
Blood Phenylalanine Levels in Patients with Phenylketonuria from Europe between 2012 and 2018: Is It a Changing Landscape?


 , ,
, ,  , , ,
, , ,  , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Participating Centres
2.2. Patient Selection
2.3. Study Design
2.4. Procedures
2.5. Statistical Analysis
2.6. Ethical Aspects
3. Results
3.1. Treatment Centre Characteristics
3.2. Subjects’ Characteristics
3.3. Blood Phe Control Per Centre
3.4. Blood Phe and Tyr Levels by Age Group
3.5. Blood Phe Control by Sex
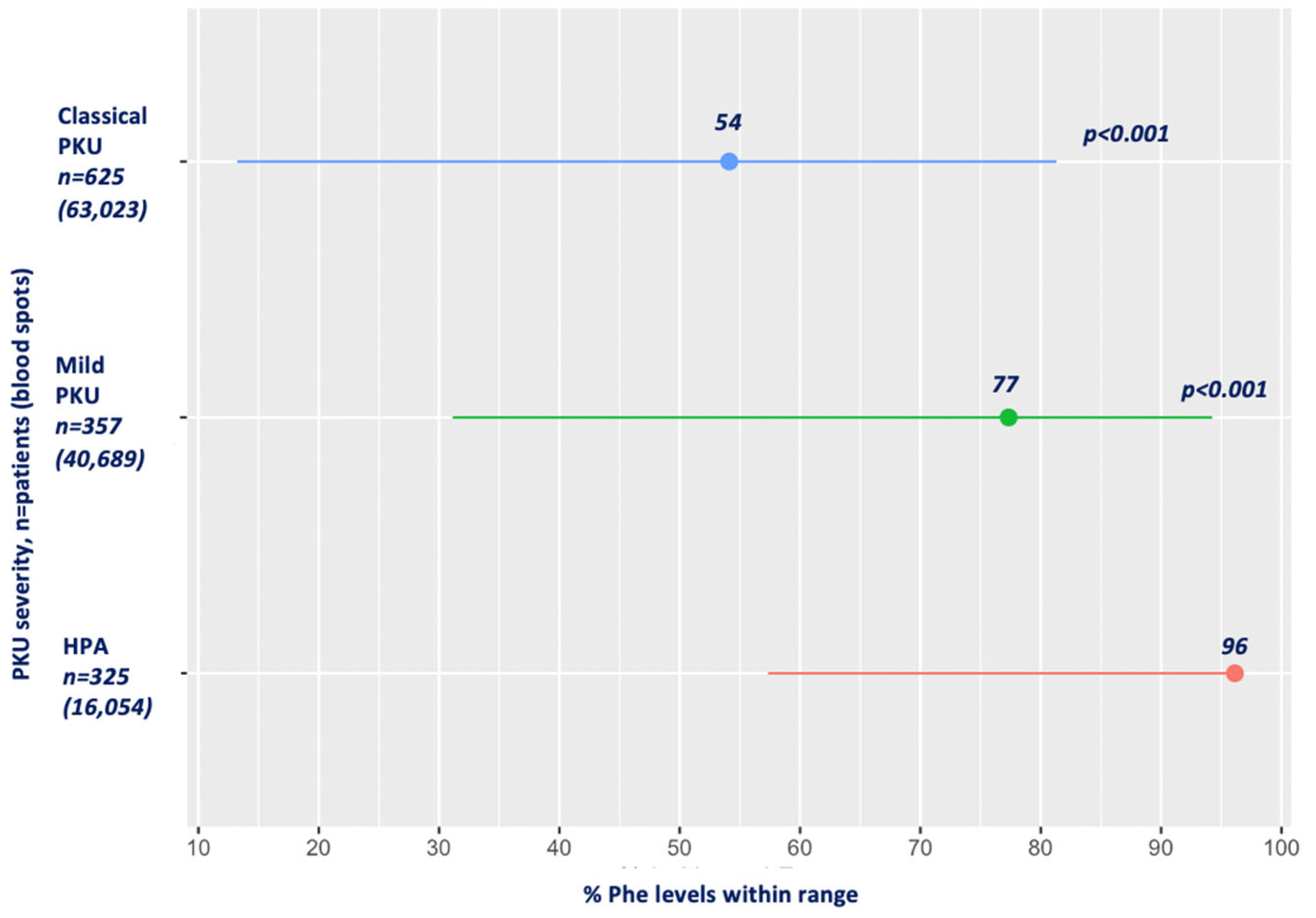
3.6. Blood Phe Control by PKU Severity
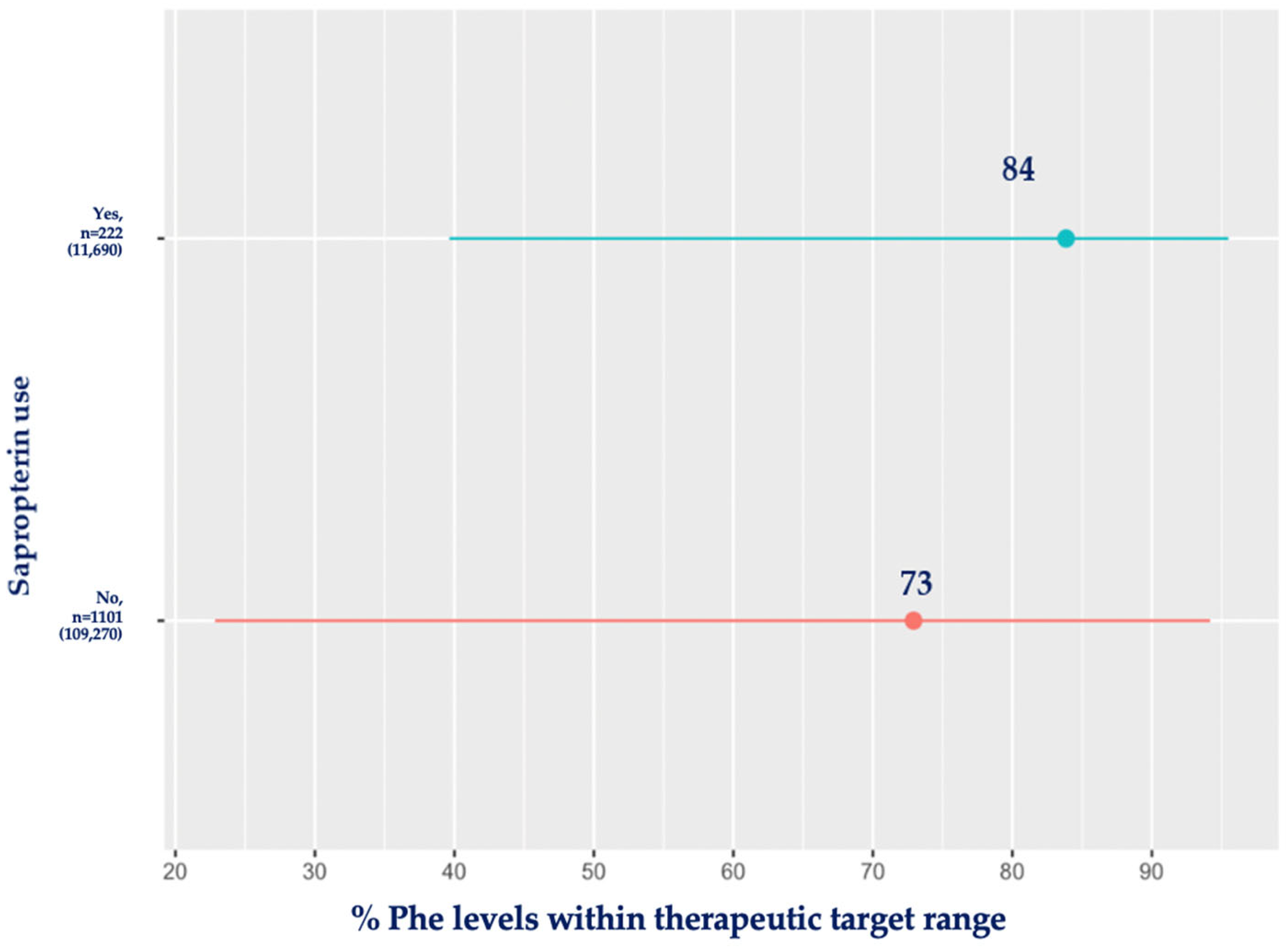
3.7. Sapropterin and Blood Phe Control
3.8. Blood Phe Variability
3.9. Frequency of Blood Phe Monitoring
4. Discussion
4.1. Limitations
4.2. Strengths
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Cox, S.R.; Deary, I.J. Brain and cognitive ageing: The present, and some predictions (…about the future). Aging Brain 2022, 2, 100032. [Google Scholar] [CrossRef] [PubMed]
- Rovelli, V.; Longo, N. Phenylketonuria and the brain. Mol. Genet. Metab. 2023, 139, 107583. [Google Scholar] [CrossRef]
- Manti, F.; Nardecchia, F.; De Leo, S.; Carducci, C.; Romani, C.; Palermo, L.; Angeloni, A.; Leuzzi, V. Towards precision medicine for phenylketonuria: The effect of restoring a strict metabolic control in adult patients with early-treated phenylketonuria. Mol. Genet. Metab. 2023, 140, 107666. [Google Scholar] [CrossRef]
- Romani, C.; Olson, A.; Aitkenhead, L.; Baker, L.; Patel, D.; Spronsen, F.V.; MacDonald, A.; Wegberg, A.V.; Huijbregts, S. Meta-analyses of cognitive functions in early-treated adults with phenylketonuria. Neurosci. Biobehav. Rev. 2022, 143, 104925. [Google Scholar] [CrossRef]
- Grant, M.L.; Jurecki, E.R.; McCandless, S.E.; Stahl, S.M.; Bilder, D.A.; Sanchez-Valle, A.; Dimmock, D. Neuropsychiatric Function Improvement in Pediatric Patients with Phenylketonuria. J. Pediatr. 2023, 260, 113526. [Google Scholar] [CrossRef] [PubMed]
- Altman, G.; Hussain, K.; Green, D.; Strauss, B.J.G.; Wilcox, G. Mental health diagnoses in adults with phenylketonuria: A retrospective systematic audit in a large UK single centre. Orphanet J. Rare Dis. 2021, 16, 520. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.A.; Kobori, J.A.; Cohen-Pfeffer, J.L.; Johnson, E.M.; Jurecki, E.R.; Grant, M.L. Neuropsychiatric comorbidities in adults with phenylketonuria: A retrospective cohort study. Mol. Genet. Metab. 2017, 121, 1–8. [Google Scholar] [CrossRef]
- Jaulent, P.; Charriere, S.; Feillet, F.; Douillard, C.; Fouilhoux, A.; Thobois, S. Neurological manifestations in adults with phenylketonuria: New cases and review of the literature. J. Neurol. 2020, 267, 531–542. [Google Scholar] [CrossRef]
- Thomas, L.; Olson, A.; Romani, C. The impact of metabolic control on cognition, neurophysiology, and well-being in PKU: A systematic review and meta-analysis of the within-participant literature. Mol. Genet. Metab. 2023, 138, 106969. [Google Scholar] [CrossRef]
- Trefz, K.F.; Muntau, A.C.; Kohlscheen, K.M.; Altevers, J.; Jacob, C.; Braun, S.; Greiner, W.; Jha, A.; Jain, M.; Alvarez, I.; et al. Clinical burden of illness in patients with phenylketonuria (PKU) and associated comorbidities—A retrospective study of German health insurance claims data. Orphanet J. Rare Dis. 2019, 14, 181. [Google Scholar] [CrossRef]
- Levy, H.; Lamppu, D.; Anastosoaie, V.; Baker, J.L.; DiBona, K.; Hawthorne, S.; Lindenberger, J.; Kinch, D.; Seymour, A.; McIlduff, M.; et al. 5-year retrospective analysis of patients with phenylketonuria (PKU) and hyperphenylalaninemia treated at two specialized clinics. Mol. Genet. Metab. 2020, 129, 177–185. [Google Scholar] [CrossRef]
- Manti, F.; Caviglia, S.; Cazzorla, C.; Dicintio, A.; Pilotto, A.; Burlina, A.P. Expert opinion of an Italian working group on the assessment of cognitive, psychological, and neurological outcomes in pediatric, adolescent, and adult patients with phenylketonuria. Orphanet J. Rare Dis. 2022, 17, 443. [Google Scholar] [CrossRef]
- Charrière, S.; Maillot, F.; Bouée, S.; Douillard, C.; Jacob, C.; Schneider, K.M.; Theil, J.; Arnoux, J.B. Health status and comorbidities of adult patients with phenylketonuria (PKU) in France with a focus on early-diagnosed patients—A nationwide study of health insurance claims data. Mol. Genet. Metab. 2023, 139, 107625. [Google Scholar] [CrossRef]
- Adams, A.D.; Fiesco-Roa, M.; Wong, L.; Jenkins, G.P.; Malinowski, J.; Demarest, O.M.; Rothberg, P.G.; Hobert, J.A. Phenylalanine hydroxylase deficiency treatment and management: A systematic evidence review of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100358. [Google Scholar] [CrossRef]
- Shintaku, H.; Ohura, T.; Takayanagi, M.; Kure, S.; Owada, M.; Matsubara, Y.; Yoshino, M.; Okano, Y.; Ito, T.; Okuyama, T.; et al. Guide for diagnosis and treatment of hyperphenylalaninemia. Pediatr. Int. 2021, 63, 8–12. [Google Scholar] [CrossRef]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef]
- MacDonald, A.; van Wegberg, A.M.J.; Ahring, K.; Beblo, S.; Bélanger-Quintana, A.; Burlina, A.; Campistol, J.; Coşkun, T.; Feillet, F.; Giżewska, M.; et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J. Rare Dis. 2020, 15, 171. [Google Scholar] [CrossRef]
- Pena, M.J.; Almeida, M.F.; van Dam, E.; Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Gokmen-Ozel, H.; Lammardo, A.M.; MacDonald, A.; Robert, M.; et al. Special low protein foods for phenylketonuria: Availability in Europe and an examination of their nutritional profile. Orphanet J. Rare Dis. 2015, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Adams, D.J.; Bélanger-Quintana, A.; Bushueva, T.V.; Cerone, R.; Chien, Y.H.; Chiesa, A.; Coşkun, T.; de Las Heras, J.; Feillet, F.; et al. International best practice for the evaluation of responsiveness to sapropterin dihydrochloride in patients with phenylketonuria. Mol. Genet. Metab. 2019, 127, 1–11. [Google Scholar] [CrossRef]
- Longo, N.; Arnold, G.L.; Pridjian, G.; Enns, G.M.; Ficicioglu, C.; Parker, S.; Cohen-Pfeffer, J.L. Long-term safety and efficacy of sapropterin: The PKUDOS registry experience. Mol. Genet. Metab. 2015, 114, 557–563. [Google Scholar] [CrossRef]
- Burton, B.K.; Clague, G.E.; Harding, C.O.; Kucuksayrac, E.; Levy, D.G.; Lindstrom, K.; Longo, N.; Maillot, F.; Muntau, A.C.; Rutsch, F.; et al. Long-term comparative effectiveness of pegvaliase versus medical nutrition therapy with and without sapropterin in adults with phenylketonuria. Mol. Genet. Metab. 2024, 141, 108114. [Google Scholar] [CrossRef]
- Scala, I.; Concolino, D.; Della Casa, R.; Nastasi, A.; Ungaro, C.; Paladino, S.; Capaldo, B.; Ruoppolo, M.; Daniele, A.; Bonapace, G.; et al. Long-term follow-up of patients with phenylketonuria treated with tetrahydrobiopterin: A seven years experience. Orphanet J. Rare Dis. 2015, 10, 14. [Google Scholar] [CrossRef]
- Rohr, F.; Burton, B.; Dee, A.; Harding, C.O.; Lilienstein, J.; Lindstrom, K.; MacLeod, E.; Rose, S.; Singh, R.; van Calcar, S.; et al. Evaluating change in diet with pegvaliase treatment in adults with phenylketonuria: Analysis of phase 3 clinical trial data. Mol. Genet. Metab. 2023, 141, 108122. [Google Scholar] [CrossRef]
- Ishige, M.; Ito, T.; Hamazaki, T.; Kuwahara, M.; Lee, L.; Shintaku, H. Two-year interim safety and efficacy of pegvaliase in Japanese adults with phenylketonuria. Mol. Genet. Metab. 2023, 140, 107697. [Google Scholar] [CrossRef]
- Krämer, J.; Baerwald, C.; Heimbold, C.; Kamrath, C.; Parhofer, K.G.; Reichert, A.; Rutsch, F.; Stolz, S.; Weinhold, N.; Muntau, A.C. Two years of pegvaliase in Germany: Experiences and best practice recommendations. Mol. Genet. Metab. 2023, 139, 107564. [Google Scholar] [CrossRef]
- Bjoraker, K.J.; Eggerding, C.; Ellenberg, E.; Hollander, S.; Holmes, B.M.; Lindstrom, K.; McNutt, M.; Miller, S.; Northrup, H.; Rogers, M.; et al. Best practice recommendations for the management of anxiety during the pegvaliase journey. Mol. Genet. Metab. 2024, 141, 107737. [Google Scholar] [CrossRef]
- Walter, J.H.; White, F.J.; Hall, S.K.; MacDonald, A.; Rylance, G.; Boneh, A.; Francis, D.E.; Shortland, G.J.; Schmidt, M.; Vail, A. How practical are recommendations for dietary control in phenylketonuria? Lancet 2002, 360, 55–57. [Google Scholar] [CrossRef]
- Jurecki, E.R.; Cederbaum, S.; Kopesky, J.; Perry, K.; Rohr, F.; Sanchez-Valle, A.; Viau, K.S.; Sheinin, M.Y.; Cohen-Pfeffer, J.L. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet. Metab. 2017, 120, 190–197. [Google Scholar] [CrossRef]
- Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Gokmen-Ozel, H.; Lammardo, A.M.; MacDonald, A.; Motzfeldt, K.; Nowacka, M.; Robert, M.; van Rijn, M. Blood phenylalanine control in phenylketonuria: A survey of 10 European centres. Eur. J. Clin. Nutr. 2011, 65, 275–278. [Google Scholar] [CrossRef]
- Kanufre, V.; Almeida, M.F.; Barbosa, C.S.; Carmona, C.; Bandeira, A.; Martins, E.; Rocha, S.; Guimas, A.; Ribeiro, R.; MacDonald, A.; et al. Metabolic Control of Patients with Phenylketonuria in a Portuguese Metabolic Centre Comparing Three Different Recommendations. Nutrients 2021, 13, 3118. [Google Scholar] [CrossRef]
- Blau, N. BIOPKU. Available online: http://www.biopku.org/home/home.asp (accessed on 19 June 2024).
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Walter, J.H.; White, F.J. Blood phenylalanine control in adolescents with phenylketonuria. Int. J. Adolesc. Med. Health 2004, 16, 41–45. [Google Scholar] [CrossRef]
- Becsei, D.; Kiss, E.; Szatmári, I.; Arató, A.; Reusz, G.; Szabó, A.J.; Bókay, J.; Zsidegh, P. A retrospective analysis of metabolic control in children with PKU in the COVID-19 era. Mol. Genet. Metab. Rep. 2022, 32, 100897. [Google Scholar] [CrossRef]
- Walkowiak, D.; Mikołuć, B.; Mozrzymas, R.; Kałużny, Ł.; Didycz, B.; Jaglowska, J.; Kurylak, D.; Walkowiak, J. The Impact of the First 2020 COVID-19 Lockdown on the Metabolic Control of Patients with Phenylketonuria. Nutrients 2021, 13, 2024. [Google Scholar] [CrossRef]
- De Giorgi, A.; Nardecchia, F.; Romani, C.; Leuzzi, V. Metabolic control and clinical outcome in adolescents with phenylketonuria. Mol. Genet. Metab. 2023, 140, 107684. [Google Scholar] [CrossRef]
- van Vliet, D.; van der Goot, E.; van Ginkel, W.G.; van Faassen, H.J.R.; de Blaauw, P.; Kema, I.P.; Heiner-Fokkema, M.R.; van der Zee, E.A.; van Spronsen, F.J. The increasing importance of LNAA supplementation in phenylketonuria at higher plasma phenylalanine concentrations. Mol. Genet. Metab. 2022, 135, 27–34. [Google Scholar] [CrossRef]
- Cazzorla, C.; Bensi, G.; Biasucci, G.; Leuzzi, V.; Manti, F.; Musumeci, A.; Papadia, F.; Stoppioni, V.; Tummolo, A.; Vendemiale, M.; et al. Living with phenylketonuria in adulthood: The PKU ATTITUDE study. Mol. Genet. Metab. Rep. 2018, 16, 39–45. [Google Scholar] [CrossRef]
- Ilgaz, F.; Ford, S.; O‘Driscoll, M.F.; MacDonald, A. Adult PKU Clinics in the UK-Users’ Experiences and Perspectives. Nutrients 2023, 15, 4352. [Google Scholar] [CrossRef]
- Lachmann, R.; Langeveld, M. Phenylketonuria in adults: What do we know? Am. J. Clin. Nutr. 2024, 119, 870–871. [Google Scholar] [CrossRef]
- Ford, S.; O‘Driscoll, M.; MacDonald, A. Living with Phenylketonuria: Lessons from the PKU community. Mol. Genet. Metab. Rep. 2018, 17, 57–63. [Google Scholar] [CrossRef]
- Beazer, J.; Breck, J.; Eggerding, C.; Gordon, P.; Hacker, S.; Thompson, A. Strategies to engage lost to follow-up patients with phenylketonuria in the United States: Best practice recommendations. Mol. Genet. Metab. Rep. 2020, 23, 100571. [Google Scholar] [CrossRef]
- Aitkenhead, L.; Krishna, G.; Ellerton, C.; Moinuddin, M.; Matcham, J.; Shiel, L.; Hossain, S.; Kiffin, M.; Foley, J.; Skeath, R.; et al. Long-term cognitive and psychosocial outcomes in adults with phenylketonuria. J. Inherit. Metab. Dis. 2021, 44, 1353–1368. [Google Scholar] [CrossRef]
- Burlina, A.; Biasucci, G.; Carbone, M.T.; Cazzorla, C.; Paci, S.; Pochiero, F.; Spada, M.; Tummolo, A.; Zuvadelli, J.; Leuzzi, V. Italian national consensus statement on management and pharmacological treatment of phenylketonuria. Orphanet J. Rare Dis. 2021, 16, 476. [Google Scholar] [CrossRef]
- Robertson, L.; Adam, S.; Ellerton, C.; Ford, S.; Hill, M.; Randles, G.; Woodall, A.; Young, C.; MacDonald, A. Dietetic Management of Adults with Phenylketonuria (PKU) in the UK: A Care Consensus Document. Nutrients 2022, 14, 576. [Google Scholar] [CrossRef] [PubMed]
- Romani, C.; Manti, F.; Nardecchia, F.; Valentini, F.; Fallarino, N.; Carducci, C.; De Leo, S.; MacDonald, A.; Palermo, L.; Leuzzi, V. Adult cognitive outcomes in phenylketonuria: Explaining causes of variability beyond average Phe levels. Orphanet J. Rare Dis. 2019, 14, 273. [Google Scholar] [CrossRef]
- Hood, A.; Grange, D.K.; Christ, S.E.; Steiner, R.; White, D.A. Variability in phenylalanine control predicts IQ and executive abilities in children with phenylketonuria. Mol. Genet. Metab. 2014, 111, 445–451. [Google Scholar] [CrossRef]
- Mütze, U.; Thiele, A.G.; Baerwald, C.; Ceglarek, U.; Kiess, W.; Beblo, S. Ten years of specialized adult care for phenylketonuria—A single-centre experience. Orphanet J. Rare Dis. 2016, 11, 27. [Google Scholar] [CrossRef]
- Peres, M.; Almeida, M.F.; Pinto, É.J.; Carmona, C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; MacDonald, A.; et al. Implementing a Transition Program from Paediatric to Adult Services in Phenylketonuria: Results After Two Years of Follow-Up with an Adult Team. Nutrients 2021, 13, 799. [Google Scholar] [CrossRef] [PubMed]
- Bilginsoy, C.; Waitzman, N.; Leonard, C.O.; Ernst, S.L. Living with phenylketonuria: Perspectives of patients and their families. J. Inherit. Metab. Dis. 2005, 28, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, A.M.; Vliet, K.E.; MacDonald, A.; Ahring, K.; Abeln, D.; Ford, S.; Hildebrandt-Karlsen, S.; van Spronsen, F.J.; Heiner-Fokkema, M.R. Satisfaction with home blood sampling methods and expectations for future point-of-care testing in phenylketonuria: Perspectives from patients and professionals. Mol. Genet. Metab. 2024, 142, 108361. [Google Scholar] [CrossRef] [PubMed]
- Carling, R.S.; Barclay, Z.; Cantley, N.; Emmett, E.C.; Hogg, S.L.; Finezilber, Y.; Schulenburg-Brand, D.; Murphy, E.; Moat, S.J. Investigation of the relationship between phenylalanine in venous plasma and capillary blood using volumetric blood collection devices. JIMD Rep. 2023, 64, 468–476. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, K.; van Ginkel, W.G.; van Dam, E.; de Blaauw, P.; Koehorst, M.; Kingma, H.A.; van Spronsen, F.J.; Heiner-Fokkema, M.R. Dried blood spot versus venous blood sampling for phenylalanine and tyrosine. Orphanet J. Rare Dis. 2020, 15, 82. [Google Scholar] [CrossRef]
- MacDonald, A.; Rylance, G.W.; Asplin, D.; Hall, S.K.; Booth, I.W. Does a single plasma phenylalanine predict quality of control in phenylketonuria? Arch. Dis. Child. 1998, 78, 122–126. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Centre | Name of Hospital/Metabolic Centre |
|---|---|
| A | İhsan Doğramacı Children’s Hospital, Hacettepe University, Ankara, Turkey |
| B | Birmingham Children’s Hospital, Birmingham, UK |
| C | Beatrix Children’s Hospital, University of Groningen, University Medical Center, Groningen, Netherlands |
| D | Department of Pediatrics Hôpital d’Enfants Brabois, CHU Nancy, Vandoeuvre les Nancy, France |
| E | Centro de Referência na área de Doenças Hereditárias do Metabolismo, Unidade Local de Saúde de Santo António—ULSSA, Porto, Portugal |
| F | Department of Pediatrics, Endocrinology, Diabetology, Metabolic Diseases and Cardiology of the Developmental Age, Pomeranian Medical University, Szczecin, Poland |
| G | Department of PKU, Copenhagen University Hospital, Denmark |
| H | Unidad Enfermedades Metabolicas Servicio de Pediatria Hospital Ramon y Cajal, Madrid, Spain |
| I | Division of Inherited Metabolic Diseases, University Hospital of Padova, Padova, Italy |
| Centre | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | |
| Number of patients (%) | 320 (24) | 97 (7) | 101 (8) | 96 (7) | 116 (9) | 59 (4) | 314 (24) | 62 (5) | 158 (12) |
| Age group | Adults and Children | Children Only | Adults and Children | Adults and Children | Adults and Children | Adults and Children | Adults and Children | Adults and Children | Adults and Children |
| Number of patients lost to follow-up | Unknown | 0 | 0 | 17 | 4 | 5 | 20 | 5 | 6 |
| Reimbursement of SLPFs | Yes * | Yes | Yes + | Yes | Yes | No | Yes | Yes | Yes |
| Reimbursement of protein substitutes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Reimbursement of sapropterin during study period | Yes | Limited (Research only) | Yes | Yes | Yes | No | Yes | Yes | Yes |
| Home DBS samples sent to the hospital for monitoring | No (Venous samples) | Yes | Yes | Yes | Yes | Yes | No (Home liquid blood) | Yes | Yes |
| Pegvaliase availability | No | No | No | No | No | No | No | No | No |
| Specialist adult team available | No | Paediatric hospital but patients referred to adult team | Partial + | No | Partial + | No | No | Yes | Yes |
| Routine psychologist | No | No | Yes from 2018 | No | Yes | Yes from 2017 | No | No | Yes |
| Support worker | No | Yes | Yes | No | No | No | No | No | No |
| Number of full-time dietitians dedicated to IMD | 3 (not specific to IMD) | 1.3 (specific to PKU) | 1.8 | 5 (not specific to IMD) | 2 | 1 | 2.7 | 0 | 2 |
| Blood Phe target range (μmol/L) | <12 y: 120–360 ≥12 y: 120–600 | <12 y: 120–360 ≥12 y: 120–600 | <12 y: 120–360 ≥12 y: 120–600 | 2012–2017: <12 y: 120–600 ≥12 y: 600–1200 2018: <12 y: 120–360 ≥12 y: 360–900 | <12 y: 120–360 ≥12 y: 120–480 | <12 y: 120–360 ≥12 y: 120–600 | 2012–2016: 0–4 y: 180–300 4–8 y: 180–400 8–10 y: 180–600 10–12 y: 180–700 ≥12 y: 180–900 2018: <12 y: 120–360 ≥12 y: 360–600 | 2012–2016: 0–6 y: 120–360 6–9 y: 120–540 10–18 y: 120–600 ≥18 y: 120–840 2018: <12 y: 120–360 ≥12 y: 360–600 | <12 y: 120–360 ≥12 y: 120–600 |
| Frequency of clinic visits | 0–1 y: monthly; 1–2 y: 2 monthly; 2–3 y: 3 monthly; 4–18 y: 4 times a year >18 y: 6 monthly | 0–1 y: 3 monthly; 1–18 y: 6 monthly | 0–1 y: 2 monthly; 1–18 y: 2/3 times a year; ≥18 y: once a year | 0–1 y: 3 monthly; 1–18 y: 6 monthly; >18 y: once a year | 0–6 m: monthly; 6–12 m: 2 monthly; 1–12 y: 3 monthly; 12–18 y: 2/3 times a year; >18 y: 1/2 times a year | 0–1 y: monthly; 2–5 y: 2 monthly; 6–12 y: 3/4 monthly; 13–18 y: 6 monthly; >18 y: once a year | 0–6 m: 2 monthly; 6–18 m: 3 monthly; 18 m–18 y: 6 monthly; >18 y: once a year | 0–1 y: monthly; 1–6 y: 3 monthly; >6 y: 6 monthly | 0–1 y: monthly; 1–12 y: 6 monthly; >12 y: once a year |
| Method of blood Phe analysis | High-performance liquid chromatography | Tandem mass spectrometry | Tandem mass spectrometry | Tandem mass spectrometry | Tandem mass spectrometry | Enzymatic assay for in vitro diagnostic determination of L-phenylalanine in newborn blood spots | Microplate-based enzymatic (PAL) | Tandem mass spectrometry | Tandem mass spectrometry |
| Centre | Total n (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A (n = 320) | B (n = 97) | C (n = 101) | D (n = 96) | E (n = 116) | F (n = 59) | G (n = 314) | H (n = 62) | I (n = 158) | |||
| Age, years, mean ± SD | 9 ± 6 | 9 ± 5 | 23 ± 13 | 18 ± 13 | 20 ± 13 | 11 ± 8 | 22 ± 13 | 17 ± 10 | 14 ± 10 | 16 ± 10 | |
| Age group, n of patients (%) | <2 y | 22 (7) | 3 (3) | 1 (1) | 3 (3) | - | 2 (3) | 5 (2) | 2 (3) | 9 (6) | 46 (3) |
| 2–5 y | 88 (28) | 22 (23) | 8 (8) | 17 (18) | 8 (7) | 14 (24) | 27 (9) | 5 (8) | 24 (15) | 213 (16) | |
| 6–12 y | 118 (37) | 46 (47) | 10 (10) | 17 (18) | 15 (13) | 20 (34) | 60 (19) | 19 (31) | 48 (30) | 354 (27) | |
| 13–18 y | 61 (19) | 26 (27) | 21 (21) | 17 (18) | 30 (26) | 11 (20) | 55 (18) | 12 (19) | 34 (22) | 268 (20) | |
| 19–30 y | 31 (10) | - | 33 (33) | 23 (24) | 53 (46) | 12 (19) | 86 (27) | 15 (24) | 28 (18) | 280 (21) | |
| 31–40 y | - | - | 19 (19) | 13 (14) | 10 (9) | - | 43 (14) | 9 (15) | 13 (8) | 107 (8) | |
| ≥41 y | - | - | 9 (9) | 6 (6) | - | - | 38 (12) | - | 2 (1) | 55 (4) | |
| Phenotype, n of patients (%) | HPA | 97 (30) | - | 17 (17) | 19 (20) | 27 (23) | 6 (10) | 68 (22) | 10 (16) | 81 (51) | 325 (25) |
| mPKU | 69 (22) | 38 (42) | 46 (46) | 21 (22) | 54 (47) | 8 (14) | 82 (26) | 13 (21) | 26 (16) | 357 (27) | |
| cPKU | 153 (48) | 56 (58) | 33 (33) | 56 (58) | 32 (28) | 45 (76) | 160 (51) | 39 (63) | 51 (32) | 625 (47) | |
| n of patients with data on diet (%) | 318 (99) | 97 (100) | 95 (94) | 81 (84) | 115 (99) | 59 (100) | 247 (79) | 62 (100) | 89 (56) | 1163 (88) | |
| Type of treatment, n of patients (%) | Diet only * | 222 (69) | 88 (91) | 53 (52) | 66 (69) | 84 (72) | 59 (100) | 186 (59) | 45 (73) | 60 (38) | 863 (65) |
| Sapropterin | 39 (12) | 9 (9) | 42 (42) | 15 (16) | 19 (16) | - | 52 (17) | 17 (27) | 29 (18) | 222 (17) | |
| Pegvaliase | - | - | - | - | - | - | - | - | - | - | |
| Centre (n of Patients) | n of Blood Spots | Overall n of Blood Spots Performed Per Patient/Year | % of Levels Performed Fasting * (n) | Mean ± SD | ||
|---|---|---|---|---|---|---|
| Blood Phe (μmol/L) | % of Blood Phe within Target Range | Blood Tyr (μmol/L) | ||||
| + A (n = 320) | 5838 | 3 | 50% (2933) | 280 ± 281 | 70 ± 48 | 66 ± 40 |
| + B (n = 97) | 22,478 | 33 | 86% (19,242) | 239 ± 157 | 83 ± 30 | 61 ± 41 |
| C (n = 101) | 10,038 | 14 | 77% (7683) | 333 ± 255 | 79 ± 53 | 65 ± 32 |
| D (n = 96) | 6071 | 9 | NA | 333 ± 253 | 67 ± 52 | NA |
| E (n = 116) | 17,106 | 21 | NA | 347 ± 180 | 87 ± 49 | 65 ± 29 |
| F (n = 59) | 8293 | 20 | NA | 391 ± 299 | 66 ± 32 | NA |
| G (n = 314) | 30,323 | 14 | 85% (25,647) | 373 ± 273 | 65 ± 54 | 76 ± 35 |
| H (n = 62) | 5540 | 13 | NA | 331 ± 283 | 71 ± 44 | NA |
| I (n = 158) | 15,273 | 14 | NA | 317 ± 220 | 88 ± 49 | 61 ± 24 |
| TOTAL | 120,960 | 13 | NA | 313 ± 304 | 71 ± 45 | 66 ± 34 |
| Age Group | n of Patients | n of Blood Spots | Mean ± SD | ||
|---|---|---|---|---|---|
| Blood Phe (μmol/L) | % of Blood Phe within Target Range | Blood Tyr (μmol/L) | |||
| <2 y | 46 | 1618 | 187 ± 161 | 89 ± 39 | 65 ± 31 |
| 2–5 y | 213 | 19,164 | 220 ± 162 | 84 ± 35 | 63 ± 38 |
| 6–12 y | 354 | 40,553 | 261 ± 165 | 73 ± 40 | 64 ± 43 |
| 13–18 y | 268 | 26,901 | 360 ± 222 | 85 ± 50 | 58 ± 39 |
| 19–30 y | 280 | 21,501 | 466 ± 325 | 64 ± 58 | 67 ± 41 |
| 31–40 y | 107 | 8644 | 409 ± 322 | 59 ± 54 | 62 ± 35 |
| ≥41 y | 55 | 2579 | 578 ± 474 | 40 ± 60 | 68 ± 37 |
| Age Group | Mean ± SD, % of Blood Phe Levels within Target Range (n of Patients; n of Blood Spots) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Centre A | Centre B | Centre C | Centre D | Centre E | Centre F | Centre G | Centre H | Centre I | |
| <2 y * | 89 ± 38% (22; 247) | 96 ± 28% (3; 331) | 75 ± 0% (1; 33) | 96 ± 50% (3; 35) | NA | 74 ± 8% (2; 193) | 84 ± 21% (5; 370) | 94 ± 50% (2; 114) | 95 ± 33% (9; 295) |
| 2–5 y | 89 ± 40% (88; 1835) | 87 ± 25% (22; 4823) | 75 ± 43% (8; 624) | 87 ± 32% (17; 1382) | 85 ± 43% (8; 1140) | 72 ± 11% (14; 2290) | 80 ± 18% (27; 4092) | 87 ± 19% (5; 517) | 88 ± 31% (24; 2461) |
| 6–12 y | 61 ± 41% (118; 2654) | 78 ± 25% (46; 11,895) | 89 ± 31% (10; 800) | 76 ± 22% (17; 1843) | 73 ± 29% (15; 3070) | 68 ± 30% (20; 3630) | 75 ± 32% (60; 10,251) | 74 ± 38% (19; 2127) | 92 ± 40% (48; 4283) |
| 13–18 y | 64 ± 50% (61; 787) | 84 ± 39% (26; 5429) | 78 ± 41% (21; 2344) | 77 ± 57% (17; 1327) | 98 ± 28% (30; 4966) | 73 ± 29% (12; 1566) | 86 ± 50% (55; 6217) | 85 ± 37% (12; 984) | 98 ± 30% (34; 3291) |
| 19–30 y | 39 ± 44% (31; 315) | NA | 78 ± 42% (33; 3228) | 36 ± 52% (23; 614) | 84 ± 52% (53; 6630) | 51 ± 50% (11; 624) | 54 ± 59% (86; 5712) | 47 ± 43% (15; 1052) | 75 ± 55% (28; 3326) |
| 31–40 y | NA | NA | 79 ± 41% (19; 2239) | 58 ± 43% (13; 618) | 57 ± 49% (10; 1300) | NA | 54 ± 57% (43; 2428) | 58 ± 49% (9; 746) | 34 ± 47% (13; 1313) |
| ≥41 y | NA | NA | 64 ± 48% (9; 770) | 33 ± 62% (6; 252) | NA | NA | 24 ± 51% (38; 1253) | NA | 36 ± 26% (2; 304) |
| Age Group | Female | Male | p | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n of Patients | n of Blood Spots | Mean ± SD | n of Patients | n of Blood Spots | Mean ± SD | ||||
| Blood Phe | % blood Phe Levels within Target | Blood Phe | % Blood Phe Levels within Target | ||||||
| <2 y | 25 | 895 | 134 ± 179 | 92 ± 27 | 21 | 723 | 179 ± 227 | 87 ± 33 | 0.712 |
| 2–5 y | 97 | 8483 | 181 ± 200 | 87 ± 34 | 116 | 10,681 | 178 ± 203 | 86 ± 34 | 0.093 |
| 6–12 y | 164 | 16,720 | 193 ± 202 | 83 ± 37 | 190 | 23,833 | 234 ± 199 | 78 ± 42 | 0.005 |
| 13–18 y | 131 | 13,876 | 324 ± 282 | 62 ± 48 | 137 | 13,025 | 330 ± 265 | 59 ± 49 | 0.034 |
| 19–30 y | 150 | 12,375 | 481 ± 386 | 44 ± 50 | 130 | 9126 | 412 ± 352 | 47 ± 50 | <0.001 |
| 31–40 y | 63 | 5830 | 319 ± 348 | 65 ± 48 | 44 | 2814 | 544 ± 457 | 41 ± 49 | <0.001 |
| ≥41 y | 30 | 1421 | 603 ± 539 | 43 ± 49 | 25 | 1158 | 712 ± 536 | 28 ± 45 | <0.001 |
| Age Group | % of Blood Phe Levels within Target Range | p | |||||
|---|---|---|---|---|---|---|---|
| Sapropterin Group | No Sapropterin Group | ||||||
| n of Patients | n of Blood Spots | Mean ± SD | n of Patients | n of Blood Spots | Mean ± SD | ||
| <2 y | 13 | 227 | 84 ± 37 | 33 | 1667 | 89 ± 39 | 0.585 |
| 2–5 y | 29 | 755 | 84 ± 28 | 184 | 19,031 | 84 ± 35 | <0.001 |
| 6–12 y | 60 | 2164 | 80 ± 27 | 294 | 39,141 | 72 ± 40 | 0.419 |
| 13–18 y | 48 | 3907 | 91 ± 47 | 220 | 22,352 | 83 ± 50 | <0.001 |
| 19–30 y | 52 | 3446 | 78 ± 45 | 228 | 17,691 | 59 ± 57 | <0.001 |
| 31–40 y | 16 | 1126 | 76 ± 44 | 91 | 7219 | 58 ± 53 | <0.001 |
| ≥41 y | 4 | 65 | 93 ± 68 | 51 | 2169 | 39 ± 58 | <0.001 |
| Estimated RSD, μmol/L (min; max) | n of Patients | n of Blood Spots | p | ||
|---|---|---|---|---|---|
| Sex | Female | 310 (0; 6773) | 660 | 59,600 | <0.001 |
| Male | 319 (0; 3666) | 663 | 61,360 | ||
| Age group | <2 y | 291 (6; 3240) | 46 | 1618 | <0.001 |
| 2–5 y | 239 (2; 2993) | 213 | 19,164 | <0.001 | |
| 6–12 y | 201 (0; 6220) | 354 | 40,553 | <0.001 | |
| 13–18 y | 340 (0; 3666) | 268 | 26,901 | <0.001 | |
| 19–30 y | 339 (1; 3000) | 280 | 21,501 | <0.001 | |
| 31–40 y | 362 (6; 2208) | 107 | 8644 | <0.001 | |
| ≥41 y | 510 (2; 2691) | 55 | 2579 | <0.001 | |
| PKU severity | HPA | 325 (0; 3666) | 325 | 16,054 | <0.001 |
| Mild PKU | 224 (0; 4181) | 357 | 40,689 | <0.001 | |
| Classical PKU | 350 (0; 6773) | 625 | 64,217 | <0.001 | |
| Type of treatment | No sapropterin | 322 (0; 3666) | 1101 | 109,270 | <0.001 |
| Sapropterin | 229 (6; 1840) | 222 | 11,690 | ||
| TOTAL | 263 (2; 2984) | 1323 | 120,960 |
| Frequency of Monitoring | n of Blood Spots | Blood Phe Level (μmol/L) Mean ± SD | p |
|---|---|---|---|
| Weekly | 14,873 | 271 ± 204 | - |
| Once every 2 weeks | 54,326 | 376 ± 262 | <0.001 |
| Once every 4 weeks | 31,599 | 426 ± 282 | <0.001 |
| >4 weeks | 20,162 | 534 ± 468 | <0.001 |
| Reference | Country | Data Collection Period | % of Blood Phe Levels within Target Range | Target Blood Phe Levels (μmol/L) | |||
|---|---|---|---|---|---|---|---|
| Age Group | n of Patients | Outcome (%) | |||||
| Current study | Turkey, UK, Netherlands, France, Portugal, Poland, Denmark, Spain, Italy | 2012–2018 | 0–1 y: 1–5 y: 6–12 y: 13–18 y: 19–30 y: 31–40 y: ≥41 y: | 47 213 353 268 280 107 55 | 89% 84% 73% 85% 64% 59% 40% | <12 y: 120–360 ≥12 y: 360–600 | |
| Becsei et al., 2022 [37] | Hungary | May 2020–October 2020 | 2–12 y: >13 y: | 51 21 | 59% (before COVID-19) 51% (during COVID-19) 57% (before COVID-19) 52% (during COVID-19) | <12 y: 120–360 ≥12 y: 360–600 | |
| Kanufre et al., 2021 [33] | Portugal | 2017 | <12 y: ≥12 y: | 19 68 | 56% 84% | <12 y: 120–360 ≥12 y: 360–600 | (aim for ≤480) |
| Levy et al., 2020 [13] | USA | November 2012–November 2017 | 10–18 y: 19–40 y: | 70 82 | 43% 22% | All ages: 120–360 | |
| Walkowiak et al., 2021 [38] | Poland | 2019 and first few months of 2020 | 0–1 y: 1–2 y: 2–6 y: 6–12 y: 12–18 y: >18 y: | 662 548 1140 474 479 933 | 90% 77% 63% 46% 79% 68% | <12 y: 120–360 ≥12 y: 360–600 | (aim for ≤240) (aim for ≤360) |
| Jurecki et al., 2014 [31] | USA | July 2014 to September 2015 (variable period, 1 y in total for each patient) | 0–4 y: 5–12 y: 13–17 y: 18–29 y: >30 y: >18 y: | 637 911 618 660 479 933 | 88% 72% 78% 61% 79% 49% | All ages: 120–360 | |
| Ahring et al., 2011 [32] | Denmark, Spain Germany, Turkey, Italy, UK, Norway, Poland, Belgium, Netherlands | 2007–2008 (1 y in total for each patient) | 0–1 y: 1–3 y: 4–10 y: 11–16 y: >16 y: | n/a | 88% 74% 74% 89% 65% | 0–3 y: 120–360 (n = 6 centres) 120–300 (n = 1 centre) 42–240 (n = 2 centres) 120–400 (n = 1 centre) 4–10 y: 120–360 (n = 4 centres) 120–400 (n = 2 centres) 100–450 (n = 1 centre) <480 (n = 1 centre) 8–10 y: 120–600 (n = 1 centre) | 11–16 y: 120–400 (n = 1 centre) ≤480 (n = 1 centre) 120–600 (n = 3 centres) ≤630 (n = 1 centre) 100–700 (n = 2 centres) <720 (n = 1 centre) <900 (n = 2 centres) >16 y: 120–400 (n = 1 centre) <600 (n = 2 centre) <630 (n = 1 centre) <700 (n = 1 centre) <720 (n = 1 centre) <900 (n = 3 centres) <1200 (n = 1 centre) |
| Walter et al., 2002 [30] | UK, Australia | 1994–2000 | 0–4 y: 5–9 y: 10–14 y: 15–19 y: | 178 137 98 911 | 72% 73% 50% 21% | <5 y: <360 5–10 y: <480 >10 y: <700 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Ahring, K.; Almeida, M.F.; Ashmore, C.; Bélanger-Quintana, A.; Burlina, A.; Coşkun, T.; Daly, A.; van Dam, E.; Dursun, A.; et al. Blood Phenylalanine Levels in Patients with Phenylketonuria from Europe between 2012 and 2018: Is It a Changing Landscape? Nutrients 2024, 16, 2064. https://doi.org/10.3390/nu16132064
Pinto A, Ahring K, Almeida MF, Ashmore C, Bélanger-Quintana A, Burlina A, Coşkun T, Daly A, van Dam E, Dursun A, et al. Blood Phenylalanine Levels in Patients with Phenylketonuria from Europe between 2012 and 2018: Is It a Changing Landscape? Nutrients. 2024; 16(13):2064. https://doi.org/10.3390/nu16132064
Chicago/Turabian StylePinto, Alex, Kirsten Ahring, Manuela Ferreira Almeida, Catherine Ashmore, Amaya Bélanger-Quintana, Alberto Burlina, Turgay Coşkun, Anne Daly, Esther van Dam, Ali Dursun, and et al. 2024. "Blood Phenylalanine Levels in Patients with Phenylketonuria from Europe between 2012 and 2018: Is It a Changing Landscape?" Nutrients 16, no. 13: 2064. https://doi.org/10.3390/nu16132064
APA StylePinto, A., Ahring, K., Almeida, M. F., Ashmore, C., Bélanger-Quintana, A., Burlina, A., Coşkun, T., Daly, A., van Dam, E., Dursun, A., Evans, S., Feillet, F., Giżewska, M., Gökmen-Özel, H., Hickson, M., Hoekstra, Y., Ilgaz, F., Jackson, R., Leśniak, A., ... MacDonald, A. (2024). Blood Phenylalanine Levels in Patients with Phenylketonuria from Europe between 2012 and 2018: Is It a Changing Landscape? Nutrients, 16(13), 2064. https://doi.org/10.3390/nu16132064