
Recent Progress on Fructose Metabolism—Chrebp, Fructolysis, and Polyol Pathway


{kind=link}
{kind=link}
Abstract
1. Introduction
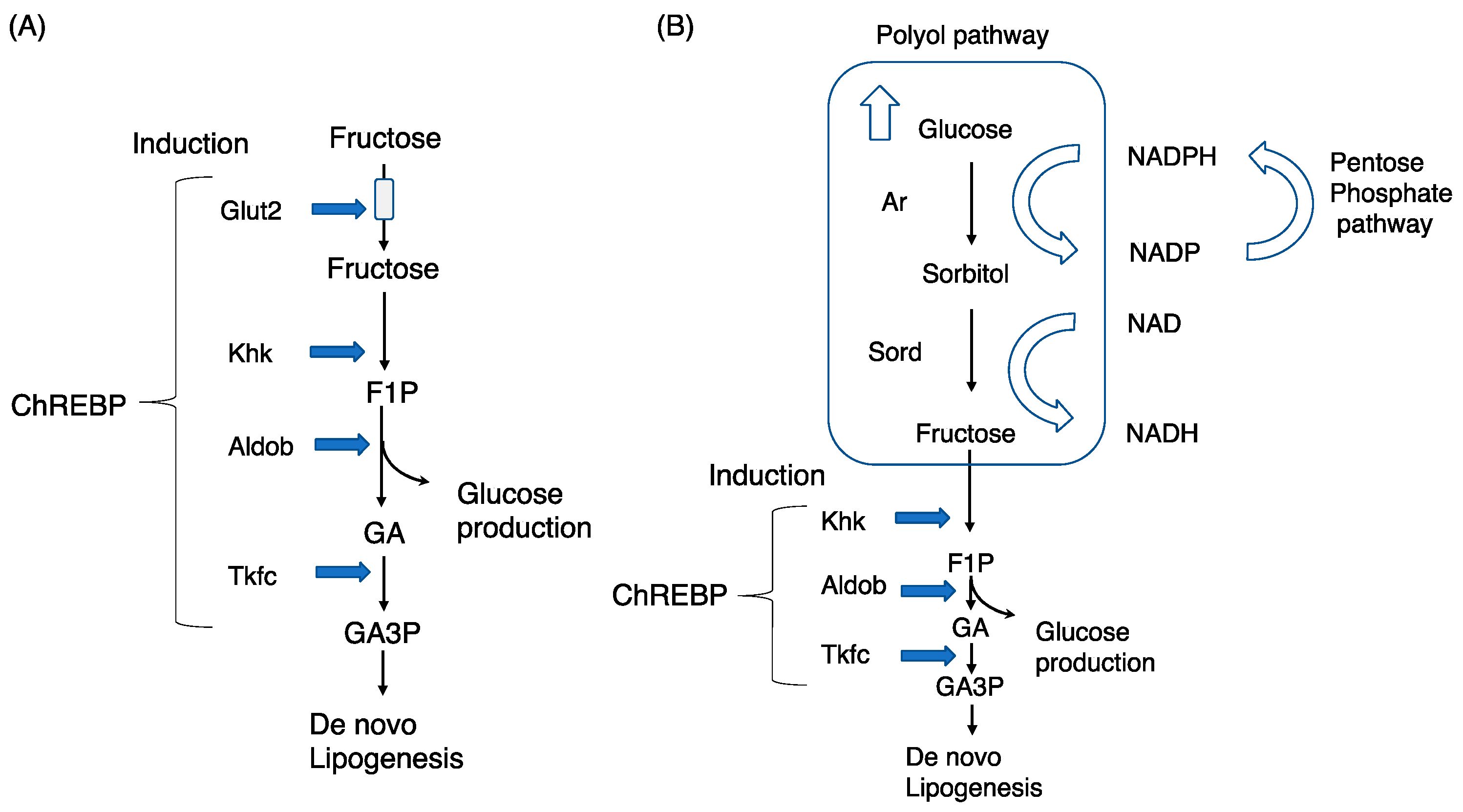
2. Exogenous Fructose Metabolism and Chrebp
2.1. Fructose, a Potent Inducer of Lipogenesis
2.2. Chrebp, a Regulator of Fructose Metabolism
2.3. Chrebp Gene Deletion and Phenotypes
2.4. Phenotypes Induced by Glut5, Khk, Aldob, and Triokinase Mutations
2.5. Comparison between Chrebp−/− and Fructose-Regulated Gene Knockout Mice
2.6. Other Transcription Factors (ATF3, Srebp1c, AhR) and Fructose
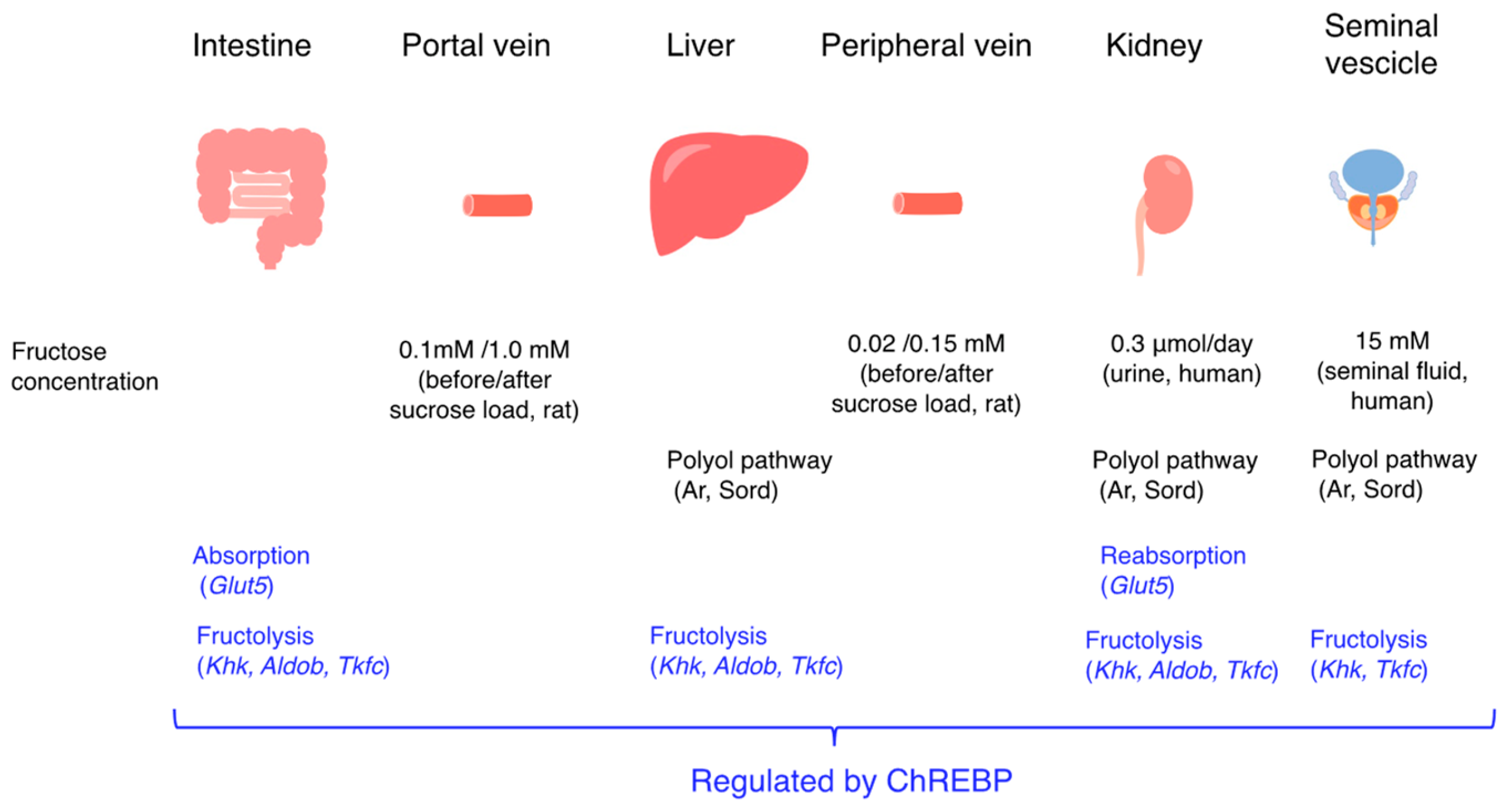
3. Endogenous Fructose Production
3.1. The Polyol Pathway and Endogenous Fructose Production
3.2. Findings in Sorbitol Dehydrogenase Knockout Mice
3.3. Findings in Aldose Reductase Knockout Mice
3.4. The Polyol Pathway and Endogenous Fructose Metabolism
3.5. Effects of Suppressing the Polyol Pathway on Glucose Tolerance
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Guidelines. Available online: https://www.who.int/news/item/04-03-2015-who-calls-on-countries-to-reduce-sugars-intake-among-adults-and-children (accessed on 5 February 2023).
- Dietary Guidelines Advisory Committee. Scientific Report of the 2020 Dietary Guidelines Advisory Committee: Advisory Report to the Secretary of Agriculture and the Secretary of Health and Human Services. US Department of Agriculture; Agricultural Research Service: Washington, DC, USA, 2020.
- Kaiser, A.; Schaefer, S.; Behrendt, I.; Eichner, G.; Fasshauer, M. Association of all-cause mortality with sugar intake from different sources in the prospective cohort of UK Biobank participants. Br. J. Nutr. 2022, 62, 727–738. [Google Scholar] [CrossRef]
- Jung, S.; Bae, H.; Song, W.S.; Jang, C. Dietary Fructose and Fructose-Induced Pathologies. Annu. Rev. Nutr. 2022, 42, 45–66. [Google Scholar] [CrossRef]
- Hotta, N.; Kawamura, T.; Umemura, T. Does the breakdown of the detoxification system for aldehydes as a result of aldose reductase upregulation lead to alcohol-induced liver injury in humans and mice? J. Diabetes Investig. 2020, 11, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K. Short- and Long-Term Adaptation to Altered Levels of Glucose: Fifty Years of Scientific Adventure. Annu. Rev. Biochem. 2021, 90, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The Roles of Carbohydrate Response Element Binding Protein in the Relationship between Carbohydrate Intake and Diseases. Int. J. Mol. Sci. 2021, 22, 12058. [Google Scholar] [CrossRef]
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef]
- Iizuka, K. The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. Nutrients 2017, 9, 181. [Google Scholar] [CrossRef]
- Lee, H.J.; Cha, J.Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef]
- Johnson, R.J.; Stenvinkel, P.; Andrews, P.; Sánchez-Lozada, L.G.; Nakagawa, T.; Gaucher, E.; Andres-Hernando, A.; Rodriguez-Iturbe, B.; Jimenez, C.R.; Garcia, G.; et al. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J. Intern. Med. 2020, 287, 252–262. [Google Scholar] [CrossRef]
- Laughlin, M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014, 6, 3117–3129. [Google Scholar] [CrossRef]
- Patel, C.; Sugimoto, K.; Douard, V.; Shah, A.; Inui, H.; Yamanouchi, T.; Ferraris, R.P. Effect of dietary fructose on portal and systemic serum fructose levels in rats and in KHK−/− and GLUT5−/− mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G779–G790. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef]
- Sillero, M.A.; Sillero, A.; Sols, A. Enzymes involved in fructose metabolism in liver and the glyceraldehyde metabolic crossroads. Eur. J. Biochem. 1969, 10, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Iizuka, K.; Takao, K.; Horikawa, Y.; Kitamura, T.; Takeda, J. ChREBP-Knockout Mice Show Sucrose Intolerance and Fructose Malabsorption. Nutrients 2018, 10, 340. [Google Scholar] [CrossRef]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef]
- Oh, A.R.; Sohn, S.; Lee, J.; Park, J.M.; Nam, K.T.; Hahm, K.B.; Kim, Y.B.; Lee, H.J.; Cha, J.Y. ChREBP deficiency leads to diarrhea-predominant irritable bowel syndrome. Metabolism 2018, 85, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Drucker, W.R.; Owens, J.E.; Craig, J.W.; Woodward, H., Jr. Metabolism of intravenous fructose and glucose in normal and diabetic subjects. J. Clin. Investig. 1952, 31, 115–125. [Google Scholar] [CrossRef]
- Koo, H.Y.; Miyashita, M.; Cho, B.H.; Nakamura, M.T. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 2009, 390, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Hochuli, M.; Aeberli, I.; Weiss, A.; Hersberger, M.; Troxler, H.; Gerber, P.A.; Spinas, G.A.; Berneis, K. Spinas, Kaspar Berneis, Sugar-Sweetened Beverages With Moderate Amounts of Fructose, but Not Sucrose, Induce Fatty Acid Synthesis in Healthy Young Men: A Randomized Crossover Study. J. Clin. Endocrinol. Metab. 2014, 99, 2164–2172. [Google Scholar] [CrossRef]
- Geidl-Flueck, B.; Hochuli, M.; Németh, Á.; Eberl, A.; Derron, N.; Köfeler, H.C.; Tappy, L.; Berneis, K.; Spinas, G.A.; Gerber, P.A. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: A randomized controlled trial. J. Hepatol. 2021, 75, 46–54. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wollheim, C.B. ChREBP rather than USF2 regulates glucose stimulation of endogenous L-pyruvate kinase expression in insulin-secreting cells. J. Biol. Chem. 2002, 277, 32746–32752. [Google Scholar] [CrossRef]
- Da Silva Xavier, G.; Rutter, G.A.; Diraison, F.; Andreolas, C.; Leclerc, I. ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic beta-cells. J. Lipid Res. 2006, 47, 2482–2491. [Google Scholar] [CrossRef]
- Zhang, P.; Kumar, A.; Katz, L.S.; Li, L.; Paulynice, M.; Herman, M.A.; Scott, D.K. Induction of the ChREBPβ Isoform Is Essential for Glucose-Stimulated β-Cell Proliferation. Diabetes 2015, 64, 4158–4170. [Google Scholar] [CrossRef]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef]
- Wei, C.; Wang, P.; Dong, Q.; Ma, X.H.; Lu, M.; Qi, S.; Shi, J.H.; Xie, Z.; Ren, A.J.; Zhang, W.J. ChREBP-regulated lipogenesis is not required for the thermogenesis of brown adipose tissue. Int. J. Obes. 2022, 46, 1068–1075. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Tang, Y.; Jespersen, N.Z.; Wallace, M.; Martinez Calejman, C.; Gujja, S.; Li, H.; Edwards, Y.J.K.; Wolfrum, C.; Metallo, C.M.; et al. Brown Fat AKT2 Is a Cold-Induced Kinase that Stimulates ChREBP-Mediated De Novo Lipogenesis to Optimize Fuel Storage and Thermogenesis. Cell Metab. 2018, 27, 195–209.e6. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Iizuka, K.; Liu, Y.; Sakurai, T.; Kubota, S.; Kubota-Okamoto, S.; Imaizumi, T.; Takahashi, Y.; Rakhat, Y.; Komori, S.; et al. Effects of ChREBP deficiency on adrenal lipogenesis and steroidogenesis. J. Endocrinol. 2021, 248, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Saito, M.; Takeda, J. Feedback looping between ChREBP and PPARα in the regulation of lipid metabolism in brown adipose tissues. Endocr. J. 2013, 60, 1145–1153. [Google Scholar] [CrossRef]
- Chen, N.; Song, S.; Yang, Z.; Wu, M.; Mu, L.; Zhou, T.; Shi, Y. ChREBP deficiency alleviates apoptosis by inhibiting TXNIP/oxidative stress in diabetic nephropathy. J. Diabetes Complicat. 2021, 35, 108050. [Google Scholar] [CrossRef]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet ChREBP-β is increased in diabetes and controls ChREBP-α and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Recazens, E.; Tavernier, G.; Dufau, J.; Bergoglio, C.; Benhamed, F.; Cassant-Sourdy, S.; Marques, M.A.; Caspar-Bauguil, S.; Brion, A.; Monbrun, L.; et al. ChREBPβ is dispensable for the control of glucose homeostasis and energy balance. JCI Insight 2022, 7, e153431. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-response Element-binding Protein (ChREBP): Role of AMP as an allosteric inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferré, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Takeda, J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic β cell line, INS-1E. Endocr. J. 2013, 60, 473–482. [Google Scholar] [CrossRef]
- Nishimura, M.; Uyeda, K. Purification and characterization of a novel xylulose 5-phosphate-activated protein phosphatase catalyzing dephosphorylation of fructose-6-phosphate,2-kinase:fructose-2,6-bisphosphatase. J. Biol. Chem. 1995, 270, 26341–26346. [Google Scholar] [CrossRef]
- Uyeda, K.; Furuya, E.; Luby, L.J. The effect of natural and synthetic D-fructose 2,6-bisphosphate on the regulatory kinetic properties of liver and muscle phosphofructokinases. J. Biol. Chem. 1981, 256, 8394–8399. [Google Scholar] [CrossRef]
- Liu, L.; Li, T.; Liao, Y.; Wang, Y.; Gao, Y.; Hu, H.; Huang, H.; Wu, F.; Chen, Y.G.; Xu, S.; et al. Triose Kinase Controls the Lipogenic Potential of Fructose and Dietary Tolerance. Cell Metab. 2020, 32, 605–618.e7. [Google Scholar] [CrossRef]
- McFerrin, L.G.; Atchley, W.R. A novel N-terminal domain may dictate the glucose response of Mondo proteins. PLoS ONE 2012, 7, e34803. [Google Scholar] [CrossRef]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Lu, J.Y.; Chen, H.Y.; Wei, C.C.; Xu, X.; Li, H.; Bai, Q.; Xia, F.Z.; Lam, S.M.; Zhang, H.; et al. Liver ChREBP Protects Against Fructose-Induced Glycogenic Hepatotoxicity by Regulating L-Type Pyruvate Kinase. Diabetes 2020, 69, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.B.; Zhang, P.; Doumen, C.; Charbonnet, M.; Lu, D.; Newgard, C.B.; Haycock, J.W.; Lange, A.J.; Scott, D.K. The promoter for the gene encoding the catalytic subunit of rat glucose-6-phosphatase contains two distinct glucose-responsive regions. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E788–E801. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; Schreurs, M.; Oosterveer, M.H.; Cortés, V.A.; Havinga, R.; Herling, A.W.; Reijngoud, D.J.; Groen, A.K.; Kuipers, F. Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1. Biochem. J. 2010, 432, 249–254. [Google Scholar] [CrossRef]
- Lei, Y.; Hoogerland, J.A.; Bloks, V.W.; Bos, T.; Bleeker, A.; Wolters, H.; Wolters, J.C.; Hijmans, B.S.; van Dijk, T.H.; Thomas, R.; et al. Hepatic Carbohydrate Response Element Binding Protein Activation Limits Nonalcoholic Fatty Liver Disease Development in a Mouse Model for Glycogen Storage Disease Type 1a. Hepatology 2020, 72, 1638–1653. [Google Scholar] [CrossRef] [PubMed]
- Rajas, F.; Dentin, R.; Cannella Miliano, A.; Silva, M.; Raffin, M.; Levavasseur, F.; Gautier-Stein, A.; Postic, C.; Mithieux, G. The absence of hepatic glucose-6 phosphatase/ChREBP couple is incompatible with survival in mice. Mol. Metab. 2021, 43, 101108. [Google Scholar] [CrossRef]
- Göğüş, S.; Koçak, N.; Ciliv, G.; Karabulut, E.; Akçören, Z.; Kale, G.; Cağlar, M. Histologic features of the liver in type Ia glycogen storage disease: Comparative study between different age groups and consecutive biopsies. Pediatr. Dev. Pathol. 2002, 5, 299–304. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, X.; VanDommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, M.B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867. [Google Scholar] [CrossRef]
- Sugimoto, K.; Hosotani, T.; Kawasaki, T.; Nakagawa, K.; Hayashi, S.; Nakano, Y.; Inui, H.; Yamanouchi, T. Eucalyptus leaf extract suppresses the postprandial elevation of portal, cardiac, and peripheral fructose concentrations after sucrose ingestion in rats. J. Clin. Biochem. Nutr. 2012, 46, 205–211. [Google Scholar] [CrossRef]
- Nakagawa, T.; Johnson, R.J.; Andres-Hernando, A.; Roncal-Jimenez, C.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Fructose Production and Metabolism in the Kidney. J. Am. Soc. Nephrol. 2020, 31, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Jones, D.I.; Aird, I.A.; Biljan, M.M.; Kingsland, C.R. Effects of sperm activity on zinc and fructose concentrations in seminal plasma. Hum. Reprod. 1996, 11, 2465–2467. [Google Scholar] [CrossRef]
- Buckett, W.M.; Lewis-Jones, D.I. Fructose concentrations in seminal plasma from men with nonobstructive azoospermia. Arch. Androl. 2002, 48, 23–27. [Google Scholar] [CrossRef]
- Mesonero, J.; Matosin, M.; Cambier, D.; Rodriguez-Yoldi, M.J.; Brot-Laroche, E. Sugar-dependent expression of the fructose transporter GLUT5 in Caco-2 cells. Biochem. J. 1995, 312 Pt 3, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Mahraoui, L.; Takeda, J.; Mesonero, J.; Chantret, I.; Dussaulx, E.; Bell, G.I.; Brot-Laroche, E. Regulation of expression of the human fructose transporter (GLUT5) by cyclic AMP. Biochem. J. 1994, 301 Pt 1, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gouyon, F.; Onesto, C.; Dalet, V.; Pages, G.; Leturque, A.; Brot-Laroche, E. Fructose modulates GLUT5 mRNA stability in differentiated Caco-2 cells: Role of cAMP-signaling pathway and PABP (polyadenylated-binding protein)-interacting protein (Paip) 2. Biochem. J. 2003, 375 Pt 1, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Matosin-Matekalo, M.; Mesonero, J.E.; Laroche, T.J.; Lacasa, M.; Brot-Laroche, E. Glucose and thyroid hormone coregulate the expression of the intestinal fructose transporter GLUT5. Biochem J. 1999, 339 Pt 2, 233–239. [Google Scholar] [CrossRef]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef]
- Taneva, I.; Grumann, D.; Schmidt, D.; Taneva, E.; von Arnim, U.; Ansorge, T.; Wex, T. Gene variants of the SLC2A5 gene encoding GLUT5, the major fructose transporter, do not contribute to clinical presentation of acquired fructose malabsorption. BMC Gastroenterol. 2022, 22, 167. [Google Scholar] [CrossRef]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2020, 3, 100217. [Google Scholar] [CrossRef] [PubMed]
- Bonthron, D.T.; Brady, N.; Donaldson, I.A.; Steinmann, B. Molecular basis of essential fructosuria: Molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum. Mol. Genet. 1994, 3, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Tran, C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients 2017, 9, 356. [Google Scholar] [CrossRef]
- Laron, Z. Essential benign fructosuria. Arch. Dis. Child. 1961, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Boesiger, P.; Buchli, R.; Meier, D.; Steinmann, B.; Gitzelmann, R. Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 31P magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 436–440. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Jang, C.; Li, N.; Milagres, T.; Kuwabara, M.; Wempe, M.F.; Rabinowitz, J.D.; et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig. 2018, 128, 2226–2238. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef]
- Oppelt, S.A.; Sennott, E.M.; Tolan, D.R. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab. 2015, 114, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, N.; Zhang, C.; Li, M.; He, X.; Yin, C.; Tu, Q.; Shen, X.; Zhang, L.; Lv, J.; et al. Fructose-1,6-Bisphosphate Aldolase B Depletion Promotes Hepatocellular Carcinogenesis Through Activating Insulin Receptor Signaling and Lipogenesis. Hepatology 2021, 74, 3037–3055. [Google Scholar] [CrossRef]
- He, X.; Li, M.; Yu, H.; Liu, G.; Wang, N.; Yin, C.; Tu, Q.; Narla, G.; Tao, Y.; Cheng, S.; et al. Loss of hepatic aldolase B activates Akt and promotes hepatocellular carcinogenesis by destabilizing the Aldob/Akt/PP2A protein complex. PLoS Biol. 2020, 18, e3000803. [Google Scholar] [CrossRef]
- Rodrigues, J.R.; Couto, A.; Cabezas, A.; Pinto, R.M.; Ribeiro, J.M.; Canales, J.; Costas, M.J.; Cameselle, J.C. Bifunctional homodimeric triokinase/FMN cyclase: Contribution of protein domains to the activities of the human enzyme and molecular dynamics simulation of domain movements. J. Biol. Chem. 2014, 289, 10620–10636. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Meunier, B.; Mestek-Boukhibar, L.; van den Broek, F.; Maldonado, E.M.; Clement, E.; Weghuber, D.; Spenger, J.; Jaros, Z.; Taha, F.; et al. Biallelic Variants in TKFC Encoding Triokinase/FMN Cyclase Are Associated with Cataracts and Multisystem Disease. Am. J. Hum. Genet. 2020, 106, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Cabezas, A.; Ng, J.C.F.; Canales, J.; Costas, M.J.; Ribeiro, J.M.; Rodrigues, J.R.; McAleer, M.A.; Castelo-Soccio, L.; Simpson, M.A.; et al. Autosomal recessive hypotrichosis with loose anagen hairs associated with TKFC mutations. Br. J. Dermatol. 2021, 184, 935–943. [Google Scholar] [CrossRef]
- Ribeiro, J.M.; Costas, M.J.; Cabezas, A.; Meunier, B.; Onoufriadis, A.; Cameselle, J.C. The TKFC Ala185Thr variant, reported as ‘null’ for fructose metabolism, is fully active as triokinase. FEBS Lett. 2022, 596, 1453–1457. [Google Scholar] [CrossRef]
- Chou, C.L.; Li, C.H.; Lin, H.; Liao, M.H.; Wu, C.C.; Chen, J.S.; Sue, Y.M.; Fang, T.C. Role of activating transcription factor 3 in fructose-induced metabolic syndrome in mice. Hypertens. Res. 2018, 41, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; Tomita, S.; Sekiya, M.; et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 2004, 53, 560–569. [Google Scholar] [CrossRef]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Löffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef]
- Batista, L.O.; Ramos, V.W.; Rosas Fernández, M.A.; Concha Vilca, C.M.; Albuquerque, K.T. Oral solution of fructose promotes SREBP-1c high-expression in the hypothalamus of Wistar rats. Nutr. Neurosci. 2019, 22, 648–654. [Google Scholar] [CrossRef]
- Pan, J.H.; Tang, J.; Beane, K.E.; Redding, M.C.; Cho, Y.J.; Kim, Y.J.; Zhao, J.; Shin, E.C.; Lee, J.H.; Kong, B.C.; et al. Hepatic transcriptomics analysis reveals that fructose intervention down-regulated xenobiotics-metabolising enzymes through aryl hydrocarbon receptor signalling suppression in C57BL/6N mice. Br. J. Nutr. 2019, 122, 769–779. [Google Scholar] [CrossRef]
- Noordeen, N.A.; Khera, T.K.; Sun, G.; Longbottom, E.R.; Pullen, T.J.; da Silva Xavier, G.; Rutter, G.A.; Leclerc, I. Carbohydrate-responsive element-binding protein (ChREBP) is a negative regulator of ARNT/HIF-1beta gene expression in pancreatic islet beta-cells. Diabetes 2010, 59, 153–160. [Google Scholar] [CrossRef]
- Clements, R.S., Jr. The polyol pathway. A historical review. Drugs 1986, 32 (Suppl. S2), 3–5. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.S.; Gupta, J. Polyol pathway and redox balance in diabetes. Pharmacol. Res. 2022, 182, 106326. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. [Google Scholar] [CrossRef]
- Sano, H.; Nakamura, A.; Yamane, M.; Niwa, H.; Nishimura, T.; Araki, K.; Takemoto, K.; Ishiguro, K.I.; Aoki, H.; Kato, Y.; et al. The polyol pathway is an evolutionarily conserved system for sensing glucose uptake. PLoS Biol. 2022, 20, e3001678. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef]
- Qiu, L.; Lin, J.; Ying, M.; Chen, W.; Yang, J.; Deng, T.; Chen, J.; Shi, D.; Yang, J.Y. Aldose Reductase Is Involved in the Development of Murine Diet-Induced Nonalcoholic Steatohepatitis. PLoS ONE 2013, 8, e73591. [Google Scholar] [CrossRef]
- Shi, C.; Wang, Y.; Gao, J.; Chen, S.; Zhao, X.; Cai, C.; Guo, C.; Qiu, L. Inhibition of aldose reductase ameliorates alcoholic liver disease by activating AMPK and modulating oxidative stress and inflammatory cytokines. Mol. Med. Rep. 2017, 16, 2767–2772. [Google Scholar] [CrossRef]
- Qiu, L.; Cai, C.; Zhao, X.; Fang, Y.; Tang, W.; Guo, C. Inhibition of aldose reductase ameliorates ethanol-induced steatosis in HepG2 cells. Mol. Med. Rep. 2017, 15, 2732–2736. [Google Scholar] [CrossRef]
- Le, Y.; Chen, L.; Zhang, Y.; Bu, P.; Dai, G.; Cheng, X. Epalrestat Stimulated Oxidative Stress, Inflammation, and Fibrogenesis in Mouse Liver. Toxicol. Sci. 2018, 163, 397–408. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Ross, R.A.; Crabb, D.W. Activation of carbohydrate response element-binding protein by ethanol. J. Investig. Med. 2013, 61, 270–277. [Google Scholar] [CrossRef]
- Liangpunsakul, S. Carbohydrate-responsive element-binding protein, Sirtuin 1, and ethanol metabolism: A complicated network in alcohol-induced hepatic steatosis. Hepatology 2015, 62, 946–994. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iizuka, K. Recent Progress on Fructose Metabolism—Chrebp, Fructolysis, and Polyol Pathway. Nutrients 2023, 15, 1778. https://doi.org/10.3390/nu15071778
Iizuka K. Recent Progress on Fructose Metabolism—Chrebp, Fructolysis, and Polyol Pathway. Nutrients. 2023; 15(7):1778. https://doi.org/10.3390/nu15071778
Chicago/Turabian StyleIizuka, Katsumi. 2023. "Recent Progress on Fructose Metabolism—Chrebp, Fructolysis, and Polyol Pathway" Nutrients 15, no. 7: 1778. https://doi.org/10.3390/nu15071778
APA StyleIizuka, K. (2023). Recent Progress on Fructose Metabolism—Chrebp, Fructolysis, and Polyol Pathway. Nutrients, 15(7), 1778. https://doi.org/10.3390/nu15071778