
Ketogenic Interventions in Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review of Current Evidence




Abstract
1. Introduction
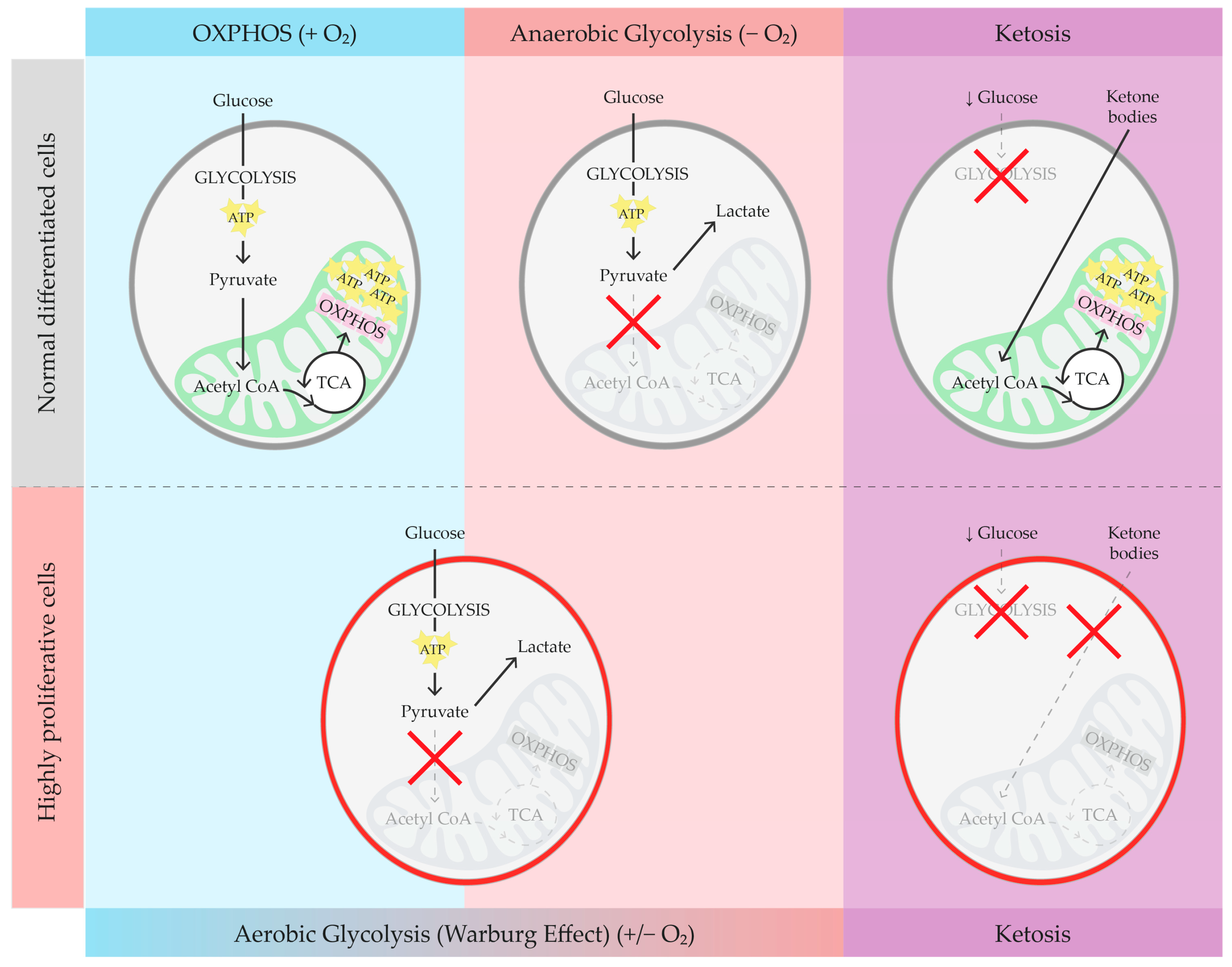
2. Brief Overview of Glucose Metabolism, Warburg Effect, and Ketosis
3. Ketosis in ADPKD Animal Models

{kind=link}
| Intervention and Study Design | Model (Orthologous: ✓/✕) | Control Group | Kidney Outcomes | Notes | |
|---|---|---|---|---|---|
| Fasting | 48 h WF [47] | ✕ Han:SPRD-Cy/+ rats aged 8 wk | 48 h WF WT | KHW ↓ * (male). CI ↓ *. Cysts apoptosis. | NK achieved. ↓ BW (both groups). |
| 24 h WF [47] | ✓ Pkd1cond/cond:NesCre mice aged 8 wk | 24 h WF WT | No TKV changes. Cysts apoptosis. | NK achieved. | |
| 72 h WF [47] | ✓ Pkd1 Persian cat aged 34.2–64.9 mo | N/A | TKV ↓ (~15%) | No NK evaluation. ↓ BW. | |
| DCR | 6 mo DCR (−40%) [45] | ✓ Pkd1RC/RC miceaged 6 wk | AL model AL WT | KHW, CI, IF, cystogenesis ↓ *. | No NK evaluation. ↓ * BW. |
| 3 mo DCR (−40%) [45] | ✓ Pkd1RC/RC miceaged 5.5 mo | AL model AL WT | KHW ↓ *. Improvement in BUN and CysC. | No NK evaluation. | |
| 2 mo DCR (−10%, −20%, −40%) [45] | ✓ Pkd1RC/RC miceaged 5.5 mo | AL model | KHW ↓ *. CI ↓ (↓ * 20%, 40%). | No NK evaluation. ↓ BW. | |
| 2 mo DCR (−40%) [45] | ✓ Pkd2WS25/- mice aged 8 wk | AL model | KHW, CI ↓ * | No NK evaluation. | |
| 12 wk DCR (−23%) [46] | ✓ Pkd1cond/cond:NesCre mice aged 35 days | AL model DCR WT | KBW ↓ *. IF ↓ | No NK evaluation. | |
| TRF | 5 wk TRF (8 h window) [47] | ✕ Han:SPRD-Cy/+ rats aged 3–8 wk | AL NC model TRF WT AL NC WT | KBW, CI, cystogenesis ↓ *. IF, SC ↓. | NK achieved. |
| DCR IMF TRF | 3 mo DCR (−40%), IMF (−80% 3 days/wk), TRF (8 h window) [53] | ✓ Pkd1RC/RC miceaged 3 mo | AL model | KBW, CI ↓ * (DCR). | ↓ * BW (DCR). |
| KD | 5 wk AL cKD [47] | ✕ Han:SPRD-Cy/+ rats aged 3–8 wk | AL NC model AL KD WT AL NC WT | KBW, KHW, CI, cystogenesis, SC ↓ *. IF ↓. | NK achieved. ↓ * BW gain. |
| 4 wk AL cKD [47] | ✕ Han:SPRD-Cy/+ rats aged 8–12 wk | AL NC model AL KD WT AL NC WT | TKV, KBW, CI ↓. IF ↓ (male). | NK achieved. ↓ BW gain. | |
| Exogenous ketosis | 5 wk Na and K BHB salt in water [47] | ✕ Han:SPRD-Cy/+ rats aged 3–8 wk | Water model Water + Na/KCl model | KBW, CI, IF ↓ *. IF ↓ *. SC, cystogenesis ↓. | No BW changes. |
| 5 wk Na and K BHB salt and/or citrate [52] | ✕ Han:SPRD-Cy/+ rats aged 3–8 wk | Water model Analogous titrations WT | KBW, SC ↓ * (BHB, BHB/citrate). CI ↓ * (80, 160 Mm BHB; 40/60, 80/60 Mm BB/citrate). | ||
| 5 wk Na and K BHB salt and citrate [52] | ✕ Han:SPRD-Cy/+ rats aged 8–12 wk | Water + Na/KCl model | KBW, CI ↓. SC ↓ *. | ||
4. Ketosis in ADPKD Patients
5. Focus on RCTs in ADPKD Patients
6. Kidney Disease, Ketosis, and Microbiota
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cordido, A.; Besada-Cerecedo, L.; García-González, M.A. The Genetic and Cellular Basis of Autosomal Dominant Polycystic Kidney Disease—A Primer for Clinicians. Front. Pediatr. 2017, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Willey, C.J.; Blais, J.D.; Hall, A.K.; Krasa, H.B.; Makin, A.J.; Czerwiec, F.S. Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol. Dial. Transplant. 2017, 32, 1356–1363. [Google Scholar] [CrossRef]
- Solazzo, A.; Testa, F.; Giovanella, S.; Busutti, M.; Furci, L.; Carrera, P.; Ferrari, M.; Ligabue, G.; Mori, G.; Leonelli, M.; et al. The prevalence of autosomal dominant polycystic kidney disease (ADPKD): A meta-analysis of European literature and prevalence evaluation in the Italian province of Modena suggest that ADPKD is a rare and underdiagnosed condition. PLoS ONE 2018, 13, e0190430. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Magistroni, R.; Boletta, A. Defective glycolysis and the use of 2-deoxy-d-glucose in polycystic kidney disease: From animal models to humans. J. Nephrol. 2017, 30, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.A.; Boletta, A. Role of the polycystins as mechanosensors of extracellular stiffness. Am. J. Physiol. Physiol. 2021, 320, F693–F705. [Google Scholar] [CrossRef] [PubMed]
- Astley, M.E.; Boenink, R.; Abd ElHafeez, S.; Trujillo-Alemán, S.; Arribas, F.; Åsberg, A.; Beckerman, P.; Bell, S.; Bouzas-Caamaño, M.E.; Farnés, J.C.; et al. The ERA Registry Annual Report 2020: A summary. Clin. Kidney J. 2023, 16, 1330–1354. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; Arici, M.; Benzing, T.; Birn, H.; Capasso, G.; Covic, A.; Devuyst, O.; Drechsler, C.; Eckardt, K.U.; Emma, F.; et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: A position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol. Dial. Transplant. 2016, 31, 337–348. [Google Scholar] [CrossRef]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef]
- Rowe, I.; Boletta, A. Defective metabolism in polycystic kidney disease: Potential for therapy and open questions. Nephrol. Dial. Transplant. 2014, 29, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Kang, J.H.; Sim, I.A.; Seong, D.Y.; Han, S.; Jang, H.; Lee, H.; Kang, S.W.; Kim, S.Y. Glucose Deprivation Induces Cancer Cell Death through Failure of ROS Regulation. Int. J. Mol. Sci. 2023, 24, 11969. [Google Scholar] [CrossRef]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell. Signal. 2019, 67, 109495. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK493179/ (accessed on 2 April 2024).
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F. Fuel Metabolism in Starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Volek, J.; Phinney, S.D. The Art and Science of Low Carbohydrate Living: An Expert Guide to Making the Life-Saving Benefits of Carbohydrate Restriction Sustainable and Enjoyable; Beyond Obesity LLC: Sacramento, CA, USA, 2011. [Google Scholar]
- Norgren, J.; Sindi, S.; Sandebring-Matton, A.; Kåreholt, I.; Akenine, U.; Nordin, K.; Rosenborg, S.; Ngandu, T.; Kivipelto, M. Capillary blood tests may overestimate ketosis: Triangulation between three different measures of β-hydroxybutyrate. Am. J. Physiol. Metab. 2020, 318, E184–E188. [Google Scholar] [CrossRef]
- Volk, B.M.; Kunces, L.J.; Freidenreich, D.J.; Kupchak, B.R.; Saenz, C.; Artistizabal, J.C.; Fernandez, M.L.; Bruno, R.S.; Maresh, C.M.; Kraemer, W.J.; et al. Effects of Step-Wise Increases in Dietary Carbohydrate on Circulating Saturated Fatty Acids and Palmitoleic Acid in Adults with Metabolic Syndrome. PLoS ONE 2014, 9, e113605. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Zhang, W.; Gao, X.; Watts, L. Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol. Aging 2015, 36, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Mezhnina, V.; Ebeigbe, O.P.; Velingkaar, N.; Poe, A.; Sandlers, Y.; Kondratov, R.V. Circadian clock controls rhythms in ketogenesis by interfering with PPARα transcriptional network. Proc. Natl. Acad. Sci. USA 2022, 119, e2205755119. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Parker, D.C.; Bhapkar, M.; Racette, S.B.; Martin, C.K.; Redman, L.M.; Das, S.K.; Connelly, M.A.; Pieper, C.F.; Orenduff, M.; et al. Calorie restriction improves lipid-related emerging cardiometabolic risk factors in healthy adults without obesity: Distinct influences of BMI and sex from CALERIETM a multicentre, phase 2, randomised controlled trial. eClinicalMedicine 2022, 43, 101261. [Google Scholar] [CrossRef] [PubMed]
- Cabo, R.; de Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Song, D.K.; Kim, Y.W. Beneficial effects of intermittent fasting: A narrative review. J. Yeungnam Med. Sci. 2022, 40, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.L.; Yao, W.; Wang, X.Y.; Gao, S.; Varady, K.A.; Forslund, S.K.; Zhang, M.; Shi, Z.Y.; Cao, F.; Zou, B.J.; et al. Intermittent fasting and health outcomes: An umbrella review of systematic reviews and meta-analyses of randomised controlled trials. eClinicalMedicine 2024, 70, 102519. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Allison, D.B.; Fontana, L.; Harvie, M.; Longo, V.D.; Malaisse, W.J.; Mosley, M.; Notterpek, L.; Ravussin, E.; Scheer, F.A.J.L.; et al. Meal frequency and timing in health and disease. Proc. Natl. Acad. Sci. USA 2014, 111, 16647–16653. [Google Scholar] [CrossRef]
- deCampo, D.M.; Kossoff, E.H. Ketogenic dietary therapies for epilepsy and beyond. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 264. [Google Scholar] [CrossRef]
- Armeno, M.L.; Kossoff, E.H. Let food be thy medicine. The interaction between ketogenic diet therapy and anti-seizure medications: A systematic review. Epileptic Disord. 2023, 25, 18–27. [Google Scholar] [CrossRef]
- Cervenka, M.C.; Wood, S.; Bagary, M.; Balabanov, A.; Bercovici, E.; Brown, M.G.; Devinsky, O.; Di Lorenzo, C.; Doherty, C.P.; Felton, E.; et al. International Recommendations for the Management of Adults Treated with Ketogenic Diet Therapies. Neurol. Clin. Pr. 2020, 11, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Lee, H.S.; You, S.J.; Kang, D.C.; Ko, T.S.; Kim, H.D. Use of a Modified Atkins Diet in Intractable Childhood Epilepsy. Epilepsia 2007, 48, 182–186. [Google Scholar] [CrossRef]
- Kossoff, E.H. The Modified Atkins Diet for Epilepsy: Two Decades of an “Alternative” Ketogenic Diet Therapy. Pediatr. Neurol. 2023, 147, 82–87. [Google Scholar] [CrossRef]
- Caprio, M.; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; Migliaccio, S.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; El Ghoch, M.; Colao, A.; Hassapidou, M.; Yumuk, V.; Busetto, L. European Guidelines for Obesity Management in Adults with a Very Low-Calorie Ketogenic Diet: A Systematic Review and Meta-Analysis. Obes. Facts 2021, 14, 222–245. [Google Scholar] [CrossRef]
- Masood, W.; Annamaraju, P.; Khan Suheb, M.Z.; Uppaluri, K.R. Ketogenic Diet; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK499830/ (accessed on 2 April 2024).
- Galali, Y.; Zebari, S.M.S.; Jabbar, A.A.; Balaky, H.H.; Sadee, B.A.; Hassanzadeh, H. The impact of ketogenic diet on some metabolic and non-metabolic diseases: Evidence from human and animal model experiments. Food Sci. Nutr. 2024, 12, 1444–1464. [Google Scholar] [CrossRef]
- Falkenhain, K.; Islam, H.; Little, J.P. Exogenous ketone supplementation: An emerging tool for physiologists with potential as a metabolic therapy. Exp. Physiol. 2022, 108, 177–187. [Google Scholar] [CrossRef]
- Lin, T.Y.; Liu, H.W.; Hung, T.M. The Ketogenic Effect of Medium-Chain Triacylglycerides. Front. Nutr. 2021, 8, 747284. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, B.J.; Cox, P.J.; Kirk, T.; Evans, R.D.; Clarke, K. Gastrointestinal Effects of Exogenous Ketone Drinks are Infrequent, Mild, and Vary According to Ketone Compound and Dose. Int. J. Sport Nutr. Exerc. Metab. 2019, 29, 596–603. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Poff, A.M.; Koutnik, A.P.; Egan, B. Nutritional Ketosis with Ketogenic Diets or Exogenous Ketones: Features, Convergence, and Divergence. Curr. Sports Med. Rep. 2020, 19, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.S.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2015, 27, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Renal. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.e5. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Shillingford, J.M.; Torres, J.; Kruger, S.L.; Bourgeois, B.C. Emerging targeted strategies for the treatment of autosomal dominant polycystic kidney disease. Clin. Kidney J. 2018, 11 (Suppl. S1), i27–i38. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Holznecht, N.; Asplund, D.A.; Amarlkhagva, T.; Kroes, B.C.; Rebello, J.; Agrawal, S.; Weimbs, T. A combination of β-hydroxybutyrate and citrate ameliorates disease progression in a rat model of polycystic kidney disease. Am. J. Physiol. Physiol. 2024, 326, F352–F368. [Google Scholar] [CrossRef]
- Hopp, K.; Catenacci, V.A.; Dwivedi, N.; Kline, T.L.; Wang, W.; You, Z.; Nguyen, D.T.; Bing, K.; Poudyal, B.; Johnson, G.C.; et al. Weight loss and cystic disease progression in autosomal dominant polycystic kidney disease. iScience 2022, 25, 103697. [Google Scholar] [CrossRef]
- Testa, F.; Marchiò, M.; Belli, M.; Giovanella, S.; Ligabue, G.; Cappelli, G.; Biagini, G.; Magistroni, R. A pilot study to evaluate tolerability and safety of a modified Atkins diet in ADPKD patients. PharmaNutrition 2019, 9, 100154. [Google Scholar] [CrossRef]
- Bruen, D.M.; Kingaard, J.J.; Munits, M.; Paimanta, C.S.; Torres, J.A.; Saville, J.; Weimbs, T. Ren.Nu, a Dietary Program for Individuals with Autosomal-Dominant Polycystic Kidney Disease Implementing a Sustainable, Plant-Focused, Kidney-Safe, Ketogenic Approach with Avoidance of Renal Stressors. Kidney Dial. 2022, 2, 183–203. [Google Scholar] [CrossRef]
- Oehm, S.; Steinke, K.; Schmidt, J.; Arjune, S.; Todorova, P.; Heinrich Lindemann, C.; Wöstmann, F.; Meyer, F.; Siedek, F.; Weimbs, T.; et al. RESET-PKD: A pilot trial on short-term ketogenic interventions in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2023, 38, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.; Coleman, E.R.; George, D.; Farmer-Bailey, H.; Ramanathan, S.; Gregory, A.; Wang, W.; Gitomer, B.Y.; Chonchol, M.; Thomas, E.; et al. Time-Restricted Feeding and Autosomal Dominant Polycystic Kidney Disease: A Pilot, Randomized Clinical Trial: TH-PO414. J. Am. Soc. Nephrol. 2023, 34 (Suppl. S11), 206. [Google Scholar] [CrossRef]
- Cukoski, S.; Lindemann, C.H.; Arjune, S.; Todorova, P.; Brecht, T.; Kühn, A.; Oehm, S.; Strubl, S.; Becker, I.; Kämmerer, U.; et al. Feasibility and impact of ketogenic dietary interventions in polycystic kidney disease: KETO-ADPKD—A randomized controlled trial. Cell Rep. Med. 2023, 4, 101283. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Marchiò, M.; D’amico, R.; Giovanella, S.; Ligabue, G.; Fontana, F.; Alfano, G.; Cappelli, G.; Biagini, G.; Magistroni, R. GREASE II. A phase II randomized, 12-month, parallel-group, superiority study to evaluate the efficacy of a Modified Atkins Diet in Autosomal Dominant Polycystic Kidney Disease patients. PharmaNutrition 2020, 13, 100206. [Google Scholar] [CrossRef]
- University of Colorado, Denver. Daily Caloric Restriction in Overweight and Obese Adults with ADPKD. clinicaltrials.gov; 2024. Available online: https://clinicaltrials.gov/study/NCT04907799 (accessed on 1 January 2024).
- Ohio State University. Feasibility and Efficacy of a Well-Formulated Ketogenic Diet in Delaying Progression of Polycystic Kidney Disease in Patients at Risk for Rapid Progression. clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT06325644 (accessed on 1 January 2024).
- Universitair Ziekenhuis Brussel. Treat Autosomal Dominant Polycystic Kidney Disease with Oral Ketone Ester? (ADKETONE). clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT06100133 (accessed on 25 July 2024).
- Strubl, S.; Oehm, S.; Torres, J.A.; Grundmann, F.; Haratani, J.; Decker, M.; Vuong, S.; Kaur Bhandal, A.; Methot, N.; Haynie-Cion, R.; et al. Ketogenic dietary interventions in autosomal dominant polycystic kidney disease—A retrospective case series study: First insights into feasibility, safety and effects. Clin. Kidney J. 2021, 15, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 571–578. [Google Scholar] [CrossRef]
- Nowak, K.L.; Moretti, F.; Bussola, N.; Steele, C.; Gregory, A.V.; Kline, T.L.; Ramanathan, S.; Trapletti, G.; Furlanello, C.; McCormick, L.; et al. Visceral Adiposity and Progression of ADPKD: A Cohort Study of Patients from the TEMPO 3:4 Trial. Am. J. Kidney Dis. 2024; in press. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Comparison of High vs. Normal/Low Protein Diets on Renal Function in Subjects without Chronic Kidney Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e97656. [Google Scholar] [CrossRef]
- Capelli, I.; Lerario, S.; Aiello, V.; Provenzano, M.; Di Costanzo, R.; Squadrani, A.; Vella, A.; Vicennati, V.; Poli, C.; La Manna, G.; et al. Diet and Physical Activity in Adult Dominant Polycystic Kidney Disease: A Review of the Literature. Nutrients 2023, 15, 2621. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Burrowes, J.D.; Byham-Gray, L.D.; Campbell, K.L.; Carrero, J.J.; Chan, W.; Fouque, D.; Friedman, A.N.; Ghaddar, S.; Goldstein-Fuchs, D.J.; et al. KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. Am. J. Kidney Dis. 2020, 76 (Suppl. S1), S1–S107. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Acharya, C.; Thongprayoon, C.; Hansrivijit, P.; Kanduri, S.R.; Kovvuru, K.; Medaura, J.; Vaitla, P.; Garcia Anton, D.F.; Mekraksakit, P.; et al. Incidence and Characteristics of Kidney Stones in Patients on Ketogenic Diet: A Systematic Review and Meta-Analysis. Diseases 2021, 9, 39. [Google Scholar] [CrossRef]
- Dyńka, D.; Kowalcze, K.; Charuta, A.; Paziewska, A. The Ketogenic Diet and Cardiovascular Diseases. Nutrients 2023, 15, 3368. [Google Scholar] [CrossRef] [PubMed]
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of ketogenic diet and ketone bodies on the cardiovascular system: Concentration matters. World J. Diabetes 2020, 11, 584–595. [Google Scholar] [CrossRef]
- Pirola, L.; Ciesielski, O.; Balcerczyk, A. Fat not so bad? The role of ketone bodies and ketogenic diet in the treatment of endothelial dysfunction and hypertension. Biochem. Pharmacol. 2022, 206, 115346. [Google Scholar] [CrossRef]
- Popiolek-Kalisz, J. Ketogenic diet and cardiovascular risk–state of the art review. Curr. Probl. Cardiol. 2024, 49, 102402. [Google Scholar] [CrossRef]
- Joo, M.; Moon, S.; Lee, Y.S.; Kim, M.G. Effects of very low-carbohydrate ketogenic diets on lipid profiles in normal-weight (body mass index < 25 kg/m2) adults: A meta-analysis. Nutr. Rev. 2023, 81, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Cukoski, S.; Kühn, A.; Lindemann, C.H.; Arjune, S.; Meyer, F.; Schömig, T.; Große-Hokamp, N.; Schmidt, J.; Antczak, P.; Weimbs, T.; et al. #2160 Ketosis moderates the effect on kidney volume in dietary interventions for ADPKD—More insights on the KETO ADPKD trial. Nephrol. Dial. Transplant. 2024, 39, gfae069-738-2160. [Google Scholar] [CrossRef]
- Mosterd, C.; Kanbay, M.; Born, B.v.D.; van Raalte, D.; Rampanelli, E. Intestinal microbiota and diabetic kidney diseases: The Role of microbiota and derived metabolites inmodulation of renal inflammation and disease progression. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101484. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, L.; Li, J.; Zhang, X.; Xie, Y.; Li, X.; Yang, B.; Yang, H. The potential mechanism of gut microbiota-microbial metabolites-mitochondrial axis in progression of diabetic kidney disease. Mol. Med. 2023, 29, 148. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Vasileva, V.Y.; Sultanova, R.F.; Sudarikova, A.V.; Ilatovskaya, D.V. Insights into the Molecular Mechanisms of Polycystic Kidney Diseases. Front. Physiol. 2021, 12, 693130. [Google Scholar] [CrossRef]
- Cao, C.; Zhu, H.; Yao, Y.; Zeng, R. Gut Dysbiosis and Kidney Diseases. Front. Med. 2022, 9, 829349. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Wei, P.; Min, Y.; Yu, M.; Zhou, G.; Yuan, G.; Sun, J.; Dai, H.; Zhou, E.; et al. Causal Effects of Specific Gut Microbiota on Chronic Kidney Diseases and Renal Function—A Two-Sample Mendelian Randomization Study. Nutrients 2023, 15, 360. [Google Scholar] [CrossRef] [PubMed]
- Mikusic, N.L.R.; Kouyoumdzian, N.M.; Choi, M.R. Gut microbiota and chronic kidney disease: Evidences and mechanisms that mediate a new communication in the gastrointestinal-renal axis. Pflügers Arch. Eur. J. Physiol. 2020, 472, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Caldarelli, M.; Franza, L.; Rio, P.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Gut–Kidney–Heart: A Novel Trilogy. Biomedicines 2023, 11, 3063. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut–kidney axis. Pediatr. Nephrol. 2016, 32, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, J.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.; Rinninella, E.; Biruete, A.; Sumida, K.; Stanford, J.; Raoul, P.; Mele, M.C.; Wang, A.Y.M.; Mafra, D. Targeting the Gut Microbiota in Kidney Disease: The Future in Renal Nutrition and Metabolism. J. Ren. Nutr. 2023, 33, S30–S39. [Google Scholar] [CrossRef]
- Yacoub, R.; Nadkarni, G.N.; McSkimming, D.I.; Chaves, L.D.; Abyad, S.; Bryniarski, M.A.; Honan, A.M.; Thomas, S.A.; Gowda, M.; He, J.C.; et al. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: A pilot study. Exp. Biol. Med. 2018, 244, 505–513. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, H.; Shi, K.; Ren, Y.; Zhang, P.; Cheng, S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology 2012, 17, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Strubl, S.; Woestmann, F.; Todorova, P.; Arjune, S.; Farowski, F.; Baar, T.; Brodesser, S.; Grundmann, F.; Vehreschild, M.J.G.T.; Mueller, R.H. #395 Gut dysbiosis in ADPKD patients: A controlled pilot study. Nephrol. Dial. Transplant. 2024, 39 (Suppl. S1), gfae069-0255-395. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2010, 5, 220–230. [Google Scholar] [CrossRef]
- Kern, L.; Kviatcovsky, D.; He, Y.; Elinav, E. Impact of caloric restriction on the gut microbiota. Curr. Opin. Microbiol. 2023, 73, 102287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, X.; Loh, Y.J.; Yang, X.; Zhang, C. The effect of calorie intake, fasting, and dietary composition on metabolic health and gut microbiota in mice. BMC Biol. 2021, 19, 51. [Google Scholar] [CrossRef]
- Popa, A.D.; Niță, O.; Gherasim, A.; Enache, A.I.; Caba, L.; Mihalache, L.; Arhire, L.I. A Scoping Review of the Relationship between Intermittent Fasting and the Human Gut Microbiota: Current Knowledge and Future Directions. Nutrients 2023, 15, 2095. [Google Scholar] [CrossRef]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; de Giorgis, V.; Veggiotti, P.; Elli, M. Short-term impact of a classical ketogenic diet on gut microbiota in GLUT1 Deficiency Syndrome: A 3-month prospective observational study. Clin. Nutr. ESPEN 2016, 17, 33–37. [Google Scholar] [CrossRef]
- Lindefeldt, M.; Eng, A.; Darban, H.; Bjerkner, A.; Zetterström, C.K.; Allander, T.; Andersson, B.; Borenstein, E.; Dahlin, M.; Prast-Nielsen, S. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhou, Q.; Qiu, C.-Z.; Dai, W.K.; Wang, H.P.; Li, Y.H.; Liao, J.X.; Lu, X.G.; Lin, S.F.; Ye, J.H.; et al. Ketogenic diet poses a significant effect on imbalanced gut microbiota in infants with refractory epilepsy. World J. Gastroenterol. 2017, 23, 6164–6171. [Google Scholar] [CrossRef] [PubMed]
- Linsalata, M.; Russo, F.; Riezzo, G.; D'Attoma, B.; Prospero, L.; Orlando, A.; Ignazzi, A.; Di Chito, M.; Sila, A.; De Nucci, S.; et al. The Effects of a Very-Low-Calorie Ketogenic Diet on the Intestinal Barrier Integrity and Function in Patients with Obesity: A Pilot Study. Nutrients 2023, 15, 2561. [Google Scholar] [CrossRef]
- Attaye, I.; van Oppenraaij, S.; Warmbrunn, M.V.; Nieuwdorp, M. The Role of the Gut Microbiota on the Beneficial Effects of Ketogenic Diets. Nutrients 2021, 14, 191. [Google Scholar] [CrossRef]
| KDI | Overview | Evidence | Comments |
|---|---|---|---|
| DCR [23,24,25,26] |
|
| Challenging long-term. Unclear evidence on achieving NK. |
| IMF [27,28,29] |
|
| Different fasting schedules: reduced or normocaloric intake, and fasting period varying from hours (e.g., 16/8) to days (e.g., 5:2). |
| TRF [27,29,30] |
|
| Different eating window duration. |
| cKD [31,32] |
|
| Requires the use of specifically calculated recipes measured in grams to meet the patient’s needs. |
| MCT [31,32,33] |
|
| Requires the use of specifically calculated recipes measured in grams. Greater food variety compared to cKD. |
| MAD [31,32,33,34,35] |
|
| Food can be measured using standard household measurements. |
| LGIT [31,32,33] |
|
| Food can be measured using standard household measurements |
| VLCKD [36,37] |
|
| Rapid weight loss effects but requires monitoring to avoid nutritional deficiencies. |
| KDI | Study Design | Length and Sample | BMI | Weight Loss Outcomes | Kidney Outcomes | Status Completed: ✓, Recruiting: ● |
|---|---|---|---|---|---|---|
| DCR vs. IMF (both with ~34% weekly energy deficit) | 2 EAs, masked (Inv, OA) | 12 mo (n = 29) | 25–45 | ↓ BW, SAT, VAT, TAT (MRI) (independent of KDI) | ↓ htTKV correlated with BW VAT and TAT loss independent of KDI. No changes in eGFR. | ✓ [53] |
| TRF (8 h window) vs. HE | 1 EA, 1 CA, masked (Inv, OA) | 12 mo (n = 29) | 25–45 | ↓ BW (independent of KDI) | ↓ htTKV correlated with BW and VAT loss independent of KDI. | ✓ [57] |
| KD (carbohydrate < 30 g/day, 0.8 g/kg BW protein intake) and 3-days WF vs. AL diet | 2 EAs, 1 CA, no masking | 3 mo (n = 63) | 18.6–34.9 | ↓ BW and significant ↓ WC (KD and WF), significant ↓ fat mass (KD), ↓ lean mass (KD and WF) | Significant creatinine-based and CysC-based eGFR ↑ with KD. Significant htTKV ↓ in a subset of KD patients reaching NK at 2/3 study visits. | ✓ [58,75] |
| DCR (30%) and increased physical activity vs. SNC | 1 EA, 1 CA, masked (Inv, OA) | 24 mo (n = 126) | 25–45 | N/A | N/A | ● [60] |
| MAD (<20 g/day of carbohydrates) vs. BND | 1 EA, 1 CA, masked (Inv, OA) | 24 mo (n = 92) | 20–30 | N/A | N/A | ● [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezzuoli, C.; Biagini, G.; Magistroni, R. Ketogenic Interventions in Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review of Current Evidence. Nutrients 2024, 16, 2676. https://doi.org/10.3390/nu16162676
Pezzuoli C, Biagini G, Magistroni R. Ketogenic Interventions in Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review of Current Evidence. Nutrients. 2024; 16(16):2676. https://doi.org/10.3390/nu16162676
Chicago/Turabian StylePezzuoli, Carla, Giuseppe Biagini, and Riccardo Magistroni. 2024. "Ketogenic Interventions in Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review of Current Evidence" Nutrients 16, no. 16: 2676. https://doi.org/10.3390/nu16162676
APA StylePezzuoli, C., Biagini, G., & Magistroni, R. (2024). Ketogenic Interventions in Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review of Current Evidence. Nutrients, 16(16), 2676. https://doi.org/10.3390/nu16162676