
The Interaction between Human Microbes and Advanced Glycation End Products: The Role of Klebsiella X15 on Advanced Glycation End Products’ Degradation

Abstract
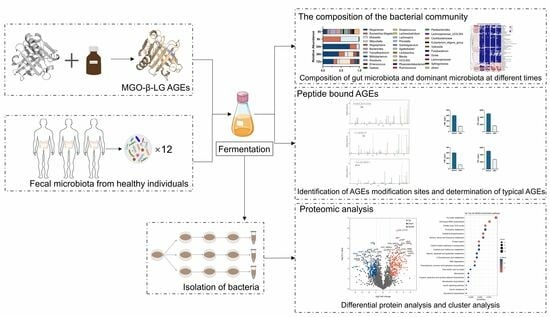
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Preparation of MGO-β-LG AGEs
2.3. In Vitro Gastrointestinal Digestion of MGO-β-LG AGEs
2.4. Anaerobic Incubations of Fecal Slurries with Digested MGO-β-LG AGEs
2.5. Gut Microbiota Analysis during In Vitro Fermentation
2.6. Isolation of Bacteria from Fermentation Broth
2.7. Analysis of CML and CEL by HPLC-MS/MS
2.8. Analysis of Modification of Digested MGO-β-LG AGEs Pre- and Host Fermentation by High-Resolution Mass Spectrometry
2.9. Proteomic Analysis of Klebsiella X15 Responses to Sole Utilization of AGEs as Nitrogen Source
2.10. Data Analysis
3. Results
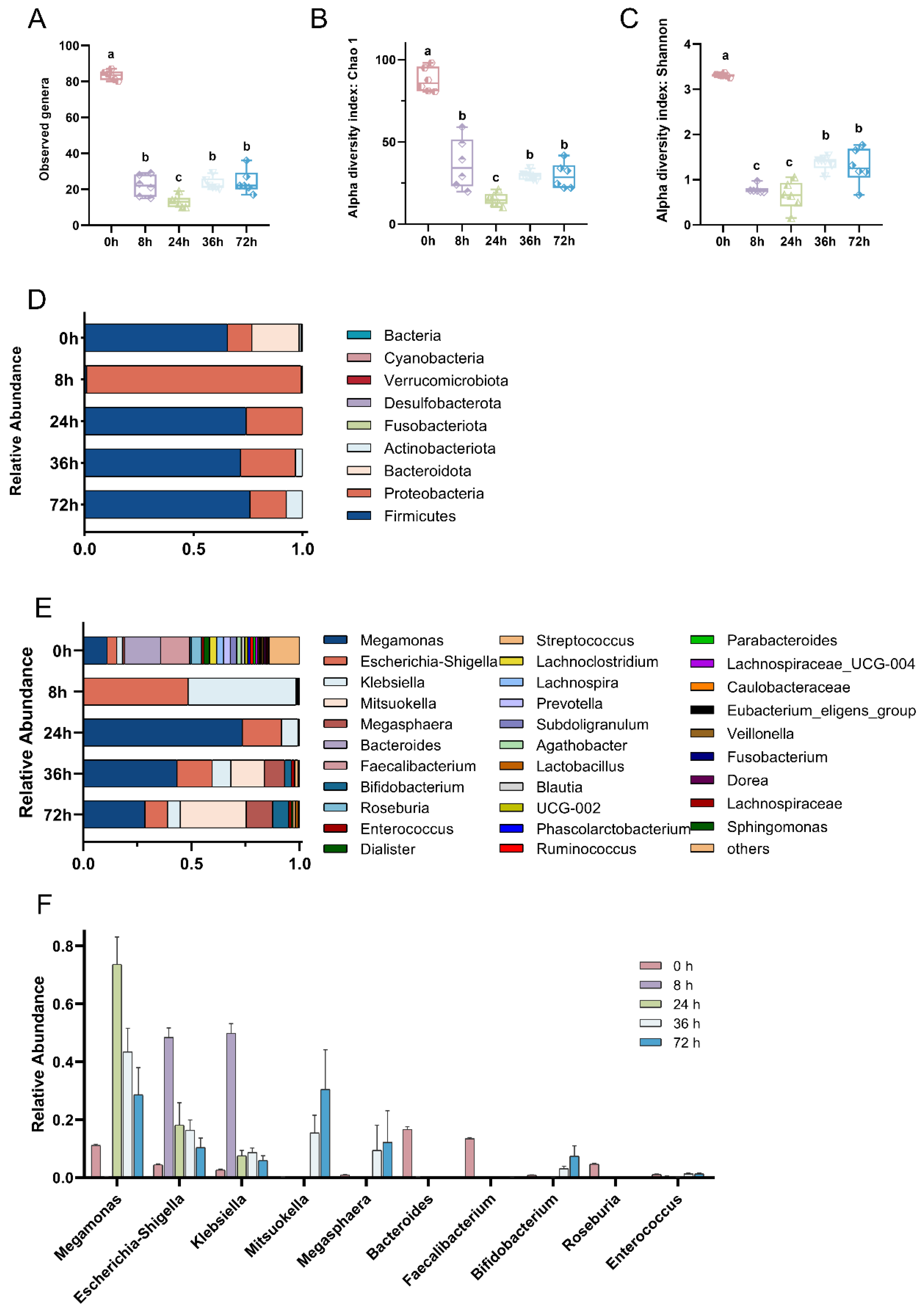
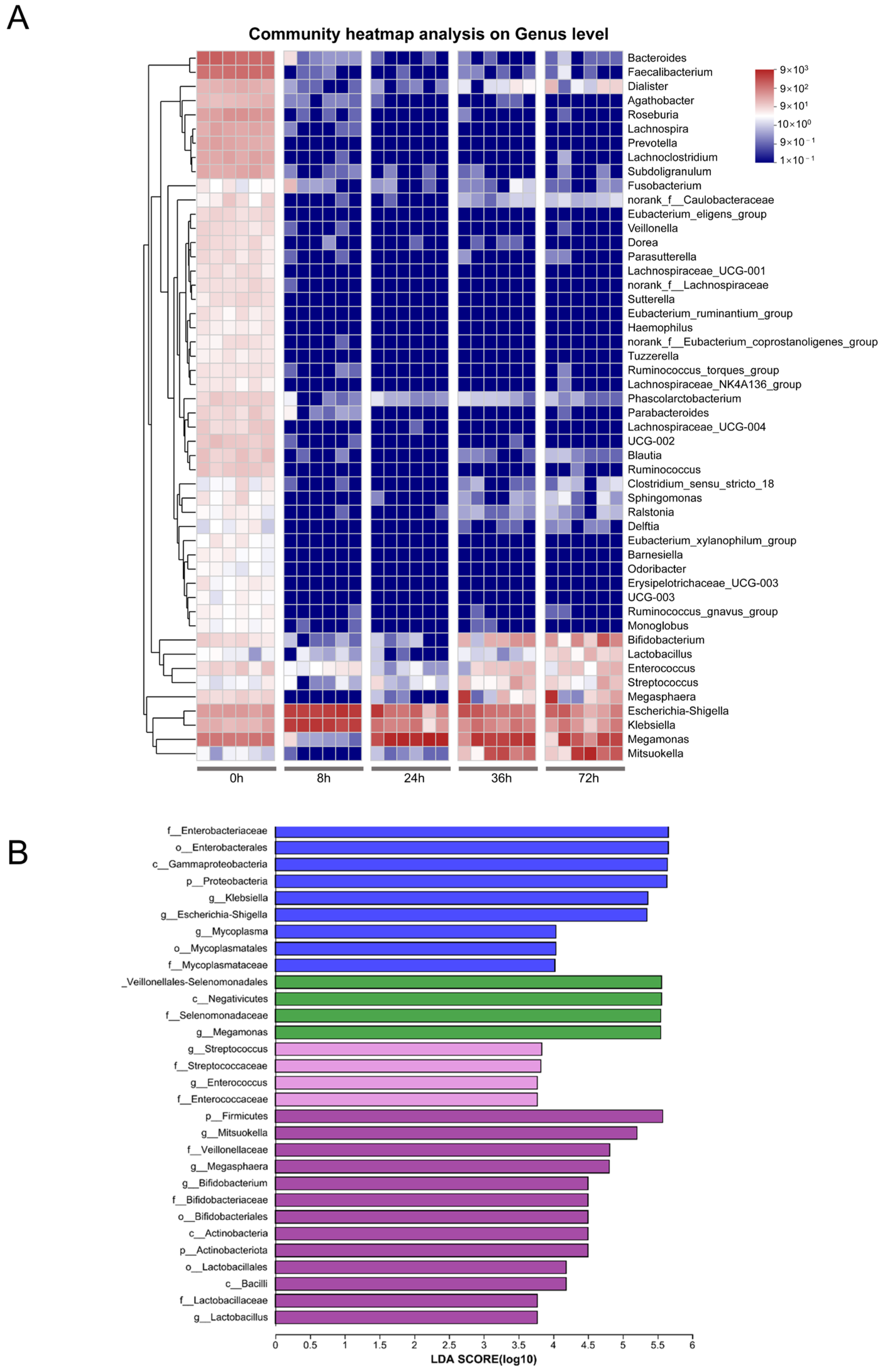
3.1. Changes in Gut Microbial Community during In Vitro Fecal Fermentation
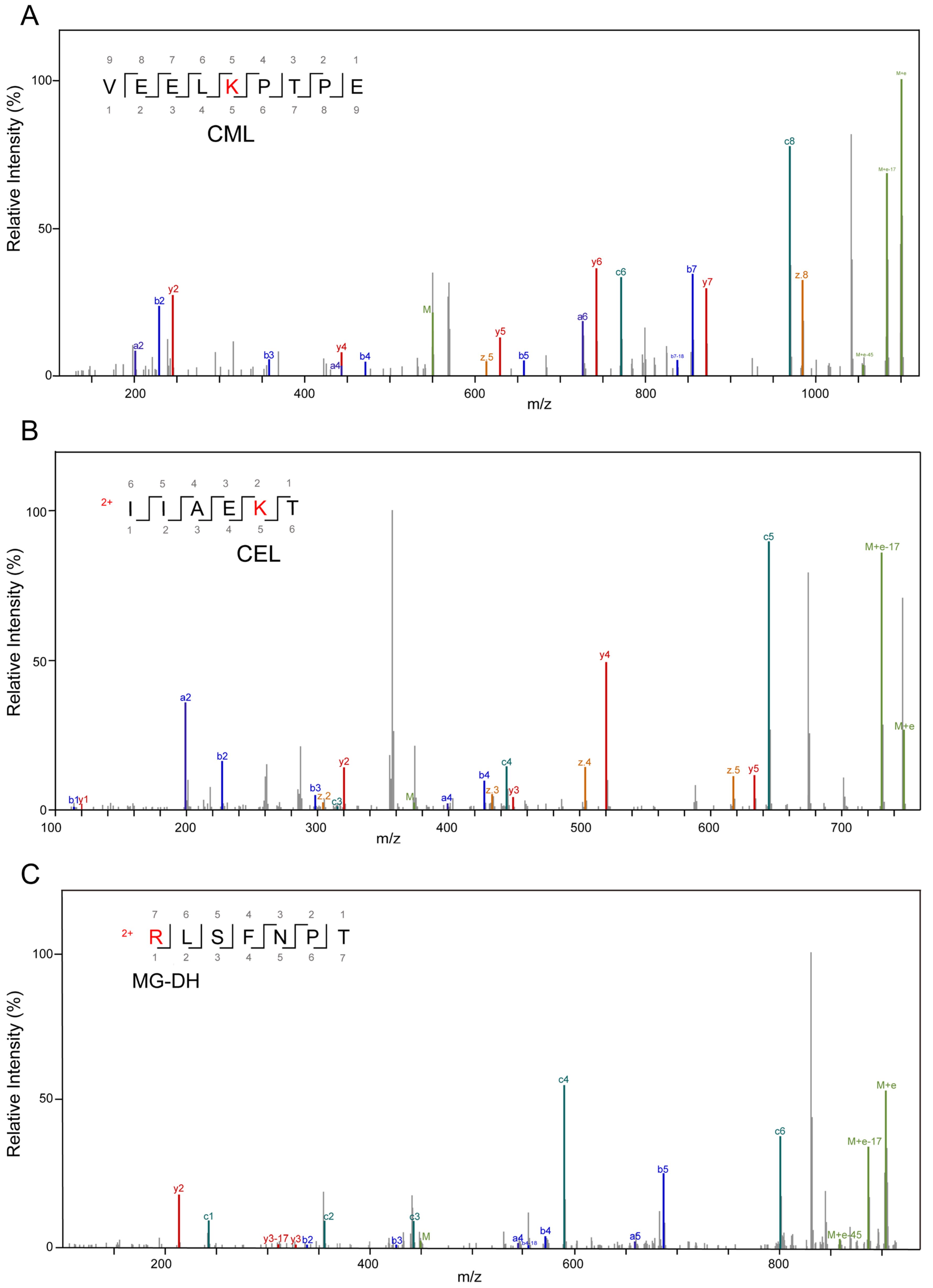
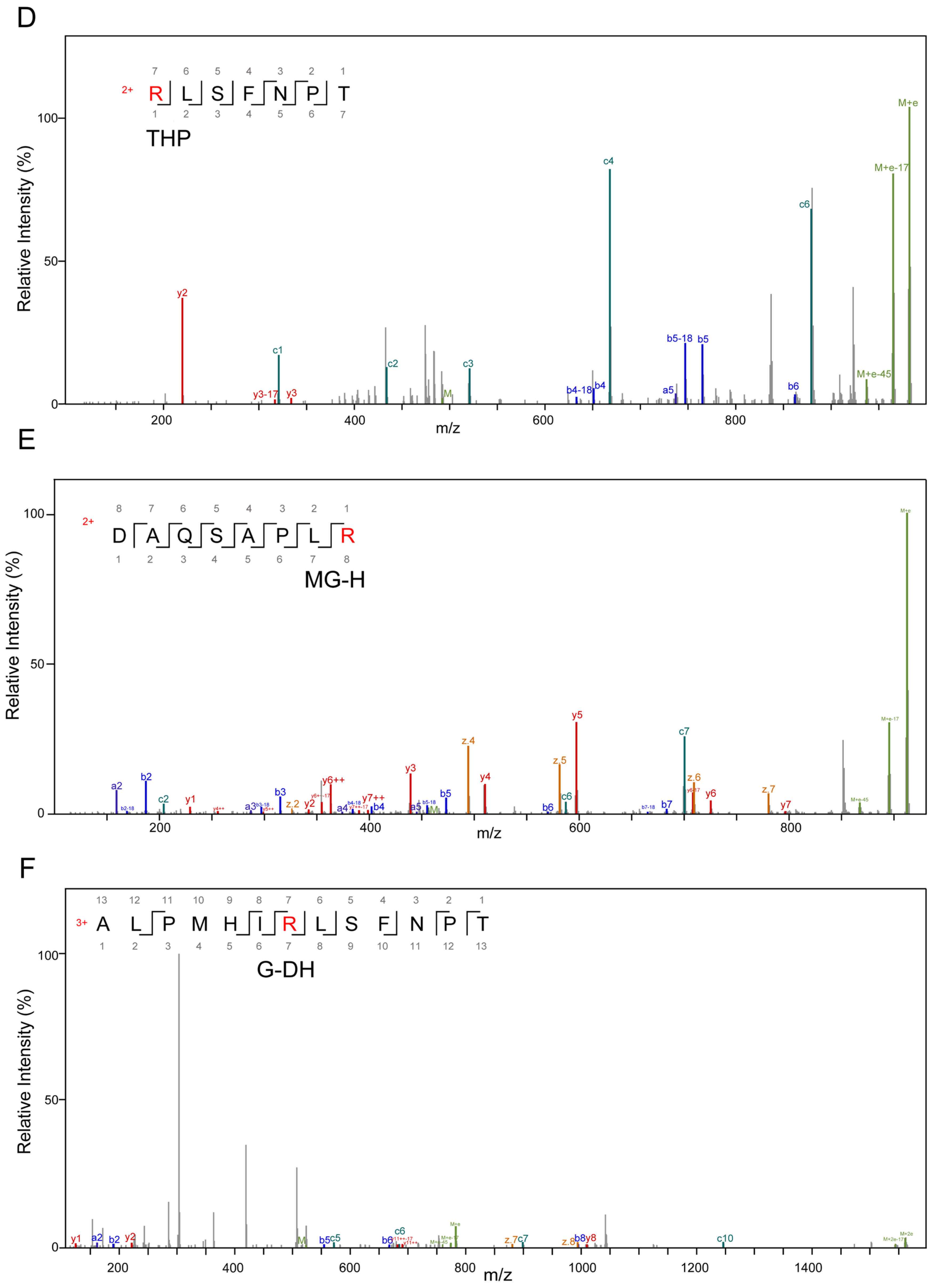
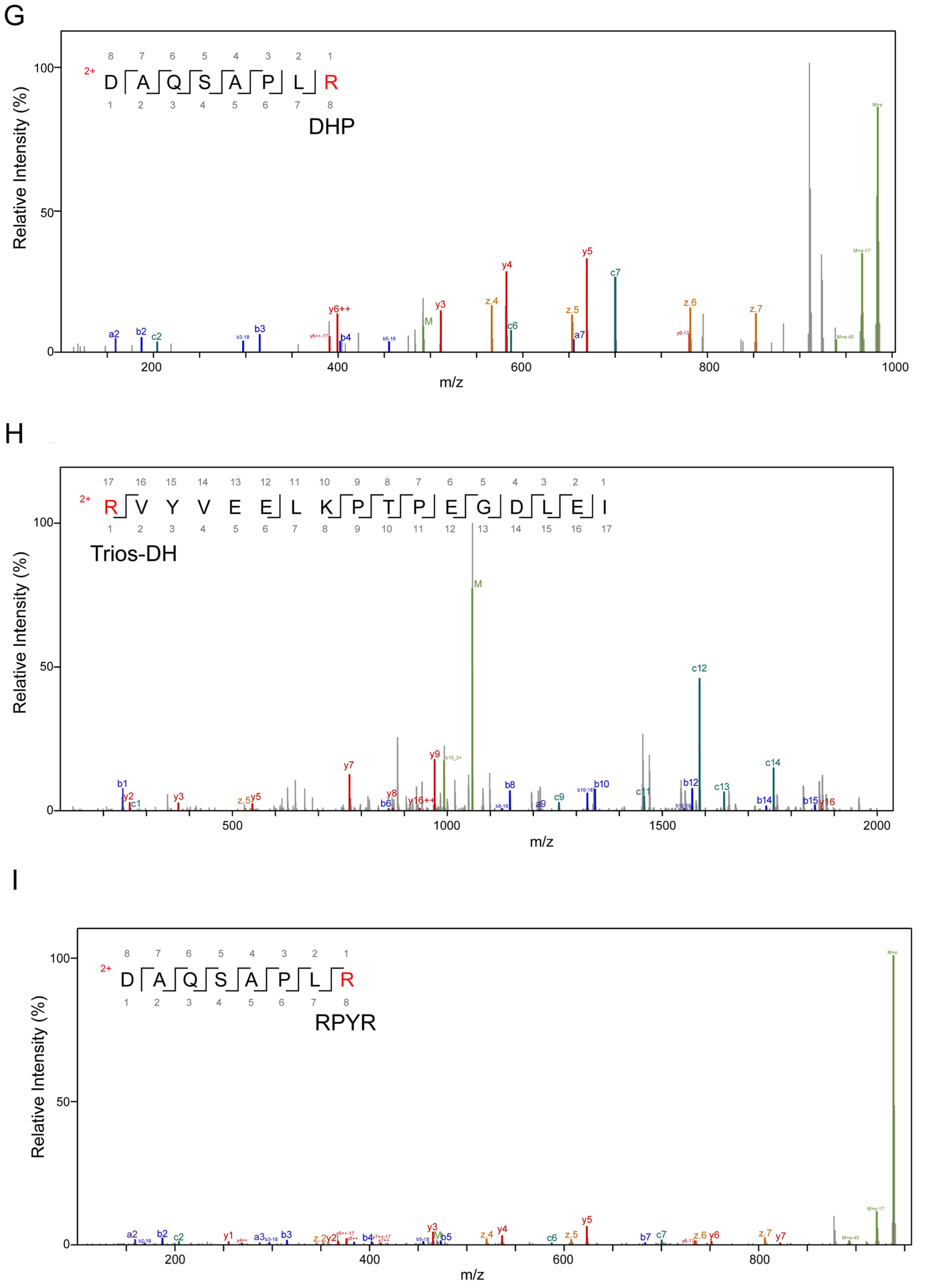
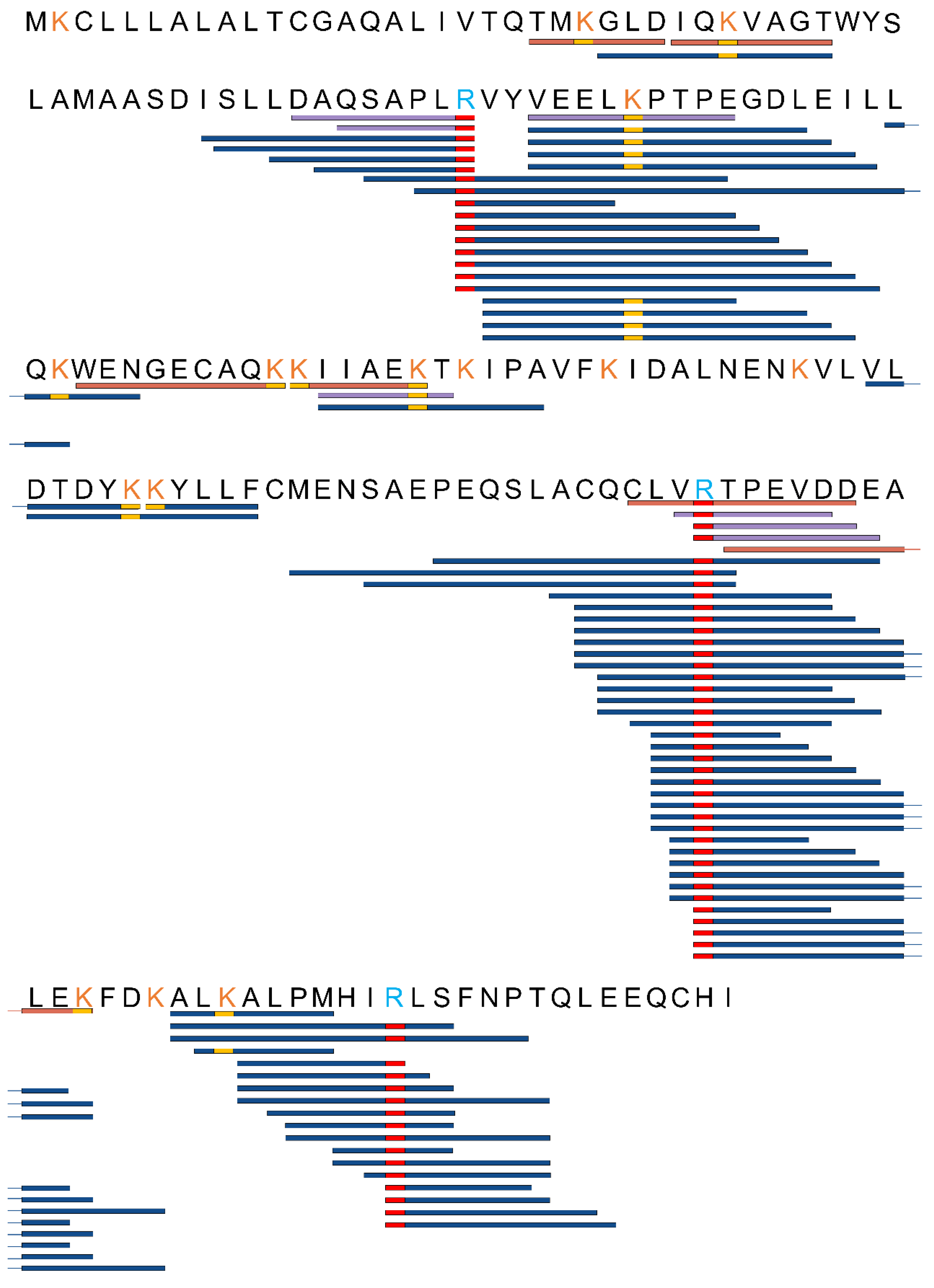
3.2. Alterations in AGE-Modified Polypeptides before and after Fermentation
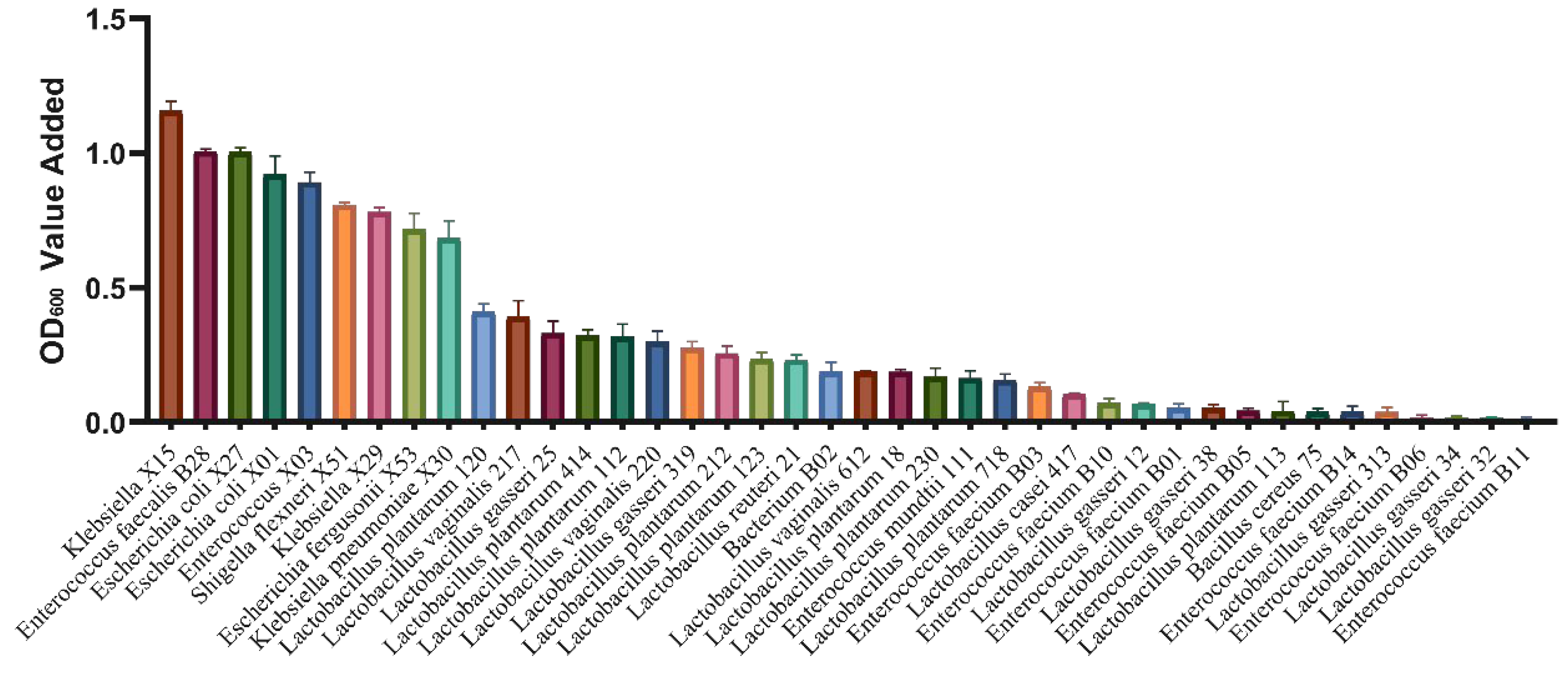
3.3. Isolation and Screening of Potentially AGE-Metabolizing Bacteria from Fermentation Broth
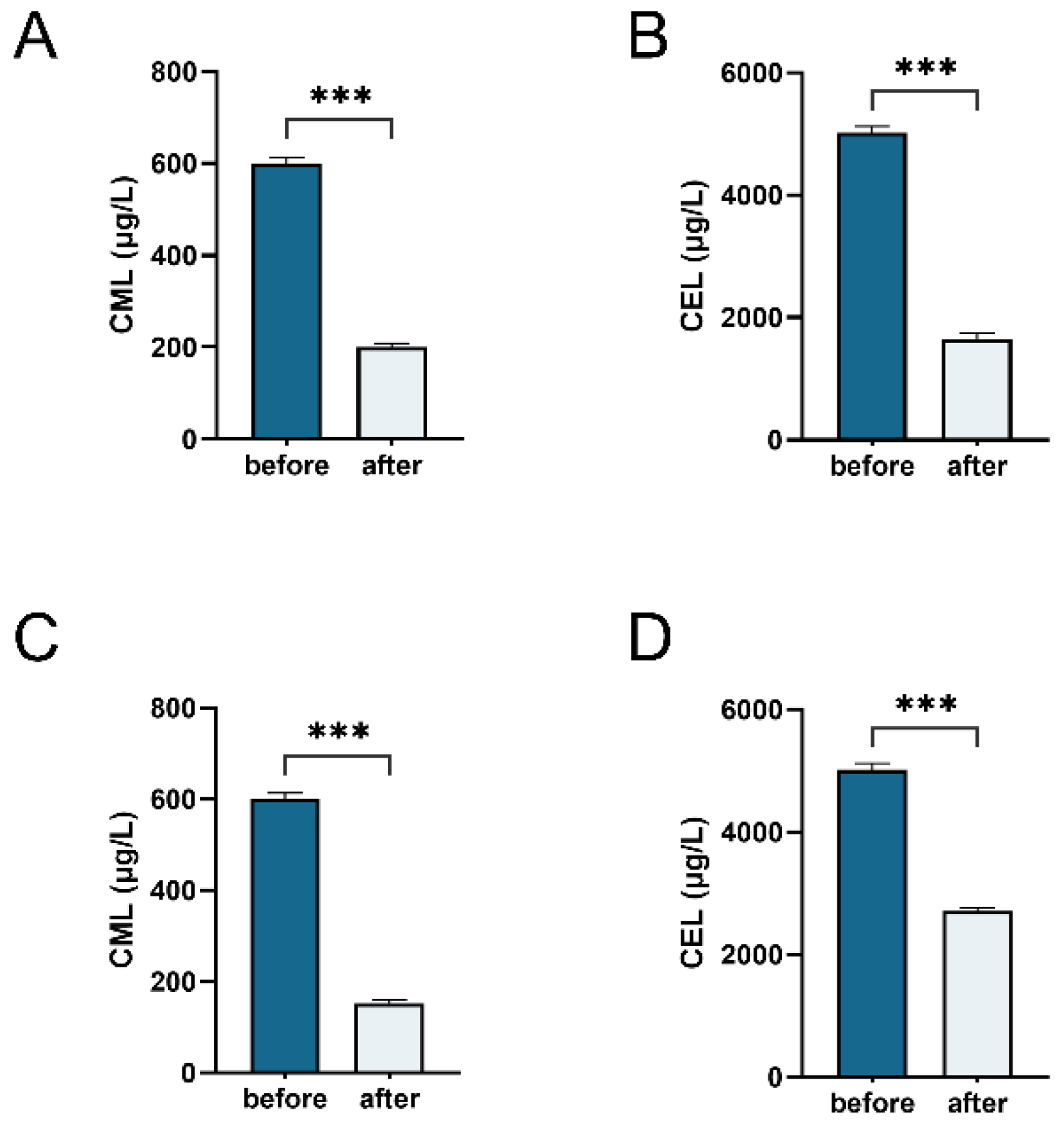
3.4. Degradation of CML and CEL during Gut Microbiota and Klebsiella Fermentation
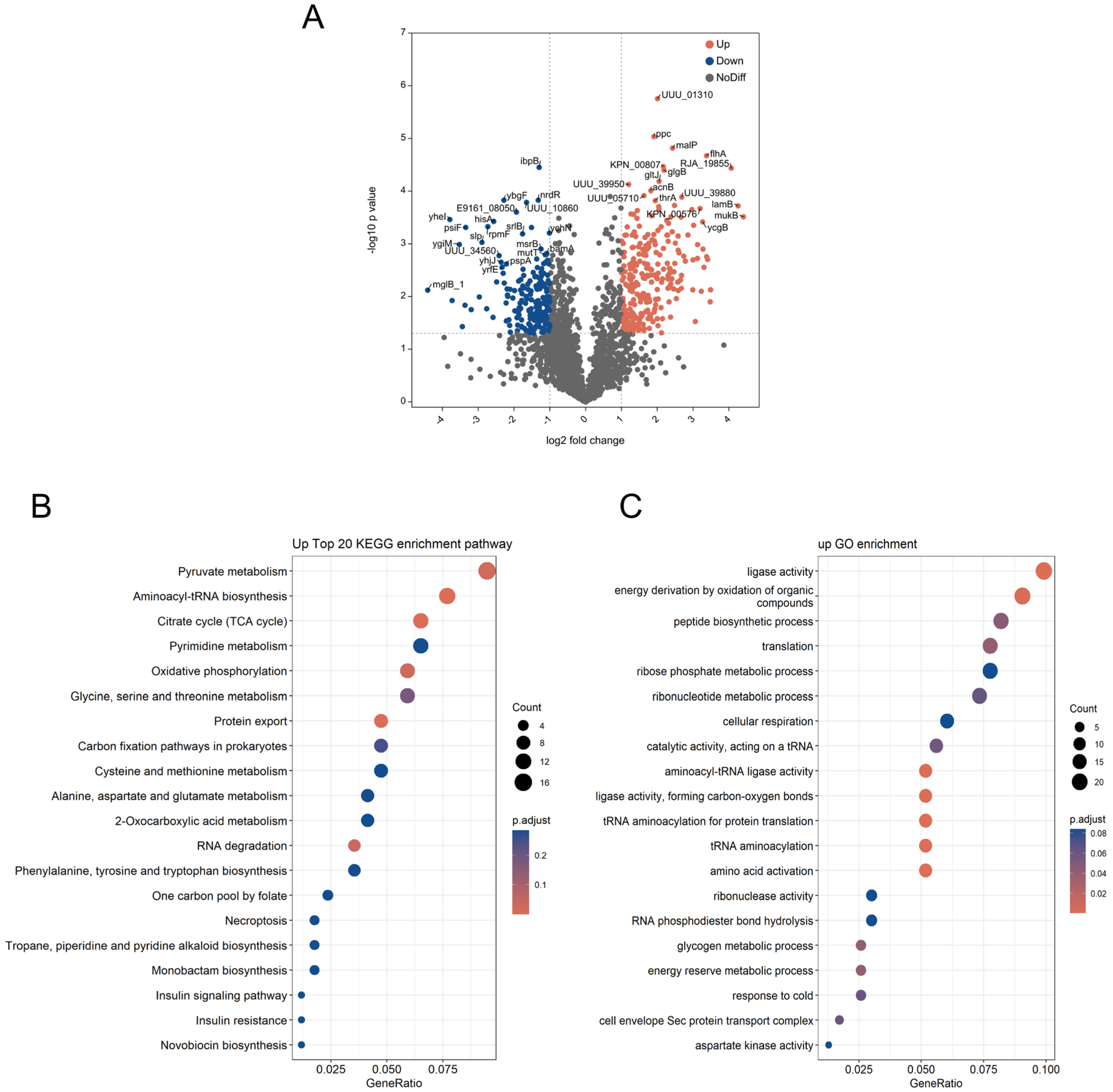
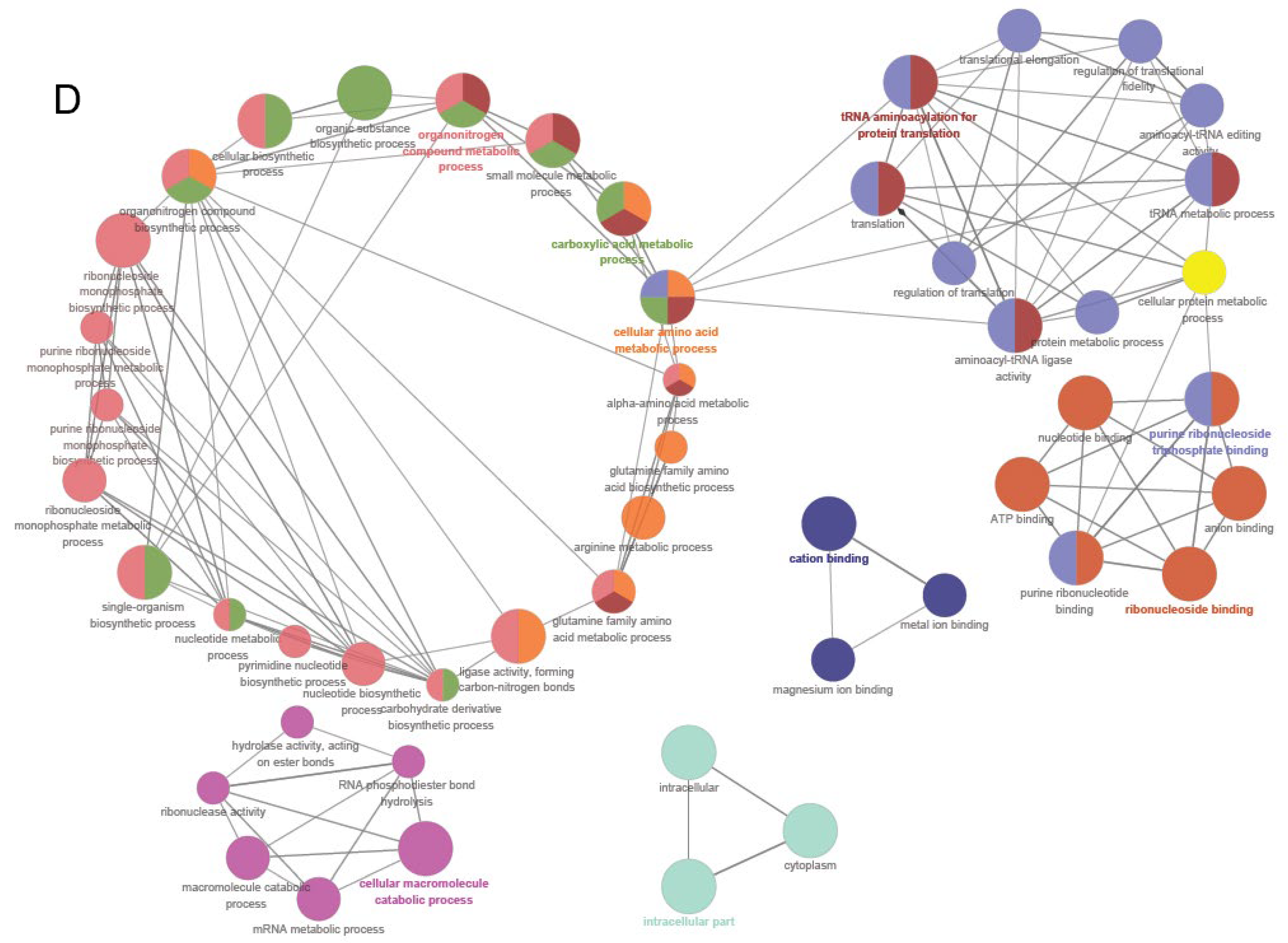
3.5. Proteomic Analysis of Klebsiella X15
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, H.; Gao, Y.; Zeng, C.; Hua, R.; Guo, Y.; Wang, Y.; Wang, Z. Effects of Maillard reaction and its product AGEs on aging and age-related diseases. Food Sci. Hum. Wellness 2024, 13, 1118–1134. [Google Scholar] [CrossRef]
- Yuan, X.; Nie, C.; Liu, H.; Ma, Q.; Peng, B.; Zhang, M.; Chen, Z.; Li, J. Comparison of metabolic fate, target organs, and microbiota interactions of free and bound dietary advanced glycation end products. Crit. Rev. Food Sci. Nutr. 2023, 63, 3612–3633. [Google Scholar] [CrossRef] [PubMed]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and progression of non-alcoholic fatty liver disease: The role of advanced glycation end products. Int. J. Mol. Sci. 2019, 20, 5037. [Google Scholar] [CrossRef] [PubMed]
- Parveen, A.; Sultana, R.; Lee, S.M.; Kim, T.H.; Kim, S.Y. Phytochemicals against anti-diabetic complications: Targeting the advanced glycation end product signaling pathway. Arch. Pharm. Res. 2021, 44, 378–401. [Google Scholar] [CrossRef]
- Prasad, K. AGE–RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell. Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef]
- Vasan, S.; Foiles, P.G.; Founds, H.W. Therapeutic potential of AGE inhibitors and breakers of AGE protein cross-links. Expert Opin. Investig. Drugs 2001, 10, 1977–1987. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.M. Determination of N epsilon-(carboxymethyl)lysine in foods and related systems. Ann. N. Y. Acad. Sci. 2008, 1126, 20–24. [Google Scholar] [CrossRef]
- Zhao, D.; Li, L.; Le, T.T.; Larsen, L.B.; Su, G.Y.; Liang, Y.; Li, B. Digestibility of Glyoxal-Glycated beta-Casein and beta-Lactoglobulin and Distribution of Peptide-Bound Advanced Glycation End Products in Gastrointestinal Digests. J. Agric. Food Chem. 2017, 65, 5778–5788. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, X.; Li, L.; Li, B.; Yang, Z. The fate of dietary advanced glycation end products in the body: From oral intake to excretion. Crit. Rev. Food Sci. Nutr. 2019, 60, 3475–3491. [Google Scholar] [CrossRef]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; María Pilar Navarro, C.D.-A. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef]
- Qu, W.; Nie, C.; Zhao, J.; Ou, X.; Zhang, Y.; Yang, S.; Bai, X.; Wang, Y.; Wang, J.; Li, J. Microbiome–Metabolomics Analysis of the Impacts of Long-Term Dietary Advanced-Glycation-End-Product Consumption on C57BL/6 Mouse Fecal Microbiota and Metabolites. J. Agric. Food Chem. 2018, 66, 8864–8875. [Google Scholar] [CrossRef]
- Shi, A.; Dong, L.; Wu, Y.; Liu, X.; Li, W.; Wang, J.; Ji, X.; Lv, H.; Zhang, Y.; Wang, S. Long-term ingestion of β-lactoglobulin-bound AGEs induces colonic inflammation by modulating RAGE (TLR4)/MYD88/NF-κB signaling pathway and gut microbiota in mice. J. Funct. Foods 2023, 107, 105690. [Google Scholar] [CrossRef]
- Hellwig, M.; Auerbach, C.; Müller, N.; Samuel, P.; Kammann, S.; Beer, F.; Gunzer, F.; Henle, T. Metabolization of the Advanced Glycation End Product N-ε-Carboxymethyllysine (CML) by Different Probiotic E. coli Strains. J. Agric. Food Chem. 2019, 67, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.P.N.; Troise, A.D.; Fogliano, V.; de Vos, W.M. Anaerobic Degradation of N-ε-Carboxymethyllysine, a Major Glycation End-Product, by Human Intestinal Bacteria. J. Agric. Food Chem. 2019, 67, 6594–6602. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wu, Y.; Wang, W.; Wu, Y.; Zhang, Y.; Wang, S. Structural modification and digestibility change of beta-lactoglobulin modified by methylglyoxal with the simulated reheating of dairy products. Food Chem. 2019, 288, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Brodkorb, A.; Egger, L.; Alminger, M.; Alvito, P.; Assuncao, R.; Ballance, S.; Bohn, T.; Bourlieu-Lacanal, C.; Boutrou, R.; Carriere, F.; et al. INFOGEST static in vitro simulation of gastrointestinal food digestion. Nat. Protoc. 2019, 14, 991–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S.; et al. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, D1522–D1527. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Hansen, R.; El-Omar, E.M.; Hold, G.L. IBD-what role do Proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B.; Wolters, M.; Weyh, C.; Kruger, K.; Ticinesi, A. The Effects of Lifestyle and Diet on Gut Microbiota Composition, Inflammation and Muscle Performance in Our Aging Society. Nutrients 2021, 13, 2045. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, S.H.; Lee, M.J.; Kim, B.K.; Song, W.J.; Park, H.W.; Cho, S.H.; Hong, S.J.; Chang, Y.S.; Kim, B.S. Different upper airway microbiome and their functional genes associated with asthma in young adults and elderly individuals. Allergy 2019, 74, 709–719. [Google Scholar] [CrossRef]
- Thorpe, H.A.; Booton, R.; Kallonen, T.; Gibbon, M.J.; Couto, N.; Passet, V.; Loez-Fernadez, S.; Rodrigues, C.; Matthews, L.; Mitchell, S.; et al. A large-scale genomic snapshot of Klebsiella spp. isolates in Northern Italy reveals limited transmission between clinical and non-clinical settings. Nat. Microbiol. 2022, 7, 2054–2067. [Google Scholar] [CrossRef] [PubMed]
- Dorman, M.J.; Short, F.L. GENOME WATCH Klebsiella pneumoniae: When a colonizer turns bad. Nat. Rev. Microbiol. 2017, 15, 384. [Google Scholar] [CrossRef]
- Saleem, A.F.; Qamar, F.N.; Shahzad, H.; Qadir, M.; Zaidi, A.K. Trends in antibiotic susceptibility and incidence of late-onset Klebsiella pneumoniae neonatal sepsis over a six-year period in a neonatal intensive care unit in Karachi, Pakistan. Int. J. Infect. Dis. 2013, 17, e961–e965. [Google Scholar] [CrossRef] [PubMed]
- Potgens, S.A.; Brossel, H.; Sboarina, M.; Catry, E.; Cani, P.D.; Neyrinck, A.M.; Delzenne, N.M.; Bindels, L.B. Klebsiella oxytoca expands in cancer cachexia and acts as a gut pathobiont contributing to intestinal dysfunction. Sci. Rep. 2018, 8, 12321. [Google Scholar] [CrossRef]
- Van Oudenhove, L.; Devreese, B. A review on recent developments in mass spectrometry instrumentation and quantitative tools advancing bacterial proteomics. Appl. Microbiol. Biotechnol. 2013, 97, 4749–4762. [Google Scholar] [CrossRef]
- Hui, S.; Silverman, J.M.; Chen, S.S.; Erickson, D.W.; Basan, M.; Wang, J.; Hwa, T.; Williamson, J.R. Quantitative proteomic analysis reveals a simple strategy of global resource allocation in bacteria. Mol. Syst. Biol. 2015, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Gokarn, R.R.; Eiteman, M.A.; Altman, E. Metabolic analysis of Escherichia coli in the presence and absence of the carboxylating enzymes phosphoenolpyruvate carboxylase and pyruvate carboxylase. Appl. Environ. Microbiol. 2000, 66, 1844–1850. [Google Scholar] [CrossRef]
- Patel, A.T.; Akhani, R.C.; Patel, M.J.; Dedania, S.R.; Patel, D.H. Bioproduction of L-Aspartic Acid and Cinnamic Acid by L-Aspartate Ammonia Lyase from Pseudomonas aeruginosa PAO1. Appl. Biochem. Biotechnol. 2017, 182, 792–803. [Google Scholar] [CrossRef]
- Huang, Y.-H.; You, W.-C.; Chen, Y.-J.; Ciou, J.-Y.; Hsieh, L.-S. Insight into the Substrate Specificity of Lactobacillus paracasei Aspartate Ammonia-Lyase. Fermentation 2023, 9, 49. [Google Scholar] [CrossRef]
- Jansen, R.S.; Mandyoli, L.; Hughes, R.; Wakabayashi, S.; Pinkham, J.T.; Selbach, B.; Guinn, K.M.; Rubin, E.J.; Sacchettini, J.C.; Rhee, K.Y. Aspartate aminotransferase Rv3722c governs aspartate-dependent nitrogen metabolism in Mycobacterium tuberculosis. Nat. Commun. 2020, 11, 1960. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification Sites | Types | Added MW (Da) | Added Composition |
|---|---|---|---|
| K | CML | 58.01 | C(2)O(2)H(2) |
| CEL | 72.02 | C(3)O(2)H(4) | |
| PYR | 108.02 | C(6)O(2)H(4) | |
| R | G-H | 39.99 | C(2)O(1) |
| MG-H | 54.01 | C(3)O(1)H(2) | |
| G-DH | 58.01 | C(2)O(2)H(2) | |
| Trios-H | 70.01 | C(3)O(2)H(2) | |
| MG-DH | 72.02 | C(3)O(3)H(4) | |
| RPYR | 80.03 | C(5)O(1)H(4) | |
| Trios-DH | 88.02 | C(3)O(3)H(4) | |
| DHP | 126.03 | C(6)O(3)H(6) | |
| THP | 144.04 | C(6)O(4)H(8) |
| Bacterial Genus and Species | No. of Isolates | Comments |
|---|---|---|
| Lactobacillus gasseri | 57 | From 72 h sample |
| Lactobacillus plantarum | 13 | From 72 h sample |
| Lactobacillus vaginalis | 13 | From 72 h sample |
| Enterococcus | 75 | From 8 h and 72 h sample |
| Escherichia coli | 17 | From 8 h sample |
| Klebsiella | 3 | From 8 h sample |
| Bacterium T15 | 4 | From 8 h sample |
| Bacillus cereus | 1 | From 72 h sample |
| Lactobacillus casei | 1 | From 72 h sample |
| Lactobacillus reuteri | 1 | From 72 h sample |
| Shigella flexneri | 1 | From 8 h sample |
| Escherichia fergusonii | 1 | From 8 h sample |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, A.; Ji, X.; Li, W.; Dong, L.; Wu, Y.; Zhang, Y.; Liu, X.; Zhang, Y.; Wang, S. The Interaction between Human Microbes and Advanced Glycation End Products: The Role of Klebsiella X15 on Advanced Glycation End Products’ Degradation. Nutrients 2024, 16, 754. https://doi.org/10.3390/nu16050754
Shi A, Ji X, Li W, Dong L, Wu Y, Zhang Y, Liu X, Zhang Y, Wang S. The Interaction between Human Microbes and Advanced Glycation End Products: The Role of Klebsiella X15 on Advanced Glycation End Products’ Degradation. Nutrients. 2024; 16(5):754. https://doi.org/10.3390/nu16050754
Chicago/Turabian StyleShi, Aiying, Xuemeng Ji, Wanhua Li, Lu Dong, Yuekun Wu, Yunhui Zhang, Xiaoxia Liu, Yan Zhang, and Shuo Wang. 2024. "The Interaction between Human Microbes and Advanced Glycation End Products: The Role of Klebsiella X15 on Advanced Glycation End Products’ Degradation" Nutrients 16, no. 5: 754. https://doi.org/10.3390/nu16050754
APA StyleShi, A., Ji, X., Li, W., Dong, L., Wu, Y., Zhang, Y., Liu, X., Zhang, Y., & Wang, S. (2024). The Interaction between Human Microbes and Advanced Glycation End Products: The Role of Klebsiella X15 on Advanced Glycation End Products’ Degradation. Nutrients, 16(5), 754. https://doi.org/10.3390/nu16050754