

Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Sample Collection
2.3. Commercial Lipidomic Preparation and Detection
2.4. Lipidomic Data Processing
2.5. RNA Extraction and Quantitative Real-Time PCR
2.6. Hepatic Malondialdehyde Measurements
2.7. Serum Alanine Transaminase (ALT) Measurements
2.8. Statistical Analysis
3. Results
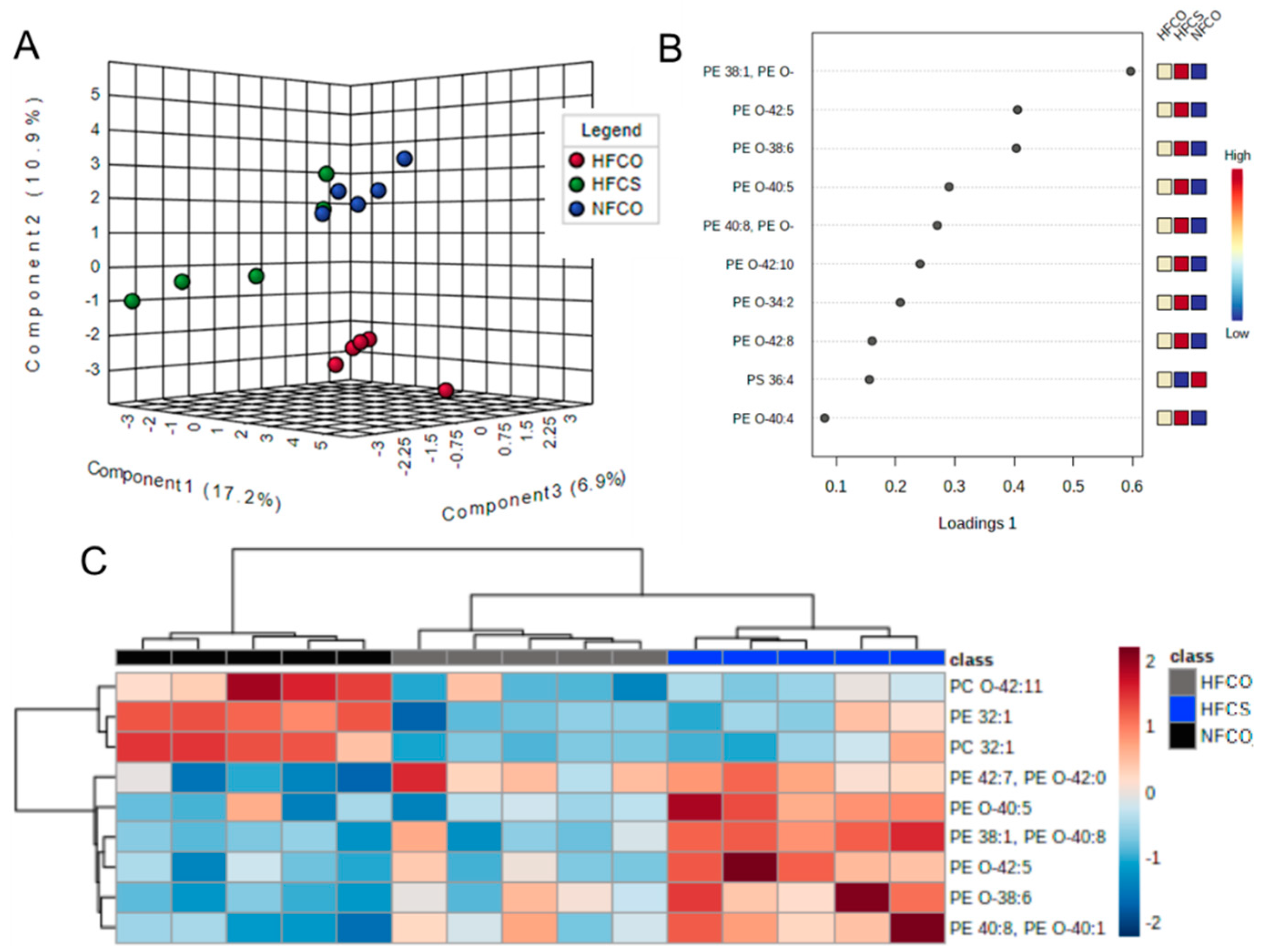
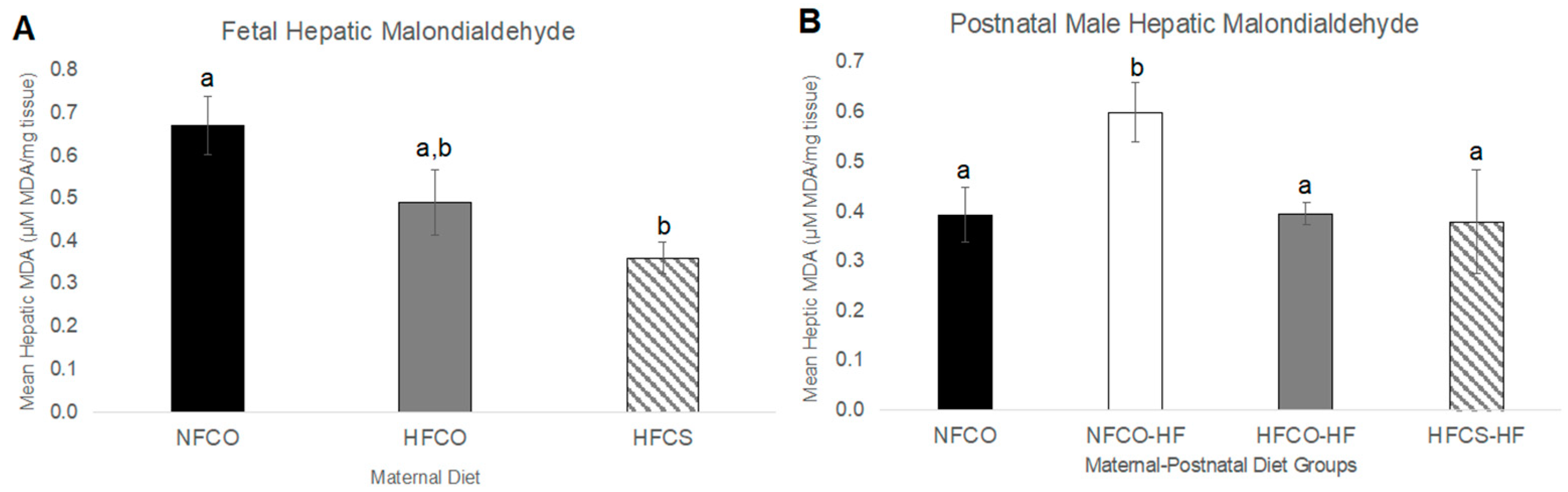
3.1. Prenatal Choline Supplement during Maternal Obesity Modulates Fetal Hepatic Phospholipidomics and Lowers Oxidative Stress
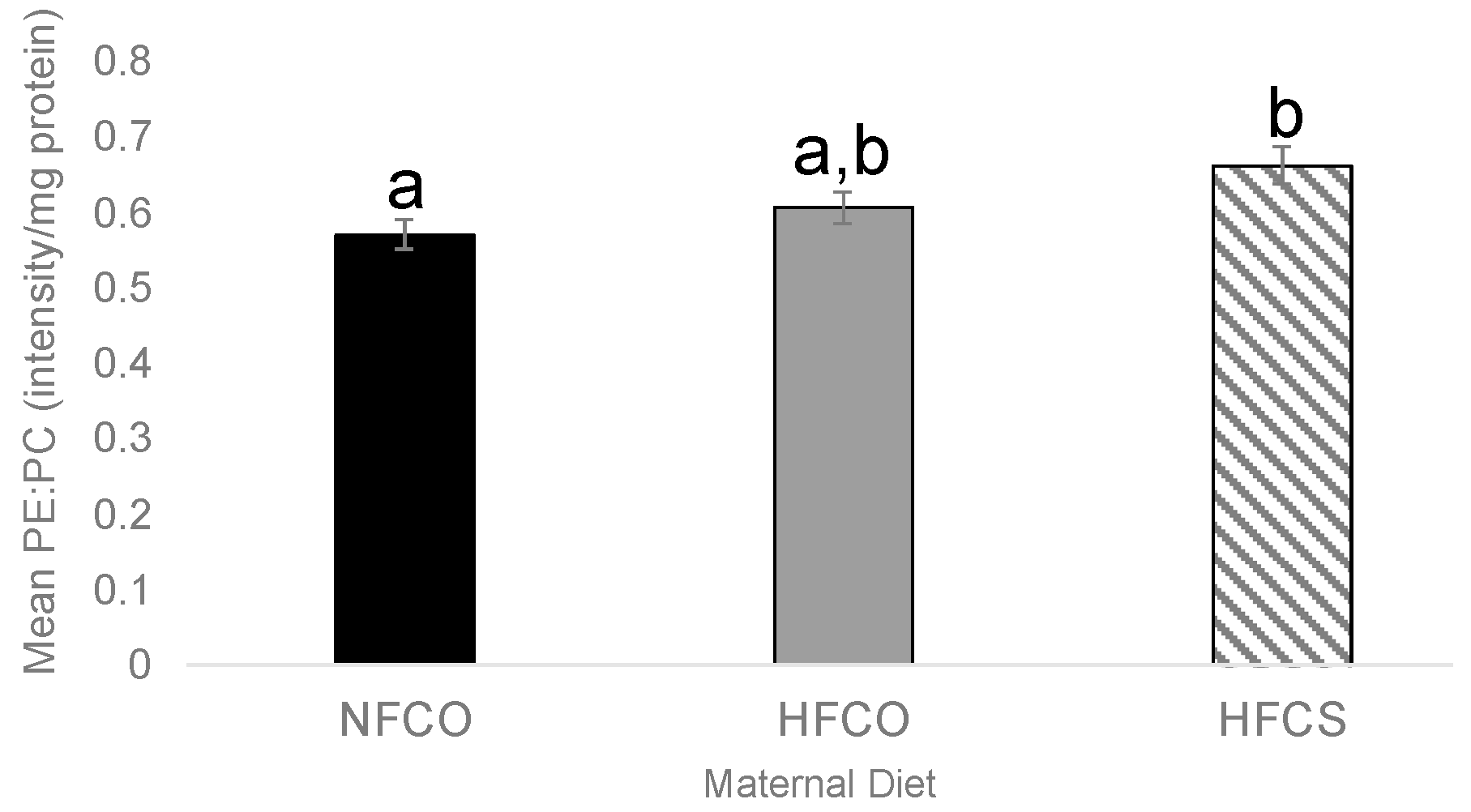
3.2. Prenatal Choline Supplement during Maternal Obesity Lowers Fetal Hepatic PE:PC Ratio
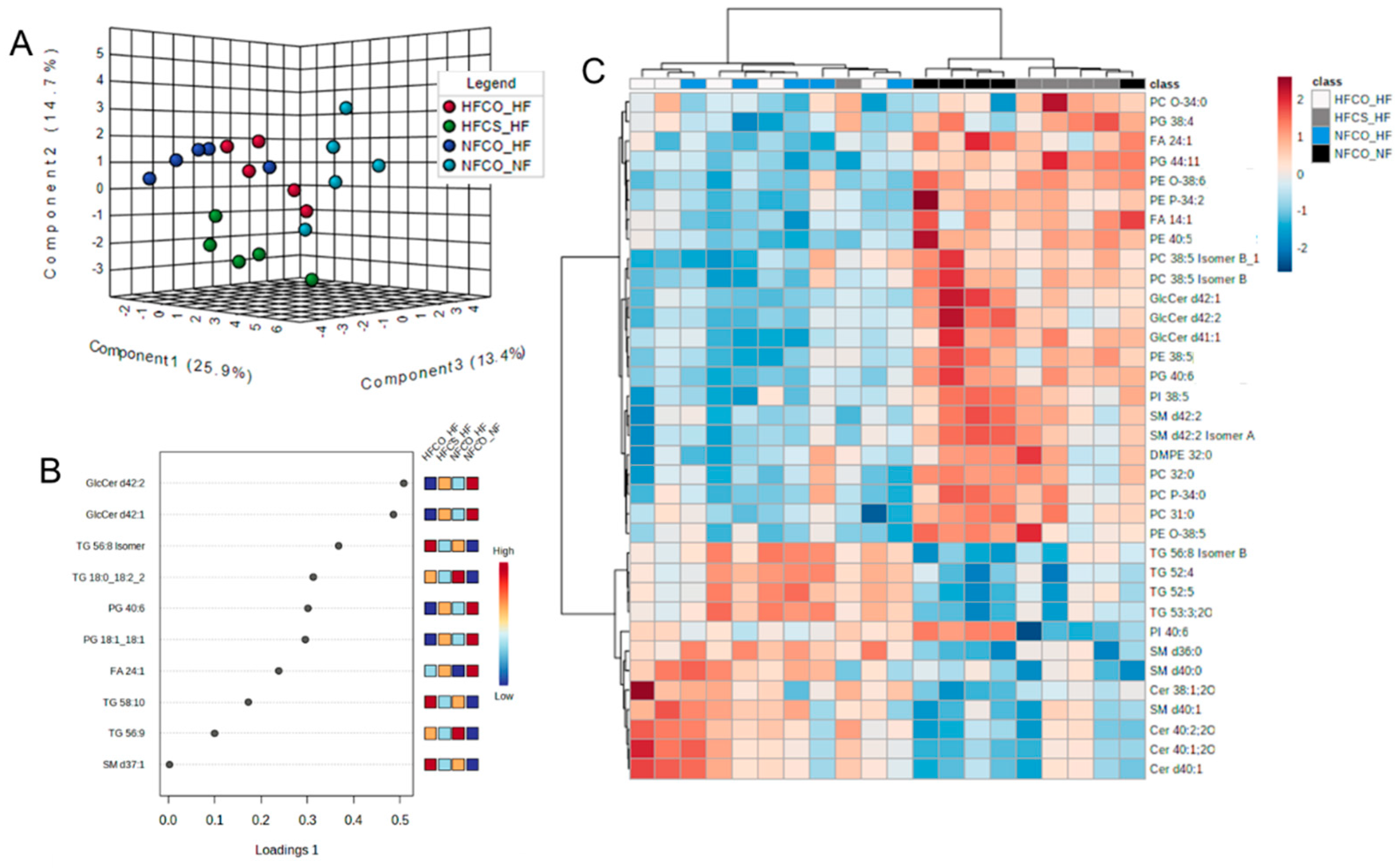
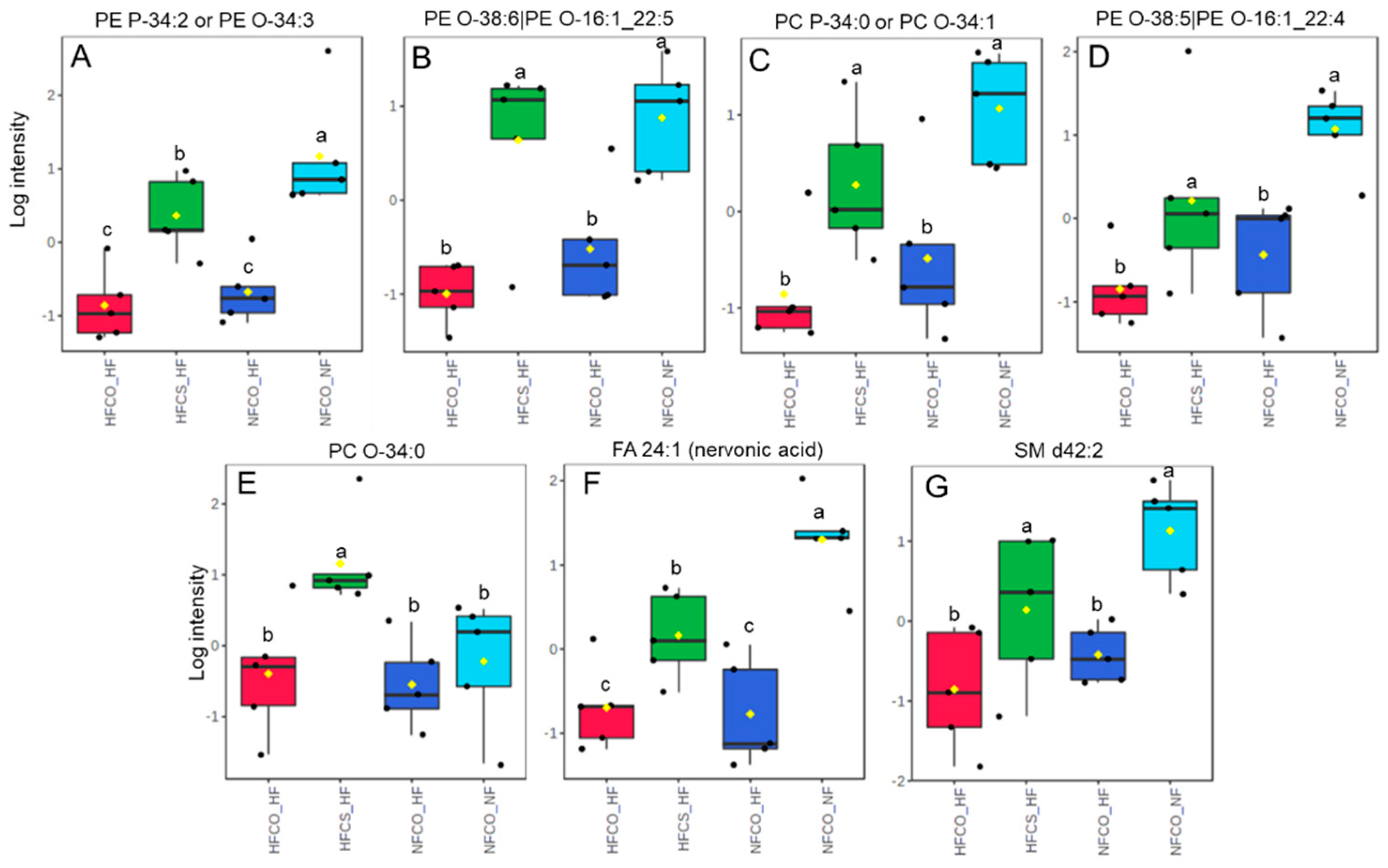
3.3. Prenatal Choline Supplement during Maternal Obesity Modulates the Complex Lipidomic Profile of Male Offspring Livers
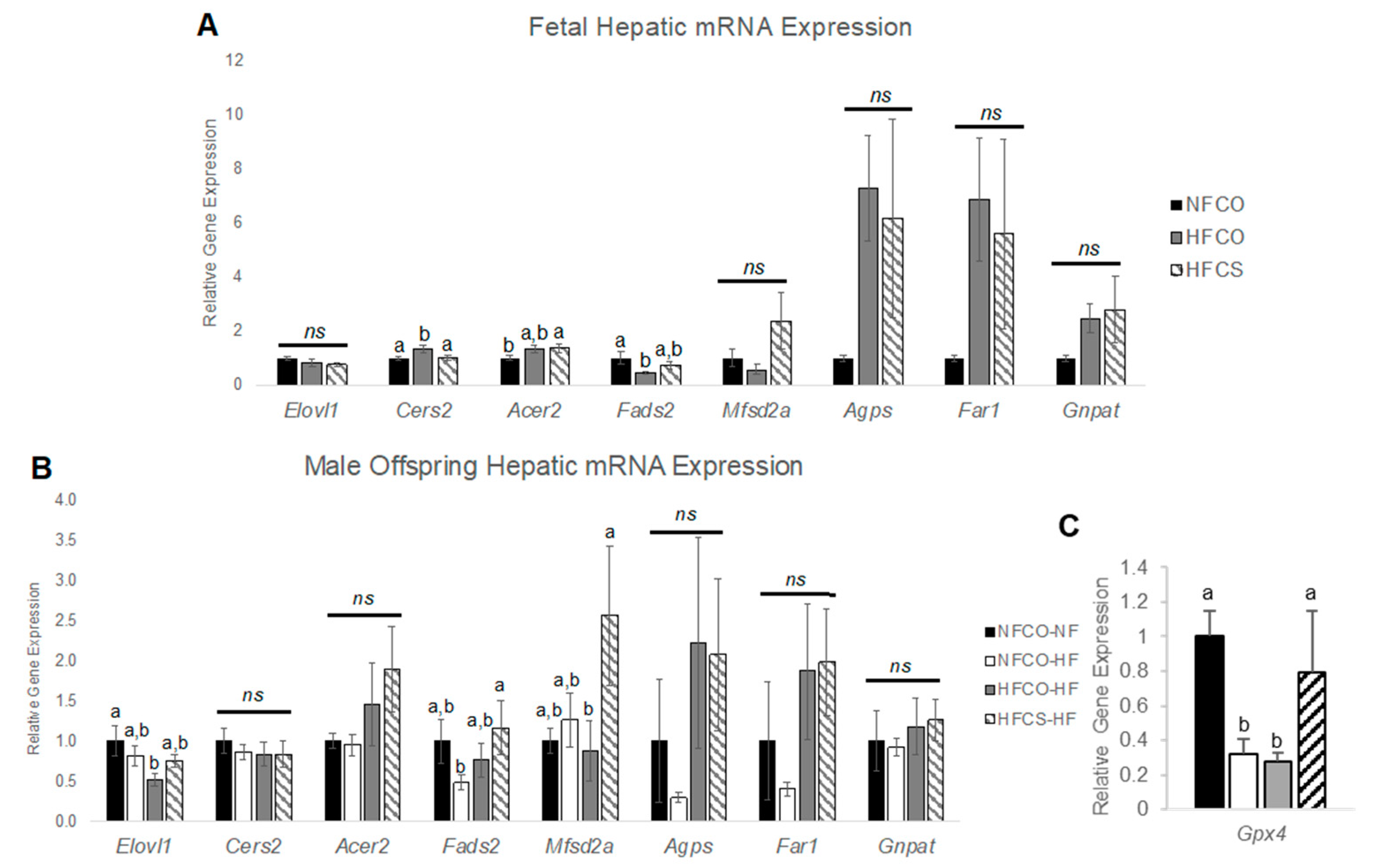
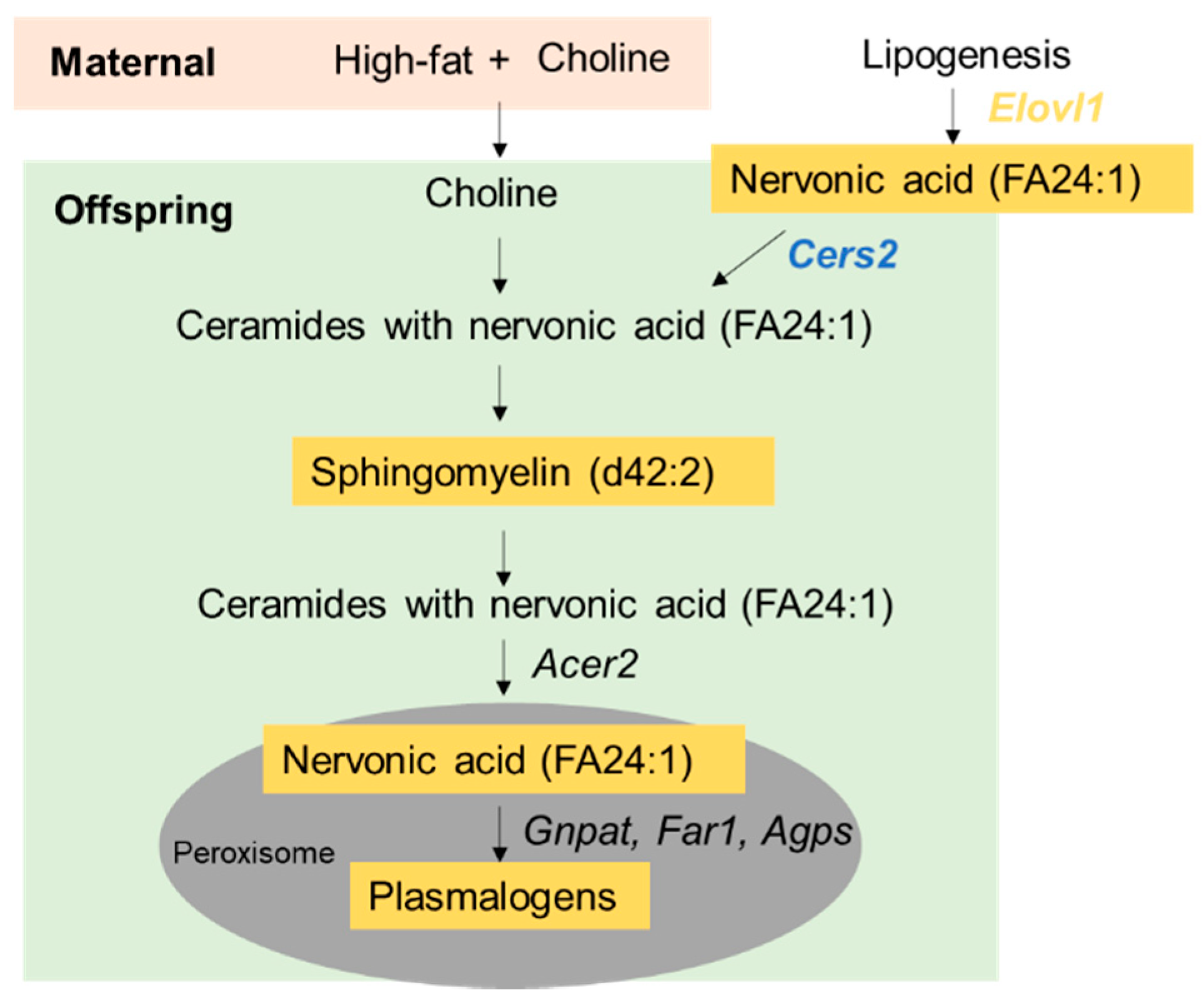
3.4. Prenatal Choline Supplement during Maternal Obesity Modulates Fetal and Male Offspring Hepatic mRNA Expression Related to Plasmalogen Synthesis and Lipid Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef]
- Nash, M.J.; Dobrinskikh, E.; Newsom, S.A.; Messaoudi, I.; Janssen, R.C.; Aagaard, K.M.; McCurdy, C.E.; Gannon, M.; Kievit, P.; Friedman, J.E.; et al. Maternal Western diet exposure increases periportal fibrosis beginning in utero in nonhuman primate offspring. JCI Insight 2021, 6, e154093. [Google Scholar] [CrossRef]
- Thompson, M.D. Developmental Programming of NAFLD by Parental Obesity. Hepatol. Commun. 2020, 4, 1392–1403. [Google Scholar] [CrossRef]
- Gutierrez Sanchez, L.H.; Tomita, K.; Guo, Q.; Furuta, K.; Alhuwaish, H.; Hirsova, P.; Baheti, S.; Alver, B.; Hlady, R.; Robertson, K.D.; et al. Perinatal Nutritional Reprogramming of the Epigenome Promotes Subsequent Development of Nonalcoholic Steatohepatitis. Hepatol. Commun. 2018, 2, 1493–1512. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Panel, I.C. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Cheng, J.; Williams, J.; Brown, S.; dela Pena, A.; Bell-Anderson, K.S.; Farrell, G.C. Activation of peroxisome proliferator-activated receptor alpha by dietary fish oil attenuates steatosis, but does not prevent experimental steatohepatitis because of hepatic lipoperoxide accumulation. J. Gastroenterol. Hepatol. 2008, 23, 267–275. [Google Scholar] [CrossRef]
- Wiering, L.; Tacke, F. Treating inflammation to combat non-alcoholic fatty liver disease. J. Endocrinol. 2022, 256, e220194. [Google Scholar] [CrossRef]
- Zisser, A.; Ipsen, D.H.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis-Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Kartsoli, S.; Kostara, C.E.; Tsimihodimos, V.; Bairaktari, E.T.; Christodoulou, D.K. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kondo, K.; Maeba, R.; Nishimukai, M.; Nezu, T.; Hara, H. Proportion of nervonic acid in serum lipids is associated with serum plasmalogen levels and metabolic syndrome. J. Oleo Sci. 2014, 63, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Öhman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef]
- Konstantynowicz-Nowicka, K.; Berk, K.; Chabowski, A.; Kasacka, I.; Bielawiec, P.; Łukaszuk, B.; Harasim-Symbor, E. High-Fat Feeding in Time-Dependent Manner Affects Metabolic Routes Leading to Nervonic Acid Synthesis in NAFLD. Int. J. Mol. Sci. 2019, 20, 3829. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012, 26, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Strupp, B.J.; Muscalu, L.; Nevins, J.E.H.; Canfield, R.L. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB J. 2018, 32, 2172–2180. [Google Scholar] [CrossRef]
- Meck, W.H.; Williams, C.L. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 2003, 27, 385–399. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Mellott, T.J. Choline nutrition programs brain development via DNA and histone methylation. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 82–94. [Google Scholar] [CrossRef]
- Kwan, S.T.C.; King, J.H.; Grenier, J.K.; Yan, J.; Jiang, X.; Roberson, M.S.; Caudill, M.A. Maternal Choline Supplementation during Normal Murine Pregnancy Alters the Placental Epigenome: Results of an Exploratory Study. Nutrients 2018, 10, 417. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Dave, B.; Trasino, S.; Saxena, A.; Liu, J.; Caviglia, J.M.; Edwards, K.; Dembitzer, M.; Sheeraz, S.; Khaldi, S.; et al. Maternal Choline Supplementation and High-Fat Feeding Interact to Influence DNA Methylation in Offspring in a Time-Specific Manner. Front. Nutr. 2022, 9, 841787. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Jiang, X.; Caudill, M.A. Choline: Exploring the Growing Science on Its Benefits for Moms and Babies. Nutrients 2019, 11, 1823. [Google Scholar] [CrossRef] [PubMed]
- Korsmo, H.W.; Jiang, X. One carbon metabolism and early development: A diet-dependent destiny. Trends Endocrinol. Metab. 2021, 32, 579–593. [Google Scholar] [CrossRef] [PubMed]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.H.; Chan, J.P.; Cazenave-Gassiot, A.; Poh, R.W.; Foo, J.C.; Galam, D.L.; Ghosh, S.; Nguyen, L.N.; Barathi, V.A.; Yeo, S.W.; et al. Mfsd2a Is a Transporter for the Essential ω-3 Fatty Acid Docosahexaenoic Acid (DHA) in Eye and Is Important for Photoreceptor Cell Development. J. Biol. Chem. 2016, 291, 10501–10514. [Google Scholar] [CrossRef] [PubMed]
- Thomas Rajarethnem, H.; Megur Ramakrishna Bhat, K.; Jc, M.; Kumar Gopalkrishnan, S.; Mugundhu Gopalram, R.B.; Rai, K.S. Combined Supplementation of Choline and Docosahexaenoic Acid during Pregnancy Enhances Neurodevelopment of Fetal Hippocampus. Neurol. Res. Int. 2017, 2017, 8748706. [Google Scholar] [CrossRef]
- Nam, J.; Greenwald, E.; Jack-Roberts, C.; Ajeeb, T.T.; Malysheva, O.V.; Caudill, M.A.; Axen, K.; Saxena, A.; Semernina, E.; Nanobashvili, K.; et al. Choline prevents fetal overgrowth and normalizes placental fatty acid and glucose metabolism in a mouse model of maternal obesity. J. Nutr. Biochem. 2017, 49, 80–88. [Google Scholar] [CrossRef]
- Jack-Roberts, C.; Joselit, Y.; Nanobashvili, K.; Bretter, R.; Malysheva, O.V.; Caudill, M.A.; Saxena, A.; Axen, K.; Gomaa, A.; Jiang, X. Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity. Nutrients 2017, 9, 899. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Edwards, K.; Dave, B.; Jack-Roberts, C.; Yu, H.; Saxena, A.; Salvador, M.; Dembitzer, M.; Phagoora, J.; Jiang, X. Prenatal Choline Supplementation during High-Fat Feeding Improves Long-Term Blood Glucose Control in Male Mouse Offspring. Nutrients 2020, 12, 144. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid. Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- Barupal, D.K.; Zhang, Y.; Shen, T.; Fan, S.; Roberts, B.S.; Fitzgerald, P.; Wancewicz, B.; Valdiviez, L.; Wohlgemuth, G.; Byram, G.; et al. A Comprehensive Plasma Metabolomics Dataset for a Cohort of Mouse Knockouts within the International Mouse Phenotyping Consortium. Metabolites 2019, 9, 101. [Google Scholar] [CrossRef]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, R.A.; Lake, A.C.; Nagan, N.; Gaposchkin, D.P.; Legner, M.A.; Lieberthal, W. Plasmalogens as endogenous antioxidants: Somatic cell mutants reveal the importance of the vinyl ether. Biochem. J. 1999, 338, 769–776. [Google Scholar] [CrossRef]
- Engelmann, B. Plasmalogens: Targets for oxidants and major lipophilic antioxidants. Biochem. Soc. Trans. 2004, 32, 147–150. [Google Scholar] [CrossRef]
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Walenbergh, S.M.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Hladik, A.; Göderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2017, 65, 1181–1195. [Google Scholar] [CrossRef]
- Busch, C.J.; Binder, C.J. Malondialdehyde epitopes as mediators of sterile inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 398–406. [Google Scholar] [CrossRef]
- Hendrikx, T.; Binder, C.J. Oxidation-Specific Epitopes in Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 607011. [Google Scholar] [CrossRef]
- Ohno, Y.; Suto, S.; Yamanaka, M.; Mizutani, Y.; Mitsutake, S.; Igarashi, Y.; Sassa, T.; Kihara, A. ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18439–18444. [Google Scholar] [CrossRef]
- Sztolsztener, K.; Konstantynowicz-Nowicka, K.; Harasim-Symbor, E.; Chabowski, A. Time-Dependent Changes in Hepatic Sphingolipid Accumulation and PI3K/Akt/mTOR Signaling Pathway in a Rat Model of NAFLD. Int. J. Mol. Sci. 2021, 22, 12478. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Sun, H.; Liu, X.; Zheng, Y.; Xu, D.; Wang, J.; Jia, D.; Han, X.; Liu, F.; et al. Defective Phosphatidylglycerol Remodeling Causes Hepatopathy, Linking Mitochondrial Dysfunction to Hepatosteatosis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 763–781. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A. Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem. 2012, 152, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Jin, J.; Xu, R.; Hu, W.; Szulc, Z.M.; Bielawski, J.; Obeid, L.M.; Mao, C. Substrate specificity, membrane topology, and activity regulation of human alkaline ceramidase 2 (ACER2). J. Biol. Chem. 2010, 285, 8995–9007. [Google Scholar] [CrossRef]
- Pu, W.; Zhang, H.; Huang, X.; Tian, X.; He, L.; Wang, Y.; Zhang, L.; Liu, Q.; Li, Y.; Zhao, H.; et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat. Commun. 2016, 7, 13369. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Bozelli, J.C.; Azher, S.; Epand, R.M. Plasmalogens and Chronic Inflammatory Diseases. Front. Physiol. 2021, 12, 730829. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Stadelmann-Ingrand, S.; Favreliere, S.; Fauconneau, B.; Mauco, G.; Tallineau, C. Plasmalogen degradation by oxidative stress: Production and disappearance of specific fatty aldehydes and fatty alpha-hydroxyaldehydes. Free Radic. Biol. Med. 2001, 31, 1263–1271. [Google Scholar] [CrossRef]
- Stadelmann-Ingrand, S.; Pontcharraud, R.; Fauconneau, B. Evidence for the reactivity of fatty aldehydes released from oxidized plasmalogens with phosphatidylethanolamine to form Schiff base adducts in rat brain homogenates. Chem. Phys. Lipids 2004, 131, 93–105. [Google Scholar] [CrossRef]
- Jang, J.E.; Park, H.S.; Yoo, H.J.; Baek, I.J.; Yoon, J.E.; Ko, M.S.; Kim, A.R.; Kim, H.S.; Lee, S.E.; Kim, S.W.; et al. Protective role of endogenous plasmalogens against hepatic steatosis and steatohepatitis in mice. Hepatology 2017, 66, 416–431. [Google Scholar] [CrossRef]
- Almsherqi, Z.A. Potential Role of Plasmalogens in the Modulation of Biomembrane Morphology. Front. Cell Dev. Biol. 2021, 9, 673917. [Google Scholar] [CrossRef] [PubMed]
- Sindelar, P.J.; Guan, Z.; Dallner, G.; Ernster, L. The protective role of plasmalogens in iron-induced lipid peroxidation. Free Radic. Biol. Med. 1999, 26, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campión, J.; Martínez, J.A. Weight gain induced by high-fat feeding involves increased liver oxidative stress. Obes. 2006, 14, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E. Effect of High-Fat Diets on Oxidative Stress, Cellular Inflammatory Response and Cognitive Function. Nutrients 2019, 11, 2579. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Gill, R.; Tsung, A.; Billiar, T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic. Biol. Med. 2010, 48, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Sassa, T.; Kihara, A. Metabolism of very long-chain Fatty acids: Genes and pathophysiology. Biomol. Ther. 2014, 22, 83–92. [Google Scholar] [CrossRef]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef]
- Chaurasia, B.; Summers, S.A. Ceramides-Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Sphingomyelinases and Liver Diseases. Biomolecules 2020, 10, 1497. [Google Scholar] [CrossRef] [PubMed]
- Marei, W.F.A.; Smits, A.; Mohey-Elsaeed, O.; Pintelon, I.; Ginneberge, D.; Bols, P.E.J.; Moerloose, K.; Leroy, J.L.M.R. Differential effects of high fat diet-induced obesity on oocyte mitochondrial functions in inbred and outbred mice. Sci. Rep. 2020, 10, 9806. [Google Scholar] [CrossRef]
- Monthé-Drèze, C.; Penfield-Cyr, A.; Smid, M.C.; Sen, S. Maternal Pre-Pregnancy Obesity Attenuates Response to Omega-3 Fatty Acids Supplementation During Pregnancy. Nutrients 2018, 10, 1908. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Wang, Y.F.; Xu, Q.H.; Chen, S.S. Omega-3 fatty acids as a treatment for non-alcoholic fatty liver disease in children: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2018, 37, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Ferchaud-Roucher, V.; Kramer, A.; Silva, E.; Pantham, P.; Weintraub, S.T.; Jansson, T.; Powell, T.L. A potential role for lysophosphatidylcholine in the delivery of long chain polyunsaturated fatty acids to the fetal circulation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 394–402. [Google Scholar] [CrossRef]
- Yan, J.; Jiang, X.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Brenna, J.T.; Stabler, S.P.; Allen, R.H.; Gregory, J.F.; Caudill, M.A. Pregnancy alters choline dynamics: Results of a randomized trial using stable isotope methodology in pregnant and nonpregnant women. Am. J. Clin. Nutr. 2013, 98, 1459–1467. [Google Scholar] [CrossRef]
- Yan, J.; Ginsberg, S.D.; Powers, B.; Alldred, M.J.; Saltzman, A.; Strupp, B.J.; Caudill, M.A. Maternal choline supplementation programs greater activity of the phosphatidylethanolamine N-methyltransferase (PEMT) pathway in adult Ts65Dn trisomic mice. FASEB J. 2014, 28, 4312–4323. [Google Scholar] [CrossRef]
- West, A.A.; Yan, J.; Jiang, X.; Perry, C.A.; Innis, S.M.; Caudill, M.A. Choline intake influences phosphatidylcholine DHA enrichment in nonpregnant women but not in pregnant women in the third trimester. Am. J. Clin. Nutr. 2013, 97, 718–727. [Google Scholar] [CrossRef]
- Taesuwan, S.; McDougall, M.Q.; Malysheva, O.V.; Bender, E.; Nevins, J.E.H.; Devapatla, S.; Vidavalur, R.; Caudill, M.A.; Klatt, K.C. Choline metabolome response to prenatal choline supplementation across pregnancy: A randomized controlled trial. FASEB J. 2021, 35, e22063. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, J.N.; Lingrell, S.; da Silva, R.P.; Jacobs, R.L.; Vance, D.E. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 2014, 63, 2620–2630. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Species | NFCO | HFCO | HFCS | FDR Adjusted p Value |
|---|---|---|---|---|
| PE 38:1, PE O-40:8 | 1 a | 1.29 a | 4.01 b | 0.005 |
| PE 40:8, PE O-40:1 | 1 a | 1.94 b | 3.90 c | 0.033 |
| PE O-38:6 | 1 a | 1.82 b | 4.05 c | 0.025 |
| PE O-42:5 | 1 a | 1.30 a | 3.69 b | 0.033 |
| PE O-40:5 | 1 a | 1.02 a | 3.28 b | 0.033 |
| Lipid Species | NFCO-NF | NFCO-HF | HFCO-HF | HFCS-HF | FDR Adjusted p Value |
|---|---|---|---|---|---|
| PG 40:6|PG 18:1_22:5 | 1 a | 0.26 c | 0.23 c | 0.59 b | 0.002 |
| GlcCer d42:2 | 1 a | 0.25 c | 0.22 c | 0.41 b | 0.002 |
| GlcCer d42:1 | 1 a | 0.24 c | 0.23 c | 0.44 b | 0.002 |
| PE 38:5|PE 18:0_20:5 | 1 a | 0.28 b | 0.24 b | 0.65 a | 0.003 |
| FA 24:1 (nervonic acid) | 1 a | 0.24 c | 0.25 c | 0.45 b | 0.003 |
| PE P-34:2 or PE O-34:3 | 1 a | 0.28 c | 0.25 c | 0.57 b | 0.004 |
| GlcCer d41:1 | 1 a | 0.29 b | 0.28 b | 0.72 a | 0.006 |
| SM d36:0 | 1a | 4.17 c | 3.37 c | 1.83 b | 0.007 |
| PC 32:0 | 1 a | 0.37 b | 0.22 b | 0.56 a | 0.007 |
| TG 53:3;2O|TG 16:0_18:2_19:1;2O | 1 a | 3.75 b | 3.68 b | 1.75 a | 0.008 |
| TG 56:8 Isomer B | 1 a | 3.77 c | 3.77 c | 1.93 b | 0.008 |
| PI 38:5 | 1 a | 0.28 c | 0.26 c | 0.53 b | 0.008 |
| SM d42:2 Isomer A | 1 a | 0.31 b | 0.24 b | 0.48 a | 0.009 |
| TG 52:5 | 1 a | 3.66 c | 3.80 c | 1.95 b | 0.009 |
| PC 38:5 Isomer B_1 | 1 a | 0.41 b | 0.29 b | 0.71 a | 0.009 |
| PE O-38:6|PE O-16:1_22:5 | 1 a | 0.38 b | 0.27 b | 0.85 a | 0.01 |
| FA 14:1 (physeteric acid) | 1 a | 0.27 b | 0.35 b | 0.74 a | 0.013 |
| PE 40:5|PE 18:0_22:5 | 1 a | 0.28 b | 0.32 b | 0.67 a | 0.02 |
| PG 38:4|PG 18:0_20:4 | 1 a | 0.41 b | 0.53 a,b | 1.51 c | 0.021 |
| SM d42:2 | 1 a | 0.34 b | 0.25 b | 0.50 a | 0.021 |
| TG 52:4 | 1 a | 3.44 c | 3.08 c | 1.47 b | 0.022 |
| PC P-34:0 or PC O-34:1 | 1 a | 0.34 b | 0.26 b | 0.58 a | 0.022 |
| PC 31:0 | 1 a | 0.33 b | 0.28 b | 0.61 a | 0.022 |
| Cer d40:1 | 1 a | 2.37 b | 4.03 b | 1.73 a | 0.024 |
| Cer 40:1;2O|Cer 18:1;2O/22:0 | 1 a | 2.20 b | 4.01 b | 1.76 a | 0.025 |
| PE O-38:5|PE O-16:1_22:4 | 1 a | 0.35 b | 0.26 b | 0.55 a | 0.025 |
| PI 40:6|PI 18:0_22:6 | 1 a | 0.50 b | 0.66 a | 0.25 a | 0.026 |
| Cer 38:1;2O|Cer 18:1;2O/20:0 | 1 a | 2.25 b | 4.02 c | 1.91 a | 0.03 |
| SM d40:0 | 1 a | 2.50 b | 2.70 b | 0.96 a | 0.033 |
| Cer 40:2;2O|Cer 18:2;2O/22:0 | 1 a | 2.5 b | 3.70 b | 1.78 a | 0.04 |
| PG 44:11|PG 22:5_22:6 | 1 a | 0.37 b | 0.41 b | 1.04 a | 0.043 |
| DMPE 32:0|DMPE 16:0_16:0 | 1 a | 0.42 b | 0.30 b | 0.76 a | 0.044 |
| PC 38:5 Isomer B_1 | 1 a | 0.41 b | 0.29 b | 0.71 a | 0.046 |
| SM d40:1 | 1 a | 2.32 b | 3.64 b | 1.61 a | 0.046 |
| PC O-34:0 | 1 a | 0.80 a | 0.89 a | 2.60 b | 0.049 |
| Pathway Name | Pathway Lipids (n) | Altered Lipids (n) | p Value after Benjamini Correction |
|---|---|---|---|
| Sphingolipid metabolism | 21 | 5 | 0.004 |
| Sphingolipid signaling pathway | 9 | 3 | 0.014 |
| Autophagy–other | 3 | 2 | 0.014 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 3 | 2 | 0.014 |
| Autophagy–animal | 4 | 2 | 0.019 |
| Necroptosis | 4 | 2 | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korsmo, H.W.; Kadam, I.; Reaz, A.; Bretter, R.; Saxena, A.; Johnson, C.H.; Caviglia, J.M.; Jiang, X. Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes. Nutrients 2023, 15, 965. https://doi.org/10.3390/nu15040965
Korsmo HW, Kadam I, Reaz A, Bretter R, Saxena A, Johnson CH, Caviglia JM, Jiang X. Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes. Nutrients. 2023; 15(4):965. https://doi.org/10.3390/nu15040965
Chicago/Turabian StyleKorsmo, Hunter W., Isma’il Kadam, Aziza Reaz, Rachel Bretter, Anjana Saxena, Caroline H. Johnson, Jorge Matias Caviglia, and Xinyin Jiang. 2023. "Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes" Nutrients 15, no. 4: 965. https://doi.org/10.3390/nu15040965
APA StyleKorsmo, H. W., Kadam, I., Reaz, A., Bretter, R., Saxena, A., Johnson, C. H., Caviglia, J. M., & Jiang, X. (2023). Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes. Nutrients, 15(4), 965. https://doi.org/10.3390/nu15040965