort, A.; Vara-Ciruelos, D.; Díaz-Laviada, I. Androgen Deprivation Induces Reprogramming of Prostate Cancer Cells to Stem-Like Cells. Cells 2020, 9, 1441. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Sellers, W.R. Akt-Regulated Pathways in Prostate Cancer. Oncogene 2005, 24, 7465–7474. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.; Parsons, R. The Role of PTEN Signaling Perturbations in Cancer and in Targeted Therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liu, H.; Yao, Y.; Geng, L.; Zhang, X.; Jiang, L.; Shi, B.; Yang, F. Carnosic Acid Induces Autophagic Cell Death through Inhibition of the Akt/mTOR Pathway in Human Hepatoma Cells. J. Appl. Toxicol. 2015, 35, 485–492. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.; Fan, Y.; Li, Y. Antiproliferative Activity of Carnosic Acid Is Mediated via Inhibition of Cell Migration and Invasion, and Suppression of Phosphatidylinositol 3-Kinases (PI3K)/AKT/Mammalian Target of Rapamycin (mTOR) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 7864–7871. [Google Scholar] [CrossRef]
- El-Huneidi, W.; Bajbouj, K.; Muhammad, J.S.; Vinod, A.; Shafarin, J.; Khoder, G.; Saleh, M.A.; Taneera, J.; Abu-Gharbieh, E. Carnosic Acid Induces Apoptosis and Inhibits Akt/mTOR Signaling in Human Gastric Cancer Cell Lines. Pharmaceuticals 2021, 14, 230. [Google Scholar] [CrossRef]
- Sun, M.; Wang, G.; Paciga, J.E.; Feldman, R.I.; Yuan, Z.-Q.; Ma, X.-L.; Shelley, S.A.; Jove, R.; Tsichlis, P.N.; Nicosia, S.V.; et al. AKT1/PKBα Kinase Is Frequently Elevated in Human Cancers and Its Constitutive Activation Is Required for Oncogenic Transformation in NIH3T3 Cells. Am. J. Pathol. 2001, 159, 431–437. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. mTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.J.; Syed, D.N.; Heren, C.R.; Suh, Y.; Adhami, V.M.; Mukhtar, H. Carnosol, a Dietary Diterpene, Displays Growth Inhibitory Effects in Human Prostate Cancer PC3 Cells Leading to G2-Phase Cell Cycle Arrest and Targets the 5’-AMP-Activated Protein Kinase (AMPK) Pathway. Pharm. Res. 2008, 25, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Nie, Q.; Wang, Z.; Di, Y.; Chen, X.; Ren, K. Targeting Acetyl-CoA Carboxylase 1 for Cancer Therapy. Front. Pharmacol. 2023, 14, 1129010. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The Acetyl-CoA Carboxylase Enzyme: A Target for Cancer Therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, T. Recent Development in Acetyl-CoA Carboxylase Inhibitors and Their Potential as Novel Drugs. Future Med. Chem. 2020, 12, 533–561. [Google Scholar] [CrossRef]
- Agarwal, S.; Bell, C.M.; Rothbart, S.B.; Moran, R.G. AMP-Activated Protein Kinase (AMPK) Control of mTORC1 Is P53- and TSC2-Independent in Pemetrexed-Treated Carcinoma Cells. J. Biol. Chem. 2015, 290, 27473–27486. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 Promotes LKB1-Mediated AMPK Activation in the Ischemic Heart. FASEB J. 2015, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Y.; Shao, H.; Liu, S.; Niu, Y.; Fu, L. Globular Adiponectin Ameliorates Insulin Resistance in Skeletal Muscle by Enhancing the LKB1-Mediated AMPK Activation via SESN2. Sports Med. Health Sci. 2023, 5, 34–41. [Google Scholar] [CrossRef]
- Quan, N.; Sun, W.; Wang, L.; Chen, X.; Bogan, J.S.; Zhou, X.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Sestrin2 Prevents Age-Related Intolerance to Ischemia and Reperfusion Injury by Modulating Substrate Metabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 4153–4167. [Google Scholar] [CrossRef]
- Fu, H.; Song, W.; Wang, Y.; Deng, W.; Tang, T.; Fan, W.; Qu, S. Radiosensitizing Effects of Sestrin2 in PC3 Prostate Cancer Cells. Iran. J. Basic Med. Sci. 2018, 21, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Luo, M.; Zhang, J.; Han, F.; Hou, N.; Pan, R.; Sun, X. A Paradoxical Role for Sestrin 2 Protein in Tumor Suppression and Tumorigenesis. Cancer Cell Int. 2021, 21, 606. [Google Scholar] [CrossRef]
- Ding, B.; Haidurov, A.; Chawla, A.; Parmigiani, A.; van de Kamp, G.; Dalina, A.; Yuan, F.; Lee, J.H.; Chumakov, P.M.; Grossman, S.R.; et al. P53-Inducible SESTRINs Might Play Opposite Roles in the Regulation of Early and Late Stages of Lung Carcinogenesis. Oncotarget 2019, 10, 6997–7009. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.T.; Lee, S.H.; Kim, J.I.; Kim, Y.M. Quercetin Regulates the Sestrin 2-AMPK-P38 MAPK Signaling Pathway and Induces Apoptosis by Increasing the Generation of Intracellular ROS in a P53-Independent Manner. Int. J. Mol. Med. 2014, 33, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Cai, F.; Liu, X.; Guo, L. LKB1 Suppresses Proliferation and Invasion of Prostate Cancer through Hedgehog Signaling Pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 8480–8488. [Google Scholar]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The Dynamic Interaction of AMBRA1 with the Dynein Motor Complex Regulates Mammalian Autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef]
- Grenier, A.; Poulain, L.; Mondesir, J.; Jacquel, A.; Bosc, C.; Stuani, L.; Mouche, S.; Larrue, C.; Sahal, A.; Birsen, R.; et al. AMPK-PERK Axis Represses Oxidative Metabolism and Enhances Apoptotic Priming of Mitochondria in Acute Myeloid Leukemia. Cell Rep. 2022, 38, 110197. [Google Scholar] [CrossRef]
- Su, C.-C.; Hsieh, K.-L.; Liu, P.-L.; Yeh, H.-C.; Huang, S.-P.; Fang, S.-H.; Cheng, W.-C.; Huang, K.-H.; Chiu, F.-Y.; Lin, I.-L.; et al. AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. Int. J. Mol. Sci. 2019, 20, 1647. [Google Scholar] [CrossRef]
- Sauer, H.; Engel, S.; Milosevic, N.; Sharifpanah, F.; Wartenberg, M. Activation of AMP-Kinase by AICAR Induces Apoptosis of DU-145 Prostate Cancer Cells through Generation of Reactive Oxygen Species and Activation of c-Jun N-Terminal Kinase. Int. J. Oncol. 2012, 40, 501–508. [Google Scholar] [CrossRef]
- Ihara, M.; Shichijo, K.; Takeshita, S.; Kudo, T. Wortmannin, a Specific Inhibitor of Phosphatidylinositol-3-Kinase, Induces Accumulation of DNA Double-Strand Breaks. J. Radiat. Res. 2020, 61, 171–176. [Google Scholar] [CrossRef]
- Cleary, J.M.; Shapiro, G.I. Development of Phosphoinositide-3 Kinase Pathway Inhibitors for Advanced Cancer. Curr. Oncol. Rep. 2010, 12, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and Cancer Therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadile, M.; Sze, N.S.K.; Fajardo, V.A.; Tsiani, E. Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling. Nutrients 2024, 16, 1257. https://doi.org/10.3390/nu16091257
Nadile M, Sze NSK, Fajardo VA, Tsiani E. Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling. Nutrients. 2024; 16(9):1257. https://doi.org/10.3390/nu16091257
Chicago/Turabian StyleNadile, Matteo, Newman Siu Kwan Sze, Val A. Fajardo, and Evangelia Tsiani. 2024. "Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling" Nutrients 16, no. 9: 1257. https://doi.org/10.3390/nu16091257
APA StyleNadile, M., Sze, N. S. K., Fajardo, V. A., & Tsiani, E. (2024). Inhibition of Prostate Cancer Cell Survival and Proliferation by Carnosic Acid Is Associated with Inhibition of Akt and Activation of AMPK Signaling. Nutrients, 16(9), 1257. https://doi.org/10.3390/nu16091257