
Changes in Macronutrients during Dieting Lead to Weight Cycling and Metabolic Complications in Mouse Model
 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
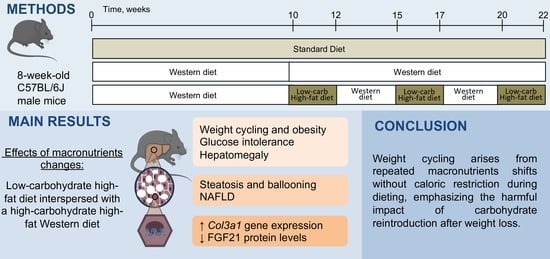
2.1. Animals and Experimental Design
2.2. Glucose Tolerance Test
2.3. Anatomical Measurements and Tissue Samples
2.4. Liver Histological Analysis
2.5. RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR
2.6. Western Blotting
2.7. Data Analyses
3. Results
3.1. Changes in Macronutrients during Dieting Induce a Weight-Cycling Pattern and Obesity
3.2. Changes in Macronutrients during Dieting Induces Glucose Intolerance and Dyslipidemia
3.3. Changes in Macronutrients during Dieting Induce Nonalcoholic Steatosis Development
3.4. An Intermittent Ketogenic Diet Does Not Activate the FGF21 Signaling Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kopp, W. How Western Diet and Lifestyle Drive the Pandemic of Obesity and Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [Google Scholar] [CrossRef]
- Jehan, S.; Zizi, F.; Pandi-Perumal, S.R.; McFarlane, S.I.; Jean-Louis, G.; Myers, A.K. Energy Imbalance: Obesity, Associated Comorbidities, Prevention, Management and Public Health Implications. Adv. Obes. Weight. Manag. Control 2020, 10, 146–161. [Google Scholar] [CrossRef]
- Johnston, B.C.; Kanters, S.; Bandayrel, K.; Wu, P.; Naji, F.; Siemieniuk, R.A.; Ball, G.D.C.; Busse, J.W.; Thorlund, K.; Guyatt, G.; et al. Comparison of Weight Loss among Named Diet Programs in Overweight and Obese Adults: A Meta-Analysis. JAMA 2014, 312, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Wing, R.R.; Phelan, S. Long-Term Weight Loss Maintenance. Am. J. Clin. Nutr. 2005, 82, 222S–225S. [Google Scholar] [CrossRef] [PubMed]
- Mackie, G.M.; Samocha-Bonet, D.; Tam, C.S. Does Weight Cycling Promote Obesity and Metabolic Risk Factors? Obes. Res. Clin. Pract. 2017, 11, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kroke, A.; Liese, A.; Schulz, M.; Bergmann, M.; Klipstein-Grobusch, K.; Hoffmann, K.; Boeing, H. Recent Weight Changes and Weight Cycling as Predictors of Subsequent Two Year Weight Change in a Middle-Aged Cohort. Int. J. Obes. 2002, 26, 403–409. [Google Scholar] [CrossRef]
- Wallner, S.J.; Luschnigg, N.; Schnedl, W.J.; Lahousen, T.; Sudi, K.; Crailsheim, K.; Möller, R.; Tafeit, E.; Horejsi, R. Body Fat Distribution of Overweight Females with a History of Weight Cycling. Int. J. Obes. 2004, 28, 1143–1148. [Google Scholar] [CrossRef]
- Delahanty, L.M.; Pan, Q.; Jablonski, K.A.; Aroda, V.R.; Watson, K.E.; Bray, G.A.; Kahn, S.E.; Florez, J.C.; Perreault, L.; Franks, P.W. Effects of Weight Loss, Weight Cycling, and Weight Loss Maintenance on Diabetes Incidence and Change in Cardiometabolic Traits in the Diabetes Prevention Program. Diabetes Care 2014, 37, 2738–2745. [Google Scholar] [CrossRef]
- Neamat-Allah, J.; Barrdahl, M.; Hüsing, A.; Katzke, V.A.; Bachlechner, U.; Steffen, A.; Kaaks, R.; Schulze, M.B.; Boeing, H.; Kühn, T. Weight Cycling and the Risk of Type 2 Diabetes in the EPIC-Germany Cohort. Diabetologia 2015, 58, 2718–2725. [Google Scholar] [CrossRef][Green Version]
- Jung, I.; Koo, D.-J.; Lee, M.Y.; Moon, S.J.; Kwon, H.; Park, S.E.; Rhee, E.-J.; Lee, W.-Y. Increased Risk of Nonalcoholic Fatty Liver Disease in Individuals with High Weight Variability. Endocrinol. Metab. 2021, 36, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Vergnaud, A.-C.; Bertrais, S.; Oppert, J.-M.; Maillard-Teyssier, L.; Galan, P.; Hercberg, S.; Czernichow, S. Weight Fluctuations and Risk for Metabolic Syndrome in an Adult Cohort. Int. J. Obes. 2008, 32, 315–321. [Google Scholar] [CrossRef]
- Kim, M.N.; Han, K.; Yoo, J.; Ha, Y.; Chon, Y.E.; Lee, J.H.; Simon, T.G.; Chan, A.T.; Hwang, S.G. Body Weight Variability and the Risk of Cardiovascular Outcomes in Patients with Nonalcoholic Fatty Liver Disease. Sci. Rep. 2021, 11, 9154. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-da-Silva, S.; Da Silva, N.C.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Liver Damage Is Not Reversed during the Lean Period in Diet-induced Weight Cycling in Mice. Hepatol. Res. 2014, 44, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Sea, M.-M.; Fong, W.P.; Huang, Y.; Chen, Z.-Y. Weight Cycling-Induced Alteration in Fatty Acid Metabolism. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2000, 279, R1145–R1155. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, A.J.; Spahn, K.; Zhou, P.; Goldberg, I.D.; Narayan, P. Collagen Characterization in a Model of Nonalcoholic Steatohepatitis with Fibrosis; A Call for Development of Targeted Therapeutics. Molecules 2021, 26, 3316. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Steensels, S.; Qiao, J.; Ersoy, B.A. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites 2020, 10, 283. [Google Scholar] [CrossRef]
- Alajmi, N.; Deighton, K.; King, J.A.; Reischak-Oliveira, A.; Wasse, L.K.; Jones, J.; Batterham, R.L.; Stensel, D.J. Appetite and Energy Intake Responses to Acute Energy Deficits in Females versus Males. Med. Sci. Sports Exerc. 2016, 48, 412–420. [Google Scholar] [CrossRef]
- Burger, K.S.; Stice, E. Relation of Dietary Restraint Scores to Activation of Reward-Related Brain Regions in Response to Food Intake, Anticipated Intake, and Food Pictures. NeuroImage 2011, 55, 233–239. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Frayo, R.S.; Melhorn, S.J.; Cummings, D.E.; Schur, E.A. Effects of Multiple Cycles of Weight Loss and Regain on the Body Weight Regulatory System in Rats. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E863–E870. [Google Scholar] [CrossRef]
- MacLean, P.S.; Higgins, J.A.; Johnson, G.C.; Fleming-Elder, B.K.; Peters, J.C.; Hill, J.O. Metabolic Adjustments with the Development, Treatment, and Recurrence of Obesity in Obesity-Prone Rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 287, R288–R297. [Google Scholar] [CrossRef] [PubMed]
- Melby, C.; Schmidt, W.; Corrigan, D. Resting Metabolic Rate in Weight-Cycling Collegiate Wrestlers Compared with Physically Active, Noncycling Control Subjects. Am. J. Clin. Nutr. 1990, 52, 409–414. [Google Scholar] [CrossRef]
- Goldsmith, R.; Joanisse, D.R.; Gallagher, D.; Pavlovich, K.; Shamoon, E.; Leibel, R.L.; Rosenbaum, M. Effects of Experimental Weight Perturbation on Skeletal Muscle Work Efficiency, Fuel Utilization, and Biochemistry in Human Subjects. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R79–R88. [Google Scholar] [CrossRef]
- Doucet, E.; St-Pierre, S.; Alméras, N.; Després, J.-P.; Bouchard, C.; Tremblay, A. Evidence for the Existence of Adaptive Thermogenesis during Weight Loss. Br. J. Nutr. 2001, 85, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Camps, S.G.; Verhoef, S.P.; Westerterp, K.R. Weight Loss, Weight Maintenance, and Adaptive Thermogenesis. Am. J. Clin. Nutr. 2013, 97, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, E.; Guo, J.; Howard, L.; Kerns, J.C.; Knuth, N.D.; Brychta, R.; Chen, K.Y.; Skarulis, M.C.; Walter, M.; Walter, P.J.; et al. Persistent Metabolic Adaptation 6 Years after “The Biggest Loser” Competition: Persistent Metabolic Adaptation. Obesity 2016, 24, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.-F.; Kahn, B.B.; Libermann, T.A.; et al. A High-Fat, Ketogenic Diet Induces a Unique Metabolic State in Mice. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef] [PubMed]
- Nasser, S.; Solé, T.; Vega, N.; Thomas, T.; Balcerczyk, A.; Strigini, M.; Pirola, L. Ketogenic Diet Administration to Mice after a High-Fat-Diet Regimen Promotes Weight Loss, Glycemic Normalization and Induces Adaptations of Ketogenic Pathways in Liver and Kidney. Mol. Metab. 2022, 65, 101578. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Medak, K.D.; Townsend, L.K.; Wright, D.C. Ketogenic Diet-induced Weight Loss Occurs Independent of Housing Temperature and Is Followed by Hyperphagia and Weight Regain after Cessation in Mice. J. Physiol. 2022, 600, 4677–4693. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, S.J.; McKenzie, A.L.; Williams, P.T.; Bhanpuri, N.H.; Peters, A.L.; Campbell, W.W.; Hazbun, T.L.; Volk, B.M.; McCarter, J.P.; Phinney, S.D.; et al. Effectiveness and Safety of a Novel Care Model for the Management of Type 2 Diabetes at 1 Year: An Open-Label, Non-Randomized, Controlled Study. Diabetes Ther. 2018, 9, 583–612. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of Low-Calorie versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- McKenzie, A.L.; Hallberg, S.J.; Creighton, B.C.; Volk, B.M.; Link, T.M.; Abner, M.K.; Glon, R.M.; McCarter, J.P.; Volek, J.S.; Phinney, S.D. A Novel Intervention Including Individualized Nutritional Recommendations Reduces Hemoglobin A1c Level, Medication Use, and Weight in Type 2 Diabetes. JMIR Diabetes 2017, 2, e5. [Google Scholar] [CrossRef]
- Schiavo, L.; Pilone, V.; Rossetti, G.; Barbarisi, A.; Cesaretti, M.; Iannelli, A. A 4-Week Preoperative Ketogenic Micronutrient-Enriched Diet Is Effective in Reducing Body Weight, Left Hepatic Lobe Volume, and Micronutrient Deficiencies in Patients Undergoing Bariatric Surgery: A Prospective Pilot Study. Obes. Surg. 2018, 28, 2215–2224. [Google Scholar] [CrossRef]
- Walton, C.M.; Perry, K.; Hart, R.H.; Berry, S.L.; Bikman, B.T. Improvement in Glycemic and Lipid Profiles in Type 2 Diabetics with a 90-Day Ketogenic Diet. J. Diabetes Res. 2019, 2019, 8681959. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. A Modern Approach to Dyslipidemia. Endocr. Rev. 2022, 43, 611–653. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Wang, H.; Pan, X.; Little, P.J.; Xu, S.; Weng, J. Mouse Models of Nonalcoholic Fatty Liver Disease (NAFLD): Pathomechanisms and Pharmacotherapies. Int. J. Biol. Sci. 2022, 18, 5681–5697. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Ebbeling, C.B. The Carbohydrate-Insulin Model of Obesity: Beyond “Calories In, Calories Out”. JAMA Intern. Med. 2018, 178, 1098. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Apovian, C.M.; Aronne, L.J.; Astrup, A.; Cantley, L.C.; Ebbeling, C.B.; Heymsfield, S.B.; Johnson, J.D.; King, J.C.; Krauss, R.M.; et al. Competing Paradigms of Obesity Pathogenesis: Energy Balance versus Carbohydrate-Insulin Models. Eur. J. Clin. Nutr. 2022, 76, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Opazo-Ríos, L.; Soto-Catalán, M.; Lázaro, I.; Sala-Vila, A.; Jiménez-Castilla, L.; Orejudo, M.; Moreno, J.A.; Egido, J.; Mas-Fontao, S. Meta-Inflammation and De Novo Lipogenesis Markers Are Involved in Metabolic Associated Fatty Liver Disease Progression in BTBR Ob/Ob Mice. Int. J. Mol. Sci. 2022, 23, 3965. [Google Scholar] [CrossRef]
- Matsumoto, K.; Kato, Y.; Hayashi, M.; Miura, R.; Monzen, S.; Chiba, M. Hepatic Gene Expression and Functional Changes Associated with Nonalcoholic Steatohepatitis. Mol. Med. Rep. 2022, 26, 325. [Google Scholar] [CrossRef]
- Okuda, T.; Morita, N. A Very Low Carbohydrate Ketogenic Diet Prevents the Progression of Hepatic Steatosis Caused by Hyperglycemia in a Juvenile Obese Mouse Model. Nutr. Diabetes 2012, 2, e50. [Google Scholar] [CrossRef]
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef]
- Sougiannis, A.T.; VanderVeen, B.N.; Cranford, T.L.; Enos, R.T.; Velazquez, K.T.; McDonald, S.; Bader, J.E.; Chatzistamou, I.; Fan, D.; Murphy, E.A. Impact of Weight Loss and Partial Weight Regain on Immune Cell and Inflammatory Markers in Adipose Tissue in Male Mice. J. Appl. Physiol. 2020, 129, 909–919. [Google Scholar] [CrossRef]
- Chiang, C.; Zhang, T.-R.; Hsu, P.; Lin, S.; Chen, C. Weight Regain, but Not Weight Loss Exacerbates Hepatic Fibrosis during Multiple Weight Cycling Events in Male Mice. Eur. J. Nutr. 2024. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.-M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de Novo Lipogenesis in Normoinsulinemic and Hyperinsulinemic Subjects Consuming High-Fat, Low-Carbohydrate and Low-Fat, High-Carbohydrate Isoenergetic Diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Williams, L.M.; Campbell, F.M.; Drew, J.E.; Koch, C.; Hoggard, N.; Rees, W.D.; Kamolrat, T.; Thi Ngo, H.; Steffensen, I.-L.; Gray, S.R.; et al. The Development of Diet-Induced Obesity and Glucose Intolerance in C57Bl/6 Mice on a High-Fat Diet Consists of Distinct Phases. PLoS ONE 2014, 9, e106159. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. In Comprehensive Physiology; Terjung, R., Ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 1–22. [Google Scholar] [CrossRef]
- Kuipers, E.N.; Held, N.M.; In Het Panhuis, W.; Modder, M.; Ruppert, P.M.M.; Kersten, S.; Kooijman, S.; Guigas, B.; Houtkooper, R.H.; Rensen, P.C.N.; et al. A Single Day of High-Fat Diet Feeding Induces Lipid Accumulation and Insulin Resistance in Brown Adipose Tissue in Mice. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E820–E830. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. A Role for Brown Adipose Tissue in Diet-Induced Thermogenesis. Nature 1979, 281, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, A.G.; Calokatisa, R. Adaptation to Low Calorie Intake in Obese Mice: Contribution of a Metabolic Component to Diminished Energy Expenditures during and after Weight Loss. Int. J. Obes. 1991, 15, 7–16. [Google Scholar]
- Tremblay, A.; Chaput, J.-P. Adaptive Reduction in Thermogenesis and Resistance to Lose Fat in Obese Men. Br. J. Nutr. 2009, 102, 488. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, A.; Girardier, L. Adaptive Changes in Energy Expenditure during Refeeding Following Low-Calorie Intake: Evidence for a Specific Metabolic Component Favoring Fat Storage. Am. J. Clin. Nutr. 1990, 52, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Flier, J.S.; Lowell, B.B. Decreased Brown Fat Markedly Enhances Susceptibility to Diet-Induced Obesity, Diabetes, and Hyperlipidemia. Endocrinology 1996, 137, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Badman, M.K.; Koester, A.; Flier, J.S.; Kharitonenkov, A.; Maratos-Flier, E. Fibroblast Growth Factor 21-Deficient Mice Demonstrate Impaired Adaptation to Ketosis. Endocrinology 2009, 150, 4931–4940. [Google Scholar] [CrossRef]
- Watanabe, M.; Singhal, G.; Fisher, F.M.; Beck, T.C.; Morgan, D.A.; Socciarelli, F.; Mather, M.L.; Risi, R.; Bourke, J.; Rahmouni, K.; et al. Liver-Derived FGF21 Is Essential for Full Adaptation to Ketogenic Diet but Does Not Regulate Glucose Homeostasis. Endocrine 2020, 67, 95–108. [Google Scholar] [CrossRef]
- Smethers, A.D.; Rolls, B.J. Dietary Management of Obesity. Med. Clin. N. Am. 2018, 102, 107–124. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Hprt | GTTGGATACAGGCCAGACTTTGTTG | GATTCAACTTGCGCTCATCTTAGGC |
| Acadl | GAAGATGTCCGATTGCCAGC | AGTTTATGCTGCACCGTCTGT |
| Col1a1 | GACCGTTGAGTCCGTCTTTG | TCATCGTGGCTTCTCTGGCT |
| Col3a1 | CTGGCCCTCCTGGTGCTTCT | CCTTGGCCCATCCTTTCCTG |
| Esch1 | GCAAAGCAGGCAGGTCTTGT | TAGCTGCCAGTTCTCAGTGG |
| Hadh | TCGTGAACCGACTCTTGGTG | ATTTCATGCCACCCGTCCAA |
| Fasn | TGCACCTCACAGGCATCAAT | GTCCCACTTGATGTGAGGGG |
| Fgf21 | GTGTCAAAGCCTCTAGGTTTCTT | GGTACACATTGTAACCGTCCTC |
| Fgfr1 | GCCAGACAACTTGCCGTATG | ATTTCCTTGTCGGTGGTATTAACT |
| Klb | GGACACAACCTGATCAAGGCAC | GAGAACTCGGGGATCATGGC |
| Tgf β | ACGTGGAAATCAACGGGATCA | GTTGGTATCCAGGGCTCTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charlot, A.; Bringolf, A.; Debrut, L.; Mallard, J.; Charles, A.-L.; Crouchet, E.; Duteil, D.; Geny, B.; Zoll, J. Changes in Macronutrients during Dieting Lead to Weight Cycling and Metabolic Complications in Mouse Model. Nutrients 2024, 16, 646. https://doi.org/10.3390/nu16050646
Charlot A, Bringolf A, Debrut L, Mallard J, Charles A-L, Crouchet E, Duteil D, Geny B, Zoll J. Changes in Macronutrients during Dieting Lead to Weight Cycling and Metabolic Complications in Mouse Model. Nutrients. 2024; 16(5):646. https://doi.org/10.3390/nu16050646
Chicago/Turabian StyleCharlot, Anouk, Anthony Bringolf, Léa Debrut, Joris Mallard, Anne-Laure Charles, Emilie Crouchet, Delphine Duteil, Bernard Geny, and Joffrey Zoll. 2024. "Changes in Macronutrients during Dieting Lead to Weight Cycling and Metabolic Complications in Mouse Model" Nutrients 16, no. 5: 646. https://doi.org/10.3390/nu16050646
APA StyleCharlot, A., Bringolf, A., Debrut, L., Mallard, J., Charles, A.-L., Crouchet, E., Duteil, D., Geny, B., & Zoll, J. (2024). Changes in Macronutrients during Dieting Lead to Weight Cycling and Metabolic Complications in Mouse Model. Nutrients, 16(5), 646. https://doi.org/10.3390/nu16050646