
Selected Aspects of the Intricate Background of Immune-Related Cholangiopathies—A Critical Overview

 ,
,  and
and
Abstract
1. Introduction
2. Clinical Characteristics of PBC and PSC
3. Genetic and Epigenetic Determinants in Susceptibility to PBC and PSC
4. Bile Acids and Their Importance in the Liver and Gut Metabolism
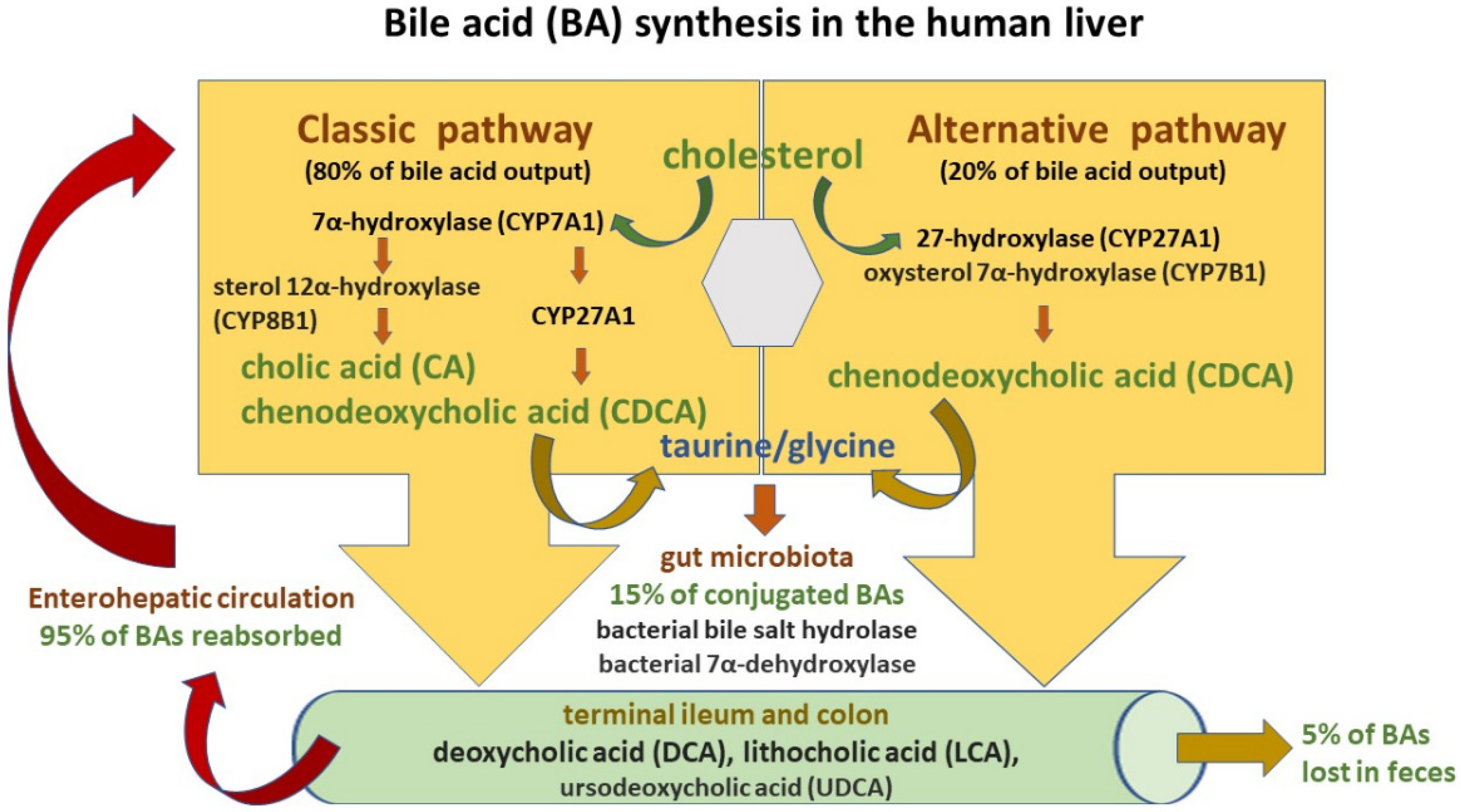
4.1. Bile Acid Synthesis and Circulation
- primary BAs, which are synthesized in the liver and secreted with bile into the small intestine: cholic acid (CA) and chenodeoxycholic acid (CDCA);
- secondary BAs, which are formed from primary BAs in the large intestine under the influence of bacterial enzymes: deoxycholic acid (DCA) originating from cholic acid, and lithocholic acid (LCA) originating from chenodeoxycholic acid;
- tertiary BAs, which are metabolites of major BAs including ursodeoxycholic acid (UDCA), which is currently used to treat liver diseases and bile duct disorders [53].
4.2. Effects of BAs in the Human Body
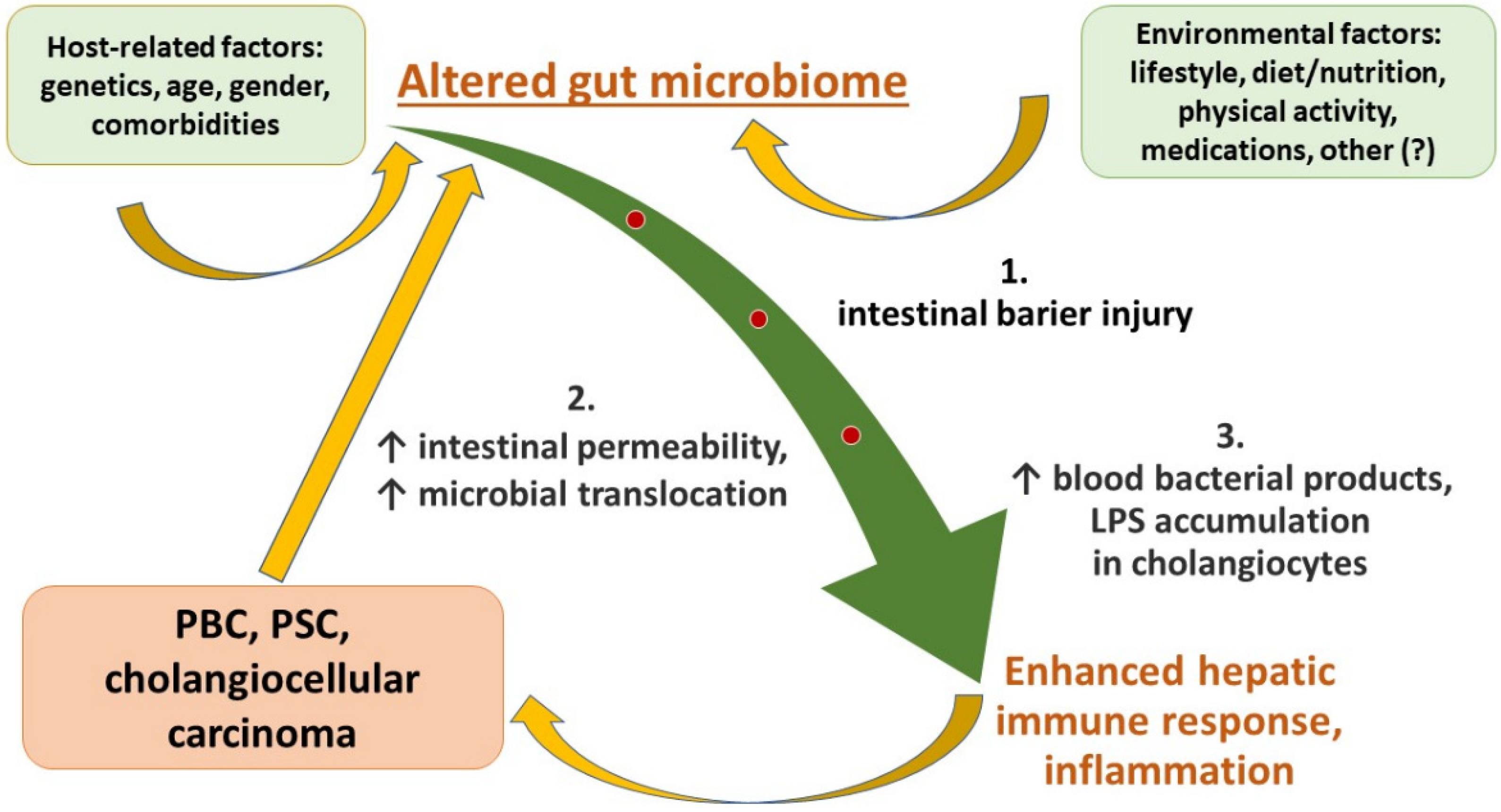
5. Role of the Gut–Liver Axis in the Development of Liver Injury and Progression to End-Stage Liver Disease
5.1. Constituents of the Gut Barrier
5.2. Leaky Gut and its Hepatic Consequences
5.3. Liver Sinusoid Endothelial Cells and Kupffer Cells as the Liver Scavenging System
6. Dysbiosis in Immune-Related Cholangiopathies
6.1. Gut Microbiome Alterations in Patients with PSC
6.2. Gut Microbiome Alterations in Patients with PBC
7. Current Treatments in PBC and PSC
7.1. Ursodeoxycholic Acid (UDCA) and Norursodeoxycholic Acid (norUDCA)
7.2. Obeticholic Acid (OCA)
7.3. Regulators of Bile Acid Homeostasis
7.3.1. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists
7.3.2. Apical Sodium-Dependent Transporter (ASBT) Inhibitors
7.3.3. Farnesoid X Receptor (FXR) Agonists
7.3.4. Other Therapeutic Options
7.4. Probiotics, Prebiotics, Postbiotics, and Synbiotics in PBC and PSC
7.5. Fecal Microbiota Transplantation (FMT) in Immune-Related Cholangiopathies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trivedi, P.J.; Hirschfield, G.M. Recent Advances in Clinical Practice: Epidemiology of Autoimmune Liver Diseases. Gut 2021, 70, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Huisman, E.J.; Trip, E.J.; Siersema, P.D.; van Hoek, B.; van Erpecum, K.J. Protein Energy Malnutrition Predicts Complications in Liver Cirrhosis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; Banales, J.M.; Medina, J.F. Primary Biliary Cholangitis: Pathogenic Mechanisms. Curr. Opin. Gastroenterol. 2021, 37, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Gallo, C.; Howardson, B.O.; Cristoferi, L.; Carbone, M.; Gershwin, M.E.; Invernizzi, P. An Update on Novel Pharmacological Agents for Primary Sclerosing Cholangitis. Expert. Opin. Ther. Targets 2022, 26, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary Sclerosing Cholangitis, Part 1: Epidemiology, Etiopathogenesis, Clinical Features, and Treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar] [PubMed]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. On Fibrates and Cholestasis: A Review. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef]
- Ali, A.H.; Carey, E.J.; Lindor, K.D. Diagnosis and Management of Primary Biliary Cirrhosis. Expert Rev. Clin. Immunol. 2014, 10, 1667–1678. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Dyson, J.K.; Alexander, G.J.M.; Chapman, M.H.; Collier, J.; Hübscher, S.; Patanwala, I.; Pereira, S.P.; Thain, C.; Thorburn, D.; et al. The British Society of Gastroenterology/UK-PBC Primary Biliary Cholangitis Treatment and Management Guidelines. Gut 2018, 67, 1568–1594. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Li, J.; Hou, S.; Ci, B.; Liu, B.; Xu, K. Prevalence and Impact of Sjögren’s Syndrome in Primary Biliary Cholangitis: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2022, 27, 100746. [Google Scholar] [CrossRef]
- Laschtowitz, A.; de Veer, R.C.; Van der Meer, A.J.; Schramm, C. Diagnosis and Treatment of Primary Biliary Cholangitis. United Eur. Gastroenterol. J. 2020, 8, 667–674. [Google Scholar] [CrossRef]
- Banales, J.M.; Sáez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-Regulation of MicroRNA 506 Leads to Decreased Cl-/HCO3- Anion Exchanger 2 Expression in Biliary Epithelium of Patients with Primary Biliary Cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; de Buy Wenniger, L.M.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Elferink, R.P.O.; Beuers, U. A Biliary HCO3-Umbrella Constitutes a Protective Mechanism against Bile Acid-Induced Injury in Human Cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. An Impaired Biliary Bicarbonate Umbrella May Be Involved in Dysregulated Autophagy in Primary Biliary Cholangitis. Lab. Investig. 2018, 98, 745–754. [Google Scholar] [CrossRef]
- Lleo, A.; Leung, P.S.C.; Hirschfield, G.M.; Gershwin, E.M. The Pathogenesis of Primary Biliary Cholangitis: A Comprehensive Review. Semin. Liver Dis. 2020, 40, 34–48. [Google Scholar] [CrossRef]
- Colapietro, F.; Lleo, A.; Generali, E. Antimitochondrial Antibodies: From Bench to Bedside. Clin. Rev. Allergy Immunol. 2022, 63, 166–177. [Google Scholar] [CrossRef]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Evolution of Our Understanding of PBC. Best Pract. Res. Clin. Gastroenterol. 2018, 34–35, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Bernstein, D.; Shiffman, M.L.; Kwo, P.; Kim, W.R.; Kowdley, K.V.; Jacobson, I.M. Diagnosis and Management of Primary Biliary Cholangitis. Off. J. Am. Coll. Gastroenterol. ACG 2019, 114, 48. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The Diagnosis and Management of Patients with Primary Biliary Cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Wu, H.; Chen, C.; Ziani, S.; Nelson, L.J.; Ávila, M.A.; Nevzorova, Y.A.; Cubero, F.J. Fibrotic Events in the Progression of Cholestatic Liver Disease. Cells 2021, 10, 1107. [Google Scholar] [CrossRef]
- Primary Biliary Cirrhosis—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1405232/ (accessed on 16 December 2022).
- Lleo, A.; Jepsen, P.; Morenghi, E.; Carbone, M.; Moroni, L.; Battezzati, P.M.; Podda, M.; Mackay, I.R.; Gershwin, M.E.; Invernizzi, P. Evolving Trends in Female to Male Incidence and Male Mortality of Primary Biliary Cholangitis. Sci. Rep. 2016, 6, 25906. [Google Scholar] [CrossRef]
- Tanaka, A. Current Understanding of Primary Biliary Cholangitis. Clin. Mol. Hepatol. 2021, 27, 1–21. [Google Scholar] [CrossRef]
- Qiu, F.; Tang, R.; Zuo, X.; Shi, X.; Wei, Y.; Zheng, X.; Dai, Y.; Gong, Y.; Wang, L.; Xu, P.; et al. A Genome-Wide Association Study Identifies Six Novel Risk Loci for Primary Biliary Cholangitis. Nat. Commun. 2017, 8, 14828. [Google Scholar] [CrossRef]
- Beuers, U.; Gershwin, M.E.; Gish, R.G.; Invernizzi, P.; Jones, D.E.J.; Lindor, K.; Ma, X.; Mackay, I.R.; Parés, A.; Tanaka, A.; et al. Changing Nomenclature for PBC: From “cirrhosis” to “Cholangitis”. Clin. Res. Hepatol. Gastroenterol. 2015, 39, e57–e59. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary Sclerosing Cholangitis—A Comprehensive Review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.W. Primary Sclerosing Cholangitis-A Long Night’s Journey into Day. Clin. Liver Dis. 2022, 20, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Rabiee, A.; Silveira, M.G. Primary Sclerosing Cholangitis. Transl. Gastroenterol. Hepatol. 2021, 6, 29. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Laupland, K.B.; Butzner, D.; Urbanski, S.J.; Lee, S.S. The Burden of Large and Small Duct Primary Sclerosing Cholangitis in Adults and Children: A Population-Based Analysis. Am. J. Gastroenterol. 2007, 102, 1042–1049. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Beuers, U.; Ponsioen, C.Y. Epidemiology of Primary Sclerosing Cholangitis and Primary Biliary Cirrhosis: A Systematic Review. J. Hepatol. 2012, 56, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; van Erpecum, K.J.; van Nieuwkerk, K.M.J.; Drenth, J.P.H.; Poen, A.C.; Witteman, B.J.M.; Tuynman, H.A.R.E.; Beuers, U.; Ponsioen, C.Y. Primary Sclerosing Cholangitis Is Associated with a Distinct Phenotype of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2012, 18, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Engel, B.; Taubert, R.; Jaeckel, E.; Manns, M.P. The Future of Autoimmune Liver Diseases—Understanding Pathogenesis and Improving Morbidity and Mortality. Liver Int. 2020, 40, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Kwo, P.Y.; Cohen, S.M.; Lim, J.K. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Off. J. Am. Coll. Gastroenterol. ACG 2017, 112, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Marchioni Beery, R.M.; Vaziri, H.; Forouhar, F. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: A Review Featuring a Women’s Health Perspective. J. Clin. Transl. Hepatol. 2014, 2, 266–284. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.M.; Pinto, C.; Maroni, L.; Benedetti, A.; Marzioni, M. Inflammation and the Gut-Liver Axis in the Pathophysiology of Cholangiopathies. Int. J. Mol. Sci. 2018, 19, 3003. [Google Scholar] [CrossRef]
- Visseren, T.; Erler, N.S.; Heimbach, J.K.; Eaton, J.E.; Selzner, N.; Gulamhusein, A.; van der Heide, F.; Porte, R.J.; van Hoek, B.; Alwayn, I.P.J.; et al. Inflammatory Conditions Play a Role in Recurrence of PSC after Liver Transplantation: An International Multicentre Study. JHEP Rep. 2022, 4, 100599. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Paraboschi, E.M.; Cappadona, C.; Caime, C.; Binatti, E.; Cristoferi, L.; Asselta, R.; Invernizzi, P. The Role of Epigenetics in Primary Biliary Cholangitis. Int. J. Mol. Sci. 2022, 23, 4873. [Google Scholar] [CrossRef]
- Marzorati, S.; Lleo, A.; Carbone, M.; Gershwin, M.E.; Invernizzi, P. The epigenetics of PBC: The link between genetic susceptibility and environment. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.J.; Hirschfield, G.M. Using GWAS to Identify Genetic Predisposition in Hepatic Autoimmunity. J. Autoimmun. 2016, 66, 25–39. [Google Scholar] [CrossRef]
- Folseraas, T.; Liaskou, E.; Anderson, C.A.; Karlsen, T.H. Genetics in PSC: What Do the “Risk Genes” Teach Us? Clin. Rev. Allergy Immunol. 2015, 48, 154–164. [Google Scholar] [CrossRef]
- Jiang, X.; Karlsen, T.H. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Hohenester, S.D.; van de Graaf, S.F.J.; Tolenaars, D.; van Lienden, K.; Verheij, J.; Marzioni, M.; Karlsen, T.H.; Oude Elferink, R.P.J.; Beuers, U. Knockout of the Primary Sclerosing Cholangitis-Risk Gene Fut2 Causes Liver Disease in Mice. Hepatology 2017, 66, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Fryett, J.J.; Ueno, K.; Darlay, R.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Nishida, N.; Khor, S.-S.; Gervais, O.; et al. An International Genome-Wide Meta-Analysis of Primary Biliary Cholangitis: Novel Risk Loci and Candidate Drugs. J. Hepatol. 2021, 75, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Joshita, S.; Umemura, T.; Tanaka, E.; Ota, M. Genetic Contribution to the Pathogenesis of Primary Biliary Cholangitis. J. Immunol. Res. 2017, 2017, 3073504. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Lammert, F. Search for Genetic Modifiers of PSC: Time to Increase the Number of Needles in the Haystack. Ann. Hepatol. 2017, 16, 830–831. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.-H.; Schrumpf, E.; Boberg, K.-M. Genetic Epidemiology of Primary Sclerosing Cholangitis. World J. Gastroenterol. 2007, 13, 5421–5431. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Biology, Pathophysiology, and Therapeutics. Clin. Liver Dis. 2020, 15, 91–94. [Google Scholar] [CrossRef]
- Kriaa, A.; Mariaule, V.; Jablaoui, A.; Rhimi, S.; Mkaouar, H.; Hernandez, J.; Korkmaz, B.; Lesner, A.; Maguin, E.; Aghdassi, A.; et al. Bile Acids: Key Players in Inflammatory Bowel Diseases? Cells 2022, 11, 901. [Google Scholar] [CrossRef]
- Min, Y.W.; Rezaie, A.; Pimentel, M. Bile Acid and Gut Microbiota in Irritable Bowel Syndrome. J. Neurogastroenterol. Motil. 2022, 28, 549–561. [Google Scholar] [CrossRef]
- Molecules | Free Full-Text | The Role of Bile Acids in the Human Body and in the Development of Diseases. Available online: https://www.mdpi.com/1420-3049/27/11/3401 (accessed on 29 December 2022).
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Kosters, A.; Jirsa, M.; Groen, A.K. Genetic Background of Cholesterol Gallstone Disease. Biochim. Biophys. Acta. 2003, 1637, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acids: Regulation of Synthesis. J. Lipid. Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/27320064/ (accessed on 29 December 2022).
- Albillos, A.; de Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Caliceti, C.; Punzo, A.; Silla, A.; Simoni, P.; Roda, G.; Hrelia, S. New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer. Nutrients 2022, 14, 2964. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Stindt, J.; Weissenberger, K.; Häussinger, D. The Bile Salt Export Pump (BSEP) in Health and Disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef]
- Murakami, Y.; Tanabe, S.; Suzuki, T. High-Fat Diet-Induced Intestinal Hyperpermeability Is Associated with Increased Bile Acids in the Large Intestine of Mice. J. Food Sci. 2016, 81, H216–H222. [Google Scholar] [CrossRef]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A Story of Liver and Gut Microbes: How Does the Intestinal Flora Affect Liver Disease? A Review of the Literature. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G889–G906. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- Baghdasaryan, A.; Fuchs, C.D.; Österreicher, C.H.; Lemberger, U.J.; Halilbasic, E.; Påhlman, I.; Graffner, H.; Krones, E.; Fickert, P.; Wahlström, A.; et al. Inhibition of Intestinal Bile Acid Absorption Improves Cholestatic Liver and Bile Duct Injury in a Mouse Model of Sclerosing Cholangitis. J. Hepatol. 2016, 64, 674–681. [Google Scholar] [CrossRef]
- Chen, M.L.; Takeda, K.; Sundrud, M.S. Emerging Roles of Bile Acids in Mucosal Immunity and Inflammation. Mucosal. Immunol. 2019, 12, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, A.; Fiorotto, R.; Strazzabosco, M. Bile Acids and Their Receptors: Modulators and Therapeutic Targets in Liver Inflammation; SpringerLink: Berlin/Heidelberg, Germany, 2022; Available online: https://link.springer.com/article/10.1007/s00281-022-00935-7 (accessed on 29 December 2022).
- Portincasa, P.; Di Ciaula, A.; Wang, H.H.; Palasciano, G.; van Erpecum, K.J.; Moschetta, A.; Wang, D.Q.-H. Coordinate Regulation of Gallbladder Motor Function in the Gut-Liver Axis. Hepatology 2008, 47, 2112–2126. [Google Scholar] [CrossRef]
- Zhou, H.; Hylemon, P.B. Bile Acids Are Nutrient Signaling Hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Režen, T.; Rozman, D.; Kovács, T.; Kovács, P.; Sipos, A.; Bai, P.; Mikó, E. The Role of Bile Acids in Carcinogenesis. Cell Mol. Life Sci. 2022, 79, 243. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-Beta-Muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial Transformations of Human Bile Acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of Antibacterial Defense in the Small Intestine by the Nuclear Bile Acid Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. The Pharmacology of Bile Acids and Their Receptors. Handb. Exp. Pharmacol. 2019, 256, 3–18. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A. Signaling from Intestine to the Host: How Bile Acids Regulate Intestinal and Liver Immunity. Handb. Exp. Pharmacol. 2019, 256, 95–108. [Google Scholar] [CrossRef]
- Fiorucci, S.; Zampella, A.; Ricci, P.; Distrutti, E.; Biagioli, M. Immunomodulatory Functions of FXR. Mol. Cell. Endocrinol. 2022, 551, 111650. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial Bile Acid Metabolites Modulate Gut RORγ+ Regulatory T Cell Homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A High-Fat Meal Induces Low-Grade Endotoxemia: Evidence of a Novel Mechanism of Postprandial Inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2020, 11, 594150. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.A.M.; Bajaj, J.S.; Sanyal, A.J. Role of Gut Microbiota in Liver Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G84–G98. [Google Scholar] [CrossRef] [PubMed]
- Broadley, S.P.; Plaumann, A.; Coletti, R.; Lehmann, C.; Wanisch, A.; Seidlmeier, A.; Esser, K.; Luo, S.; Rämer, P.C.; Massberg, S.; et al. Dual-Track Clearance of Circulating Bacteria Balances Rapid Restoration of Blood Sterility with Induction of Adaptive Immunity. Cell Host Microbe 2016, 20, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-Liver Axis at the Frontier of Host-Microbial Interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef]
- Marshall, J.C. The Gut as a Potential Trigger of Exercise-Induced Inflammatory Responses. Can. J. Physiol. Pharmacol. 1998, 76, 479–484. [Google Scholar] [CrossRef]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef]
- Takeuchi, T.; Ohno, H. IgA in Human Health and Diseases: Potential Regulator of Commensal Microbiota. Front. Immunol. 2022, 13, 1024330. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. Complications and Mortality in Hereditary Hemorrhagic Telangiectasia. Neurology 2015, 84, 1886–1893. [Google Scholar] [CrossRef]
- Floreani, A.; Baragiotta, A.; Pizzuti, D.; Martines, D.; Cecchetto, A.; Chiarelli, S. Mucosal IgA Defect in Primary Biliary Cirrhosis. Am. J. Gastroenterol. 2002, 97, 508–510. [Google Scholar] [CrossRef]
- Mandato, C.; Delli Bovi, A.P.; Vajro, P. The Gut-Liver Axis as a Target of Liver Disease Management. Hepatobiliary Surg. Nutr. 2021, 10, 100–102. [Google Scholar] [CrossRef]
- Ohtani, N.; Kawada, N. Role of the Gut–Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D. Toll-like Receptors: Activation, Signalling and Transcriptional Modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef]
- Walsh, D.; McCarthy, J.; O’Driscoll, C.; Melgar, S. Pattern Recognition Receptors--Molecular Orchestrators of Inflammation in Inflammatory Bowel Disease. Cytokine Growth Factor Rev. 2013, 24, 91–104. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Gut Microbiome and Health: Mechanistic Insights—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35105664/ (accessed on 29 December 2022).
- Chaudhry, S.; Emond, J.; Griesemer, A. Immune Cell Trafficking to the Liver. Transplantation 2019, 103, 1323–1337. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive Oxygen and Mechanisms of Inflammatory Liver Injury: Present Concepts. J. Gastroenterol. Hepatol. 2011, 26, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A. Toll-like Receptors and Adaptor Molecules in Liver Disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Jin, C.J.; Baumann, A.; Brandt, A.; Engstler, A.J.; Nier, A.; Hege, M.; Schmeer, C.; Kehm, R.; Höhn, A.; Grune, T.; et al. Aging-Related Liver Degeneration Is Associated with Increased Bacterial Endotoxin and Lipopolysaccharide Binding Protein Levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G736–G747. [Google Scholar] [CrossRef]
- Cevenini, E.; Monti, D.; Franceschi, C. Inflamm-Ageing. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 14–20. [Google Scholar] [CrossRef] [PubMed]
- d’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017, 2, e00046-17. [Google Scholar] [CrossRef]
- Anhê, F.F.; Barra, N.G.; Cavallari, J.F.; Henriksbo, B.D.; Schertzer, J.D. Metabolic Endotoxemia Is Dictated by the Type of Lipopolysaccharide. Cell Rep. 2021, 36, 109691. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver Sinusoidal Endothelial Cells - Gatekeepers of Hepatic Immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Tanoi, T.; Tamura, T.; Sano, N.; Nakayama, K.; Fukunaga, K.; Zheng, Y.-W.; Akhter, A.; Sakurai, Y.; Hayashi, Y.; Harashima, H.; et al. Protecting Liver Sinusoidal Endothelial Cells Suppresses Apoptosis in Acute Liver Damage. Hepatol. Res. 2016, 46, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Liu, Y.; Xu, J.; Liu, Y. Gut–Liver Axis: Liver Sinusoidal Endothelial Cells Function as the Hepatic Barrier in Colitis-Induced Liver Injury. Front. Cell Dev. Biol. 2021, 9, 702890. [Google Scholar] [CrossRef]
- Guan, H.; Zhang, X.; Kuang, M.; Yu, J. The Gut-Liver Axis in Immune Remodeling of Hepatic Cirrhosis. Front. Immunol. 2022, 13, 2357–2366. [Google Scholar]
- Szabo, G.; Bala, S.; Petrasek, J.; Gattu, A. Gut-Liver Axis and Sensing Microbes. Dig. Dis. 2010, 28, 737–744. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial Injury, Oxidative Stress, and Antioxidant Gene Expression Are Induced by Hepatitis C Virus Core Protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human Hepatitis C Virus NS5A Protein Alters Intracellular Calcium Levels, Induces Oxidative Stress, and Activates STAT-3 and NF-ΚB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer Cells in Host Defense and Liver Disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Hepatic Uptake and Deacylation of the LPS in Bloodborne LPS-Lipoprotein Complexes—Baomei Shao, Robert S Munford, Richard Kitchens, Alan W Varley. 2012. Available online: https://journals.sagepub.com/doi/10.1177/1753425912442431 (accessed on 29 December 2022).
- Maeshima, N.; Fernandez, R.C. Recognition of Lipid A Variants by the TLR4-MD-2 Receptor Complex. Front. Cell Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-Operation of TLR4 and Raft Proteins in LPS-Induced pro-Inflammatory Signaling. Cell Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.-O. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile Acids and Intestinal Microbiota in Autoimmune Cholestatic Liver Diseases. Autoimmun Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of Gut Microbiota with Bile Acid Metabolism and Its Influence on Disease States. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef]
- Azad, M.B.; Konya, T.; Persaud, R.R.; Guttman, D.S.; Chari, R.S.; Field, C.J.; Sears, M.R.; Mandhane, P.J.; Turvey, S.E.; Subbarao, P.; et al. CHILD Study Investigators. Impact of Maternal Intrapartum Antibiotics, Method of Birth and Breastfeeding on Gut Microbiota during the First Year of Life: A Prospective Cohort Study. BJOG 2016, 123, 983–993. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the Gut Microbiota to Distant Organs in Physiology and Disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and Correlations of the Gut Microbiome, Metabolism and Immunity in Patients with Primary Biliary Cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The Gut Microbial Profile in Patients with Primary Sclerosing Cholangitis Is Distinct from Patients with Ulcerative Colitis without Biliary Disease and Healthy Controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary Sclerosing Cholangitis Is Characterised by Intestinal Dysbiosis Independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef]
- Torres, J.; Palmela, C.; Brito, H.; Bao, X.; Ruiqi, H.; Moura-Santos, P.; Pereira da Silva, J.; Oliveira, A.; Vieira, C.; Perez, K.; et al. The Gut Microbiota, Bile Acids and Their Correlation in Primary Sclerosing Cholangitis Associated with Inflammatory Bowel Disease. United Eur. Gastroenterol. J. 2018, 6, 112–122. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct Gut Microbiota Profiles in Patients with Primary Sclerosing Cholangitis and Ulcerative Colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut Microbial Profile Is Altered in Primary Biliary Cholangitis and Partially Restored after UDCA Therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Blesl, A.; Stadlbauer, V. The Gut-Liver Axis in Cholestatic Liver Diseases. Nutrients 2021, 13, 1018. [Google Scholar] [CrossRef]
- Palmela, C.; Peerani, F.; Castaneda, D.; Torres, J.; Itzkowitz, S.H. Inflammatory Bowel Disease and Primary Sclerosing Cholangitis: A Review of the Phenotype and Associated Specific Features. Gut Liver 2018, 12, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Tornai, T.; Palyu, E.; Vitalis, Z.; Tornai, I.; Tornai, D.; Antal-Szalmas, P.; Norman, G.L.; Shums, Z.; Veres, G.; Dezsofi, A.; et al. Gut Barrier Failure Biomarkers Are Associated with Poor Disease Outcome in Patients with Primary Sclerosing Cholangitis. World J. Gastroenterol. 2017, 23, 5412–5421. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.K.; Kummen, M.; Trøseid, M.; Åkra, S.; Liaskou, E.; Moum, B.; Vesterhus, M.; Karlsen, T.H.; Seljeflot, I.; Hov, J.R. Circulating Markers of Gut Barrier Function Associated with Disease Severity in Primary Sclerosing Cholangitis. Liver Int. 2019, 39, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Meddings, J.; Heathcote, E.J. Abnormal Intestinal Permeability in Primary Biliary Cirrhosis. Dig. Dis. Sci. 2006, 51, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Sasatomi, K.; Noguchi, K.; Sakisaka, S.; Sata, M.; Tanikawa, K. Abnormal Accumulation of Endotoxin in Biliary Epithelial Cells in Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis. J. Hepatol. 1998, 29, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Saint-Antoine IBD Network; Chazouillères, O.; Housset, C.; Sokol, H. Fungi Participate in the Dysbiosis of Gut Microbiota in Patients with Primary Sclerosing Cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef]
- Kummen, M.; Hov, J.R. The Gut Microbial Influence on Cholestatic Liver Disease. Liver Int. 2019, 39, 1186–1196. [Google Scholar] [CrossRef]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology 2021, 160, 1784–1798.E0. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative Microbiome Profiling Disentangles Inflammation- and Bile Duct Obstruction-Associated Microbiota Alterations across PSC/IBD Diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef]
- Rühlemann, M.; Liwinski, T.; Heinsen, F.-A.; Bang, C.; Zenouzi, R.; Kummen, M.; Thingholm, L.; Tempel, M.; Lieb, W.; Karlsen, T.; et al. Consistent Alterations in Faecal Microbiomes of Patients with Primary Sclerosing Cholangitis Independent of Associated Colitis. Aliment. Pharmacol. Ther. 2019, 50, 580–589. [Google Scholar] [CrossRef]
- Liu Chen Kiow, J.; Vincent, C.; Sidani, S.; Bouin, M. High Occurrence of Small Intestinal Bacterial Overgrowth in Primary Biliary Cholangitis. Neurogastroenterol. Motil. 2019, 31, e13691. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The Microbiota in Cirrhosis and Its Role in Hepatic Decompensation. J. Hepatol. 2021, 75, S67–S81. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Huang, C.; Zhang, Q.; Tao, S.; Hu, X.; Xu, J.; Jiang, R.; Xu, B.; Liu, Y.; Hou, J. Alterations in Gut Microbiota and Elevated Serum Bilirubin in Primary Biliary Cholangitis Patients Treated with Ursodeoxycholic Acid. Eur. J. Clin. Investig. 2022, 52, e13714. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Vergani, D. Bacteria and Primary Biliary Cirrhosis. Clinic. Rev. Allerg. Immunol. 2009, 36, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Kim, J.-H.; Kim, S.-E.; Jung, J.H.; Jang, M.-K.; Park, S.-H.; Lee, M.-S.; Kim, H.-S.; Suk, K.T.; Kim, D.J. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics. Biomedicines 2022, 10, 1288. [Google Scholar] [CrossRef]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Water, J.V.D.; Nantz, M.H.; et al. Patients with Primary Biliary Cirrhosis React against a Ubiquitous Xenobiotic-Metabolizing Bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef]
- Padgett, K.A.; Selmi, C.; Kenny, T.P.; Leung, P.S.C.; Balkwill, D.L.; Ansari, A.A.; Coppel, R.L.; Gershwin, M.E. Phylogenetic and Immunological Definition of Four Lipoylated Proteins from Novosphingobium Aromaticivorans, Implications for Primary Biliary Cirrhosis. J. Autoimmun 2005, 24, 209–219. [Google Scholar] [CrossRef]
- Selmi, C.; Bowlus, C.L.; Gershwin, M.E.; Coppel, R.L. Primary Biliary Cirrhosis. Lancet 2011, 377, 1600–1609. [Google Scholar] [CrossRef]
- Liwinski, T.; Heinemann, M.; Schramm, C. The Intestinal and Biliary Microbiome in Autoimmune Liver Disease—Current Evidence and Concepts. Semin. Immunopathol. 2022, 44, 485–507. [Google Scholar] [CrossRef]
- Butler, P.; Hamilton-Miller, J.; Baum, H.; Burroughs, A.K. Detection of M2 Antibodies in Patients with Recurrent Urinary Tract Infection Using an ELISA and Purified PBC Specific Antigens. Evidence for a Molecular Mimicry Mechanism in the Pathogenesis of Primary Biliary Cirrhosis? Biochem. Mol. Biol. Int. 1995, 35, 473–485. [Google Scholar]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Pathogen Infections and Primary Biliary Cholangitis. Clin. Exp. Immunol. 2019, 195, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Haruta, I.; Kikuchi, K.; Hashimoto, E.; Nakamura, M.; Miyakawa, H.; Hirota, K.; Shibata, N.; Kato, H.; Arimura, Y.; Kato, Y.; et al. Long-Term Bacterial Exposure Can Trigger Nonsuppurative Destructive Cholangitis Associated with Multifocal Epithelial Inflammation. Lab. Investig. 2010, 90, 577–588. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile Acids and the Gut Microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Prokopič, M.; Beuers, U. Management of Primary Sclerosing Cholangitis and Its Complications: An Algorithmic Approach. Hepatol. Int. 2021, 15, 6–20. [Google Scholar] [CrossRef]
- Hasegawa, S.; Yoneda, M.; Kurita, Y.; Nogami, A.; Honda, Y.; Hosono, K.; Nakajima, A. Cholestatic Liver Disease: Current Treatment Strategies and New Therapeutic Agents. Drugs 2021, 81, 1181–1192. [Google Scholar] [CrossRef]
- Gerussi, A.; D’Amato, D.; Cristoferi, L.; O’Donnell, S.E.; Carbone, M.; Invernizzi, P. Multiple Therapeutic Targets in Rare Cholestatic Liver Diseases: Time to Redefine Treatment Strategies. Ann. Hepatol. 2020, 19, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Barron-Millar, B.; Ogle, L.; Mells, G.; Flack, S.; Badrock, J.; Sandford, R.; Kirby, J.; Palmer, J.; Jopson, L.; Brain, J.; et al. The Serum Proteome and Ursodeoxycholic Acid Response in Primary Biliary Cholangitis. Hepatology 2021, 74, 3269–3283. [Google Scholar] [CrossRef]
- de Vries, E.; Beuers, U. Management of Cholestatic Disease in 2017. Liver Int. 2017, 37, 123–129. [Google Scholar] [CrossRef]
- Ali, A.H.; Lindor, K.D. Obeticholic Acid for the Treatment of Primary Biliary Cholangitis. Expert Opin. Pharmacother. 2016, 17, 1809–1815. [Google Scholar] [CrossRef]
- Harms, M.H.; van Buuren, H.R.; Corpechot, C.; Thorburn, D.; Janssen, H.L.A.; Lindor, K.D.; Hirschfield, G.M.; Parés, A.; Floreani, A.; Mayo, M.J.; et al. Ursodeoxycholic Acid Therapy and Liver Transplant-Free Survival in Patients with Primary Biliary Cholangitis. J. Hepatol. 2019, 71, 357–365. [Google Scholar] [CrossRef]
- Kotb, M.A. Molecular Mechanisms of Ursodeoxycholic Acid Toxicity & Side Effects: Ursodeoxycholic Acid Freezes Regeneration & Induces Hibernation Mode. Int. J. Mol. Sci. 2012, 13, 8882–8914. [Google Scholar] [CrossRef] [PubMed]
- Paumgartner, G.; Beuers, U. Mechanisms of Action and Therapeutic Efficacy of Ursodeoxycholic Acid in Cholestatic Liver Disease. Clin. Liver Dis. 2004, 8, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Castro, R.E.; Solá, S.; Steer, C.J.; Rodrigues, C.M.P. P53 Is a Key Molecular Target of Ursodeoxycholic Acid in Regulating Apoptosis. J. Biol. Chem. 2007, 282, 34250–34259. [Google Scholar] [CrossRef]
- Tanaka, A. Emerging Novel Treatments for Autoimmune Liver Diseases. Hepatol. Res. 2019, 49, 489–499. [Google Scholar] [CrossRef]
- Hofmann, A.F. Pharmacology of Ursodeoxycholic Acid, an Enterohepatic Drug. Scand. J. Gastroenterol. Suppl. 1994, 204, 1–15. [Google Scholar] [CrossRef]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New Paradigms in the Treatment of Hepatic Cholestasis: From UDCA to FXR, PXR and Beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef]
- Adamowicz, M.; Kempinska-Podhorodecka, A.; Abramczyk, J.; Banales, J.M.; Milkiewicz, P.; Milkiewicz, M. Suppression of Hepatic PPARα in Primary Biliary Cholangitis Is Modulated by MiR-155. Cells 2022, 11, 2880. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendía, L.E.; Simental-Mendía, M.; Sánchez-García, A.; Banach, M.; Serban, M.-C.; Cicero, A.F.G.; Sahebkar, A. Impact of Ursodeoxycholic Acid on Circulating Lipid Concentrations: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. Lipids. Health Dis. 2019, 18, 88. [Google Scholar] [CrossRef]
- Sánchez-García, A.; Sahebkar, A.; Simental-Mendía, M.; Simental-Mendía, L.E. Effect of Ursodeoxycholic Acid on Glycemic Markers: A Systematic Review and Meta-Analysis of Clinical Trials. Pharmacol. Res. 2018, 135, 144–149. [Google Scholar] [CrossRef]
- Shimoyama, S.; Kawata, K.; Ohta, K.; Chida, T.; Suzuki, T.; Tsuneyama, K.; Shimoda, S.; Kurono, N.; Leung, P.S.C.; Gershwin, M.E.; et al. Ursodeoxycholic Acid Impairs Liver-Infiltrating T-Cell Chemotaxis through IFN-γ and CX3CL1 Production in Primary Biliary Cholangitis. Eur. J. Immunol. 2021, 51, 1519–1530. [Google Scholar] [CrossRef]
- Zhu, G.-Q.; Shi, K.-Q.; Huang, G.-Q.; Wang, L.-R.; Lin, Y.-Q.; Braddock, M.; Chen, Y.-P.; Zhou, M.-T.; Zheng, M.-H. A Network Meta-Analysis of the Efficacy and Side Effects of UDCA-Based Therapies for Primary Sclerosing Cholangitis. Oncotarget 2015, 6, 26757–26769. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R.E.; Bonnand, A.M.; Chrétien, Y.; Poupon, R. Ten-Year Survival in Ursodeoxycholic Acid-Treated Patients with Primary Biliary Cirrhosis. The UDCA-PBC Study Group. Hepatology 1999, 29, 1668–1671. [Google Scholar] [CrossRef]
- de Vries, E.M.G.; Wang, J.; Leeflang, M.M.G.; Boonstra, K.; Weersma, R.K.; Beuers, U.H.; Geskus, R.B.; Ponsioen, C.Y. Alkaline Phosphatase at Diagnosis of Primary Sclerosing Cholangitis and 1 Year Later: Evaluation of Prognostic Value. Liver Int. 2016, 36, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Triantos, C.K.; Koukias, N.M.; Nikolopoulou, V.N.; Burroughs, A.K. Meta-Analysis: Ursodeoxycholic Acid for Primary Sclerosing Cholangitis. Aliment. Pharmacol. Ther. 2011, 34, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Steinacher, D.; Trauner, M. Nor-Ursodeoxycholic Acid as a Novel Therapeutic Approach for Cholestatic and Metabolic Liver Diseases. Dig. Dis. 2017, 35, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. European PSC norUDCA Study Group. NorUrsodeoxycholic Acid Improves Cholestasis in Primary Sclerosing Cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef]
- Zhu, C.; Boucheron, N.; Müller, A.C.; Májek, P.; Claudel, T.; Halilbasic, E.; Baazim, H.; Lercher, A.; Viczenczova, C.; Hainberger, D.; et al. 24-Norursodeoxycholic Acid Reshapes Immunometabolism in CD8+ T Cells and Alleviates Hepatic Inflammation. J. Hepatol. 2021, 75, 1164–1176. [Google Scholar] [CrossRef]
- Chapman, R.W.; Lynch, K.D. Obeticholic Acid-a New Therapy in PBC and NASH. Br. Med. Bull. 2020, 133, 95–104. [Google Scholar] [CrossRef]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile Acid Derivatives as Ligands of the Farnesoid X Receptor. Synthesis, Evaluation, and Structure-Activity Relationship of a Series of Body and Side Chain Modified Analogues of Chenodeoxycholic Acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Salhab, A.; Amer, J.; Lu, Y.; Safadi, R. Sodium+/Taurocholate Cotransporting Polypeptide as Target Therapy for Liver Fibrosis. Gut 2022, 71, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile Acid Transporters and Regulatory Nuclear Receptors in the Liver and Beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Piccinin, E.; Garcia-Irigoyen, O.; Sabbà, C.; Moschetta, A. Nuclear Receptor FXR, Bile Acids and Liver Damage: Introducing the Progressive Familial Intrahepatic Cholestasis with FXR Mutations. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018, 1864, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Kjærgaard, K.; Frisch, K.; Sørensen, M.; Munk, O.L.; Hofmann, A.F.; Horsager, J.; Schacht, A.C.; Erickson, M.; Shapiro, D.; Keiding, S. Obeticholic Acid Improves Hepatic Bile Acid Excretion in Patients with Primary Biliary Cholangitis. J. Hepatol. 2021, 74, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR Agonist Obeticholic Acid Reduces Hepatic Inflammation and Fibrosis in a Rat Model of Toxic Cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef]
- Floreani, A.; Gabbia, D.; De Martin, S. Obeticholic Acid for Primary Biliary Cholangitis. Biomedicines 2022, 10, 2464. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.-U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Clinical Review Report: Obeticholic Acid (Ocaliva): (Intercept Pharmaceuticals Canada, Inc.): Indication: For the Treatment of Primary Biliary Cholangitis (PBC) in Combination with Ursodeoxycholic Acid (UDCA) in Adults with an Inadequate Response to UDCA or as Monotherapy in Adults Unable to Tolerate UDCA. In CADTH Common Drug Reviews; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2017.
- Cazzagon, N.; Floreani, A. Primary Biliary Cholangitis: Treatment. Curr. Opin. Gastroenterol. 2021, 37, 99–104. [Google Scholar] [CrossRef]
- Soret, P.-A.; Lam, L.; Carrat, F.; Smets, L.; Berg, T.; Carbone, M.; Invernizzi, P.; Leroy, V.; Trivedi, P.; Cazzagon, N.; et al. Combination of Fibrates with Obeticholic Acid Is Able to Normalise Biochemical Liver Tests in Patients with Difficult-to-Treat Primary Biliary Cholangitis. Aliment. Pharmacol. Ther. 2021, 53, 1138–1146. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. AESOP Study Investigators. A Randomized, Placebo-Controlled, Phase II Study of Obeticholic Acid for Primary Sclerosing Cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef]
- Soret, P.A.; Chazouillères, O.; Corpechot, C. Primary Biliary Cholangitis. Rev. Prat. 2021, 71, 885–891. [Google Scholar] [PubMed]
- Lee, Y.-M.; Kaplan, M.M. The Natural History of PBC: Has It Changed? Semin. Liver Dis. 2005, 25, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Montano-Loza, A.J.; Corpechot, C. Definition and Management of Patients With Primary Biliary Cholangitis and an Incomplete Response to Therapy. Clin. Gastroenterol. Hepatol. 2021, 19, 2241–2251.e1. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Wang, C.; Shi, Y.; Wang, X.; Ma, H.; Liu, Q.; Gao, Y.; Niu, J. Peroxisome Proliferator-Activated Receptors Regulate Hepatic Immunity and Assist in the Treatment of Primary Biliary Cholangitis. Front. Immunol. 2022, 13, 940688. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Lee, J.-H.; Kim, M.A.; Leem, G.; Kim, S.W.; Chang, Y.; Lee, H.Y.; Yoon, J.S.; Park, J.Y.; Lee, Y.B.; et al. Additional Fibrate Treatment in UDCA-Refractory PBC Patients. Liver Int. 2019, 39, 1776–1785. [Google Scholar] [CrossRef]
- Levy, C.; Lindor, K.D. Itching to Know: Role of Fibrates in PBC. Off. J. Am. Coll. Gastroenterol. ACG 2018, 113, 56. [Google Scholar] [CrossRef]
- Fenofibrate Improves GLOBE and UK-PBC Scores and Histological Features in Primary Biliary Cholangitis—Minerva Medica, 5 May 2021. Available online: https://www.minervamedica.it/en/journals/minerva-medica/article.php?cod=R10Y9999N00A21050501 (accessed on 16 December 2022).
- Tanaka, A.; Hirohara, J.; Nakanuma, Y.; Tsubouchi, H.; Takikawa, H. Biochemical Responses to Bezafibrate Improve Long-Term Outcome in Asymptomatic Patients with Primary Biliary Cirrhosis Refractory to UDCA. J. Gastroenterol. 2015, 50, 675–682. [Google Scholar] [CrossRef]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Japan PBC Study Group. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients With Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a Selective PPAR-δ Agonist, in Patients with Primary Biliary Cholangitis with an Inadequate Response to Ursodeoxycholic Acid: A Double-Blind, Randomised, Placebo-Controlled, Phase 2, Proof-of-Concept Study. Lancet. Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; Levy, C.; Bowlus, C.L.; Jones, D.E.; Steinberg, A.; McWherter, C.A.; Choi, Y.-J. Seladelpar Improved Measures of Pruritus, Sleep, and Fatigue and Decreased Serum Bile Acids in Patients with Primary Biliary Cholangitis. Liver Int. 2022, 42, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Galambos, M.R.; Aspinall, R.J.; Hirschfield, G.M.; Jones, D.E.J.; Dörffel, Y.; Gordon, S.C.; Harrison, S.A.; Kremer, A.E.; Mayo, M.J.; et al. A Phase II, Randomized, Open-Label, 52-Week Study of Seladelpar in Patients with Primary Biliary Cholangitis. J. Hepatol. 2022, 77, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. A Randomized Placebo-Controlled Trial of Elafibranor in Patients with Primary Biliary Cholangitis and Incomplete Response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Hegade, V.S.; Jones, D.E.J.; Hirschfield, G.M. Apical Sodium-Dependent Transporter Inhibitors in Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Dig. Dis. 2017, 35, 267–274. [Google Scholar] [CrossRef]
- Hegade, V.S.; Bolier, R.; Oude Elferink, R.P.; Beuers, U.; Kendrick, S.; Jones, D.E. A Systematic Approach to the Management of Cholestatic Pruritus in Primary Biliary Cirrhosis. Frontline Gastroenterol 2016, 7, 158–166. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Jha, P.; Müller, M.; Auer, N.; Deutschmann, A.; Zöhrer, C.; Påhlman, I.; Graffner, H.; Fickert, P.; Trauner, M. O135 inhibition of intestinal bile acid absorption by asbt inhibitor A4250 protects against bile acid-mediated cholestatic liver injury in mice. J. Hepatol. 2014, 60, S57. [Google Scholar] [CrossRef]
- Langedijk, J.A.G.M.; Beuers, U.H.; Oude Elferink, R.P.J. Cholestasis-Associated Pruritus and Its Pruritogens. Front. Med. 2021, 8, 639674. [Google Scholar] [CrossRef]
- GLIMMER Trial—A Randomized, Double-Blind, Placebo-Controlled Study of Linerixibat, an Inhibitor of the Ileal Bile Acid Transporter, in the Treatment of Cholestatic Pruritus in Primary Biliary Cholangitis. Gastroenterol. Hepatol. 2021, 17, 11–12.
- Halilbasic, E.; Fuchs, C.; Hofer, H.; Paumgartner, G.; Trauner, M. Therapy of Primary Sclerosing Cholangitis—Today and Tomorrow. Dig. Dis. 2015, 33, 149–163. [Google Scholar] [CrossRef]
- Wong, L.L.; Hegade, V.S.; Jones, D.E.J. What Comes after Ursodeoxycholic Acid in Primary Biliary Cholangitis? Dig. Dis. 2017, 35, 359–366. [Google Scholar] [CrossRef]
- Khanna, A.; Jopson, L.; Howel, D.; Bryant, A.; Blamire, A.; Newton, J.L.; Wilkinson, J.; Steel, A.J.; Bainbridge, J.; Stefanetti, R.; et al. Rituximab for the Treatment of Fatigue in Primary Biliary Cholangitis (Formerly Primary Biliary Cirrhosis): A Randomised Controlled Trial; Efficacy and Mechanism Evaluation; NIHR Journals Library: Southampton, UK, 2018. [Google Scholar]
- Kilavuz, H.; Turan, U.; Yoldas, A.; Tolun, F.I.; Tanriverdi, B.; Yaylali, A.; Yaman, A.; Yener, M.K.; Irkorucu, O. The Effect of Farnesoid X Receptor Agonist Tropifexor on Liver Damage in Rats with Experimental Obstructive Jaundice. Acta. Cir. Bras. 2021, 36, e360902. [Google Scholar] [CrossRef]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X Receptor Agonist Tropifexor Attenuates Cholestasis in a Randomised Trial in Patients with Primary Biliary Cholangitis. JHEP Rep. 2022, 4, 100544. [Google Scholar] [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- Angulo, P.; Jorgensen, R.A.; Keach, J.C.; Dickson, E.R.; Smith, C.; Lindor, K.D. Oral Budesonide in the Treatment of Patients with Primary Biliary Cirrhosis with a Suboptimal Response to Ursodeoxycholic Acid. Hepatology 2000, 31, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Beuers, U.; Kupcinskas, L.; Ott, P.; Bergquist, A.; Färkkilä, M.; Manns, M.P.; Parés, A.; Spengler, U.; Stiess, M.; et al. A Placebo-Controlled Randomised Trial of Budesonide for PBC Following an Insufficient Response to UDCA. J. Hepatol. 2021, 74, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.C.; Trudeau, S.; Regev, A.; Uhas, J.M.; Chakladar, S.; Pinto-Correia, A.; Gottlieb, K.; Schlichting, D. Baricitinib and Primary Biliary Cholangitis. J. Transl. Autoimmun. 2021, 4, 100107. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-J.; Gou, H.-Z.; Zhang, Y.-L.; Song, X.-J.; Zhang, L. Role of Intestinal Flora in Primary Sclerosing Cholangitis and Its Potential Therapeutic Value. World J. Gastroenterol. 2022, 28, 6213–6229. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised Clinical Trial: Vancomycin or Metronidazole in Patients with Primary Sclerosing Cholangitis—A Pilot Study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef]
- Damman, J.L.; Rodriguez, E.A.; Ali, A.H.; Buness, C.W.; Cox, K.L.; Carey, E.J.; Lindor, K.D. Review Article: The Evidence That Vancomycin Is a Therapeutic Option for Primary Sclerosing Cholangitis. Aliment. Pharmacol. Ther. 2018, 47, 886–895. [Google Scholar] [CrossRef]
- Ali, A.H.; Damman, J.; Shah, S.B.; Davies, Y.; Hurwitz, M.; Stephen, M.; Lemos, L.M.; Carey, E.J.; Lindor, K.D.; Buness, C.W.; et al. Open-Label Prospective Therapeutic Clinical Trials: Oral Vancomycin in Children and Adults with Primary Sclerosing Cholangitis. Scand. J. Gastroenterol. 2020, 55, 941–950. [Google Scholar] [CrossRef]
- Żółkiewicz, J.; Marzec, A.; Ruszczyński, M.; Feleszko, W. Postbiotics—A Step Beyond Pre- and Probiotics. Nutrients 2020, 12, 2189. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Zhou, D.-D.; Gan, R.-Y.; Huang, S.-Y.; Zhao, C.-N.; Shang, A.; Xu, X.-Y.; Li, H.-B. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef]
- Abe, K.; Takahashi, A.; Fujita, M.; Imaizumi, H.; Hayashi, M.; Okai, K.; Ohira, H. Dysbiosis of Oral Microbiota and Its Association with Salivary Immunological Biomarkers in Autoimmune Liver Disease. PLoS ONE 2018, 13, e0198757. [Google Scholar] [CrossRef] [PubMed]
- Vleggaar, F.P.; Monkelbaan, J.F.; van Erpecum, K.J. Probiotics in Primary Sclerosing Cholangitis: A Randomized Placebo-Controlled Crossover Pilot Study. Eur. J. Gastroenterol. Hepatol. 2008, 20, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Iwasaki, H.; Mase, S.; Yachie, A. Successful Treatment of Primary Sclerosing Cholangitis with a Steroid and a Probiotic. Case Rep. Gastroenterol. 2012, 6, 249–253. [Google Scholar] [CrossRef]
- Chen, Y.; Guan, W.; Zhang, N.; Wang, Y.; Tian, Y.; Sun, H.; Li, X.; Wang, Y.; Liu, J. Lactobacillus Plantarum Lp2 Improved LPS-Induced Liver Injury through the TLR-4/MAPK/NFκB and Nrf2-HO-1/CYP2E1 Pathways in Mice. Food Nutr. Res. 2022, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Tian, Y.; Wang, Y.; Fan, Y.; Zhang, Y.; Xing, X.; Nan, B.; Ai, Z.; Li, X.; Wang, Y. Ameliorative Effect of Lactobacillus Plantarum Lp2 against Cyclophosphamide-Induced Liver Injury in Mice. Food Chem. Toxicol. 2022, 169, 113433. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Borody, T.J.; Zhang, F. Encyclopedia of Fecal Microbiota Transplantation: A Review of Effectiveness in the Treatment of 85 Diseases. Chin. Med. J. Engl. 2022, 135, 1927–1939. [Google Scholar] [CrossRef]
- Konturek, P.C.; Haziri, D.; Brzozowski, T.; Hess, T.; Heyman, S.; Kwiecien, S.; Konturek, S.J.; Koziel, J. Emerging Role of Fecal Microbiota Therapy in the Treatment of Gastrointestinal and Extra-Gastrointestinal Diseases. J. Physiol. Pharmacol. 2015, 66, 483–491. [Google Scholar]
- Green, J.E.; Davis, J.A.; Berk, M.; Hair, C.; Loughman, A.; Castle, D.; Athan, E.; Nierenberg, A.A.; Cryan, J.F.; Jacka, F.; et al. Efficacy and Safety of Fecal Microbiota Transplantation for the Treatment of Diseases Other than Clostridium Difficile Infection: A Systematic Review and Meta-Analysis. Gut Microbes 2020, 12, 1–25. [Google Scholar] [CrossRef]
- Gu, X.; Lu, Q.; Zhang, C.; Tang, Z.; Chu, L. Clinical Application and Progress of Fecal Microbiota Transplantation in Liver Diseases: A Review. Semin. Liver Dis. 2021, 41, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Augustine, P.; Phadke, N. Healthy Donor Fecal Microbiota Transplantation for Recurrent Bacterial Cholangitis in Primary Sclerosing Cholangitis—A Single Case Report. J. Clin. Transl. Hepatol. 2018, 6, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mechanism of Action | Agent | Main Results | Most Common Side Effects |
|---|---|---|---|
| Agonists of peroxisome proliferator-activated receptors (PPAR) | Bezafibrate [203,204,206,207] |
|
|
| Fenofibrate [203,205] |
| ||
| Seladelpar [209,210] |
|
| |
| Elafibranor [211,212] |
|
| |
| ASBT inhibitors | Linerixibat [215,216] |
|
|
| Agonists of farnesoid X receptor (FXR) | Cilofexor [222] |
|
|
| Tropifexor [221] |
| ||
| Corticosteroids | Budesonide [223,224] |
|
|
| Janus kinase 1 and 2 inhibitors | Barticinib [225] |
|
|
| Antibiotics | Vancomycin [228,229] |
|
|
| Microbiota Profile | Bacterial Genera in PBC Patients | Bacterial Genera in PSC Patients |
|---|---|---|
| enriched ↑ | Proteobacteria [130,132,145] Veillonella [130,131,135] Lactobacillus [132,135,145] | Proteobacteria [130,132,145] Veillonella [130,131,135] Lactobacillus [132,135,145] |
| Haemophilus [130,135] Clostridiales [131,132,135] Streptococcus [130,132,135,145] Pseudomonas [135] Klebsiella [130,135] Enterobacteriaceae [130] Neisseria [130] | Enterococcus [132,145] Bacteroidetes [130,135] | |
| depleted ↓ | Firmicutes [132,145] Bacteroidetes [130,135] | Firmicutes [132,145] Bacteroidetes [130,135] |
| Sutterella [135] Oscillospira [135] Faecalibacteria [135] |
| ClinicalTrials.gov Identifier/Location | Disease | Intervention | Study Design | Status | Primary Outcome Measures | Secondary Outcome Measures |
|---|---|---|---|---|---|---|
| NCT03521297 China | PBC patients with poor UDCA response | Probiotics (Micro V Probiotics) | Randomized, placebo-controlled, interventional; Phase 2 | Not yet recruiting | percentage of patients with the biochemical response (serum ALP or GGT decreased by 20% from baseline) | |
| NCT00161148 Netherlands | PSC patients with IBD | Probiotics (not defined) | Double-blind randomized cross-over pilot study; phase 3 | Unknown | probiotics’ effect on serum liver tests | probiotics’ effect on fatigue and pruritus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasztelan-Szczerbinska, B.; Rycyk-Bojarzynska, A.; Szczerbinska, A.; Cichoz-Lach, H. Selected Aspects of the Intricate Background of Immune-Related Cholangiopathies—A Critical Overview. Nutrients 2023, 15, 760. https://doi.org/10.3390/nu15030760
Kasztelan-Szczerbinska B, Rycyk-Bojarzynska A, Szczerbinska A, Cichoz-Lach H. Selected Aspects of the Intricate Background of Immune-Related Cholangiopathies—A Critical Overview. Nutrients. 2023; 15(3):760. https://doi.org/10.3390/nu15030760
Chicago/Turabian StyleKasztelan-Szczerbinska, Beata, Anna Rycyk-Bojarzynska, Agnieszka Szczerbinska, and Halina Cichoz-Lach. 2023. "Selected Aspects of the Intricate Background of Immune-Related Cholangiopathies—A Critical Overview" Nutrients 15, no. 3: 760. https://doi.org/10.3390/nu15030760
APA StyleKasztelan-Szczerbinska, B., Rycyk-Bojarzynska, A., Szczerbinska, A., & Cichoz-Lach, H. (2023). Selected Aspects of the Intricate Background of Immune-Related Cholangiopathies—A Critical Overview. Nutrients, 15(3), 760. https://doi.org/10.3390/nu15030760