

Protective Effect of Alpha-Linolenic Acid on Human Oral Squamous Cell Carcinoma Metastasis and Apoptotic Cell Death


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Analysis
2.3. Total Protein Extraction
2.4. Nuclear and Cytosolic Protein Extraction
2.5. Mitochondrial Protein Extraction
2.6. Western Blotting
2.7. Wound Healing Assay
2.8. Cell Invasion Assay
2.9. Analysis of Cell Apoptosis
2.10. Cell Colony Formation Assay
2.11. Gelatinase Zymography Analysis
2.12. Statistical Analysis
3. Results
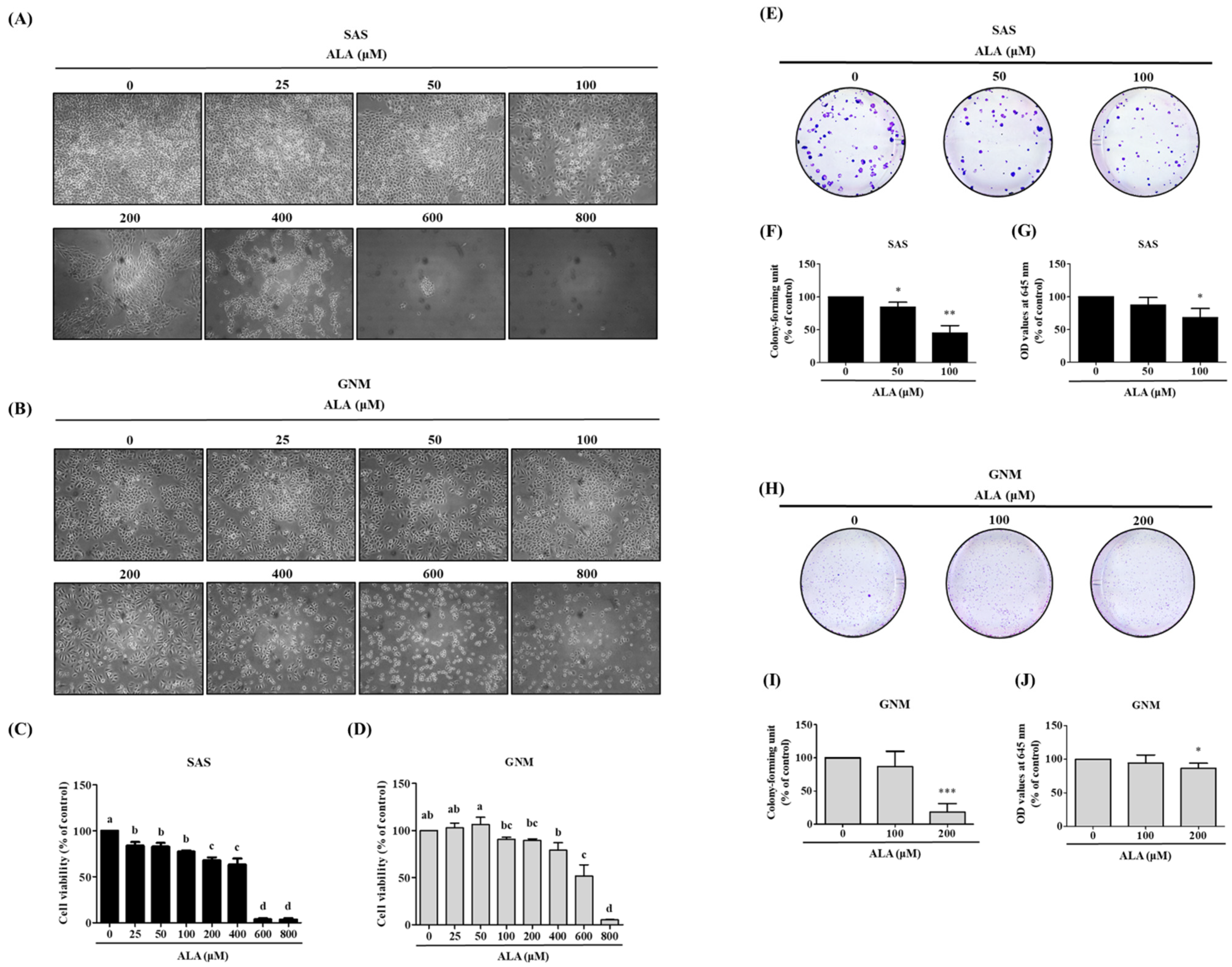
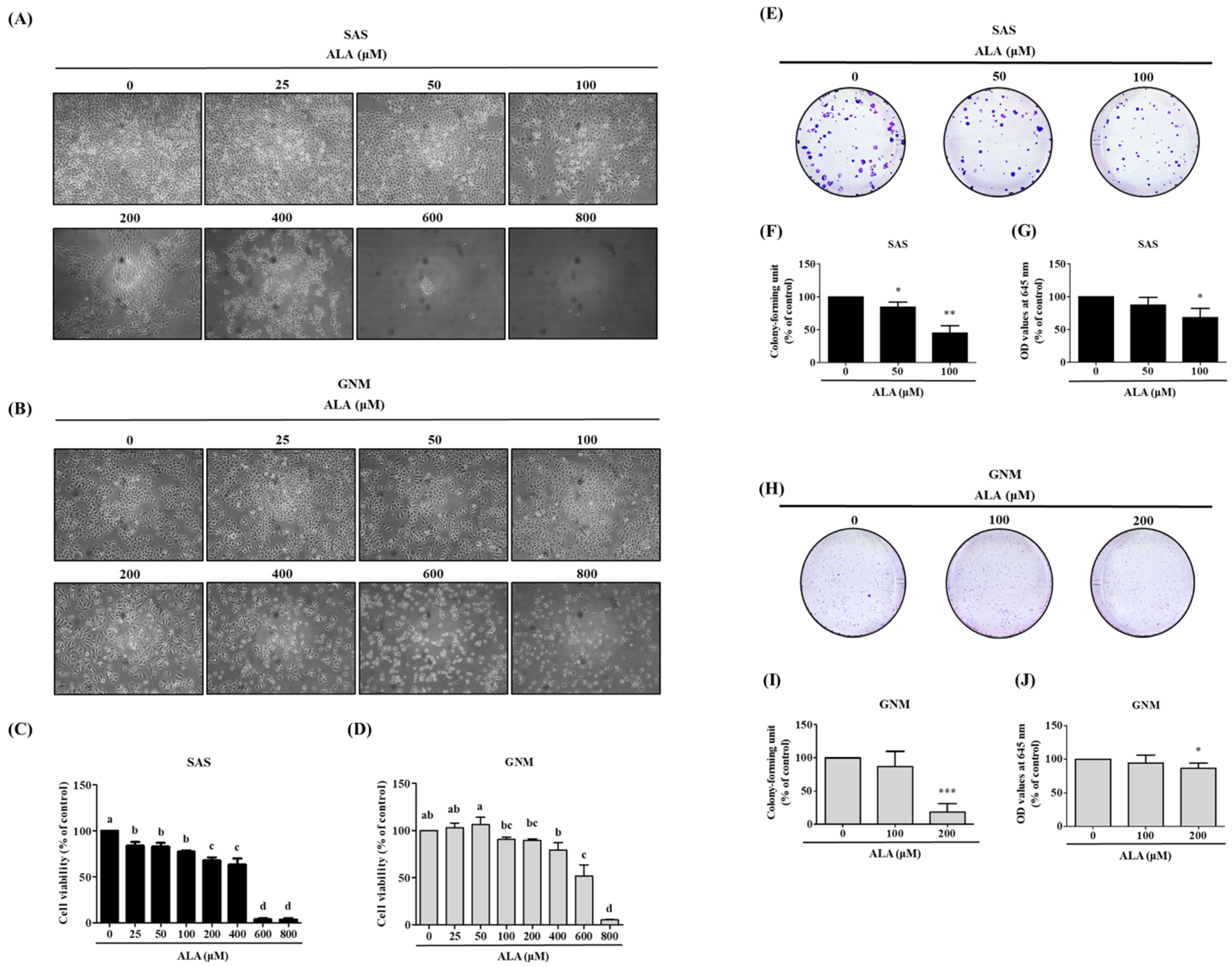
3.1. Effects of ALA on Cell Morphology, Cell Viability, and Cell Colony Formation in SAS and GNM Cells
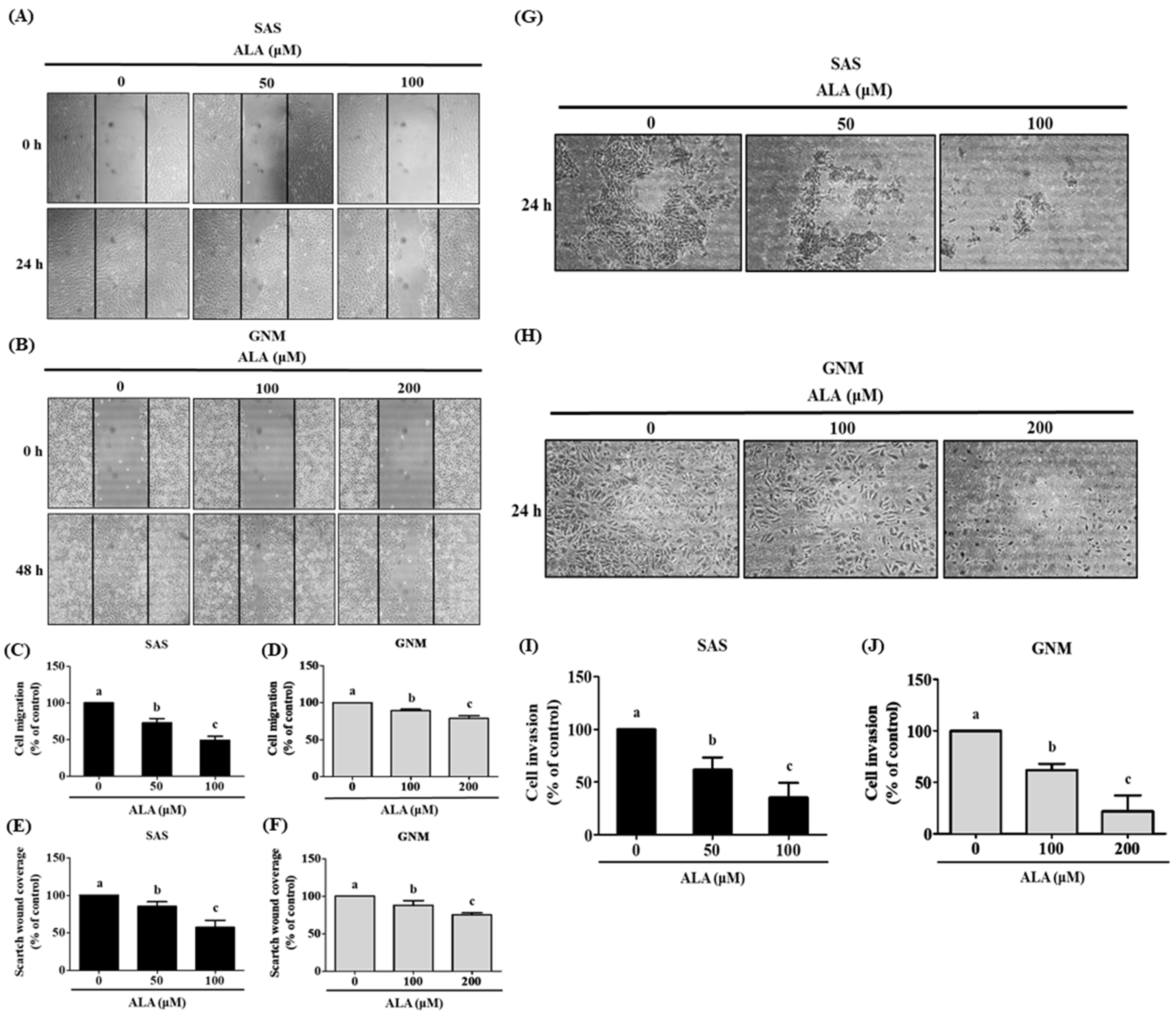
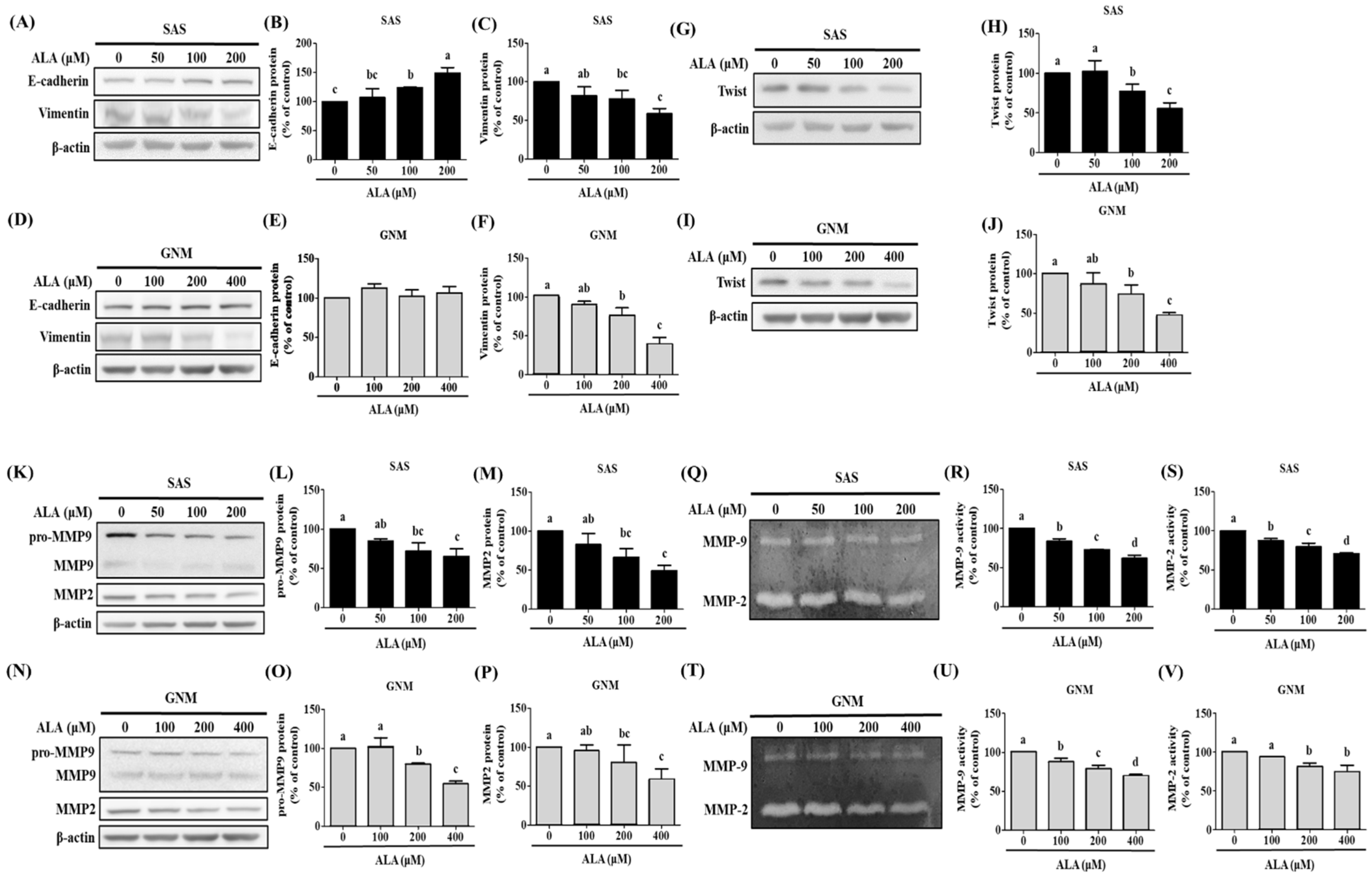
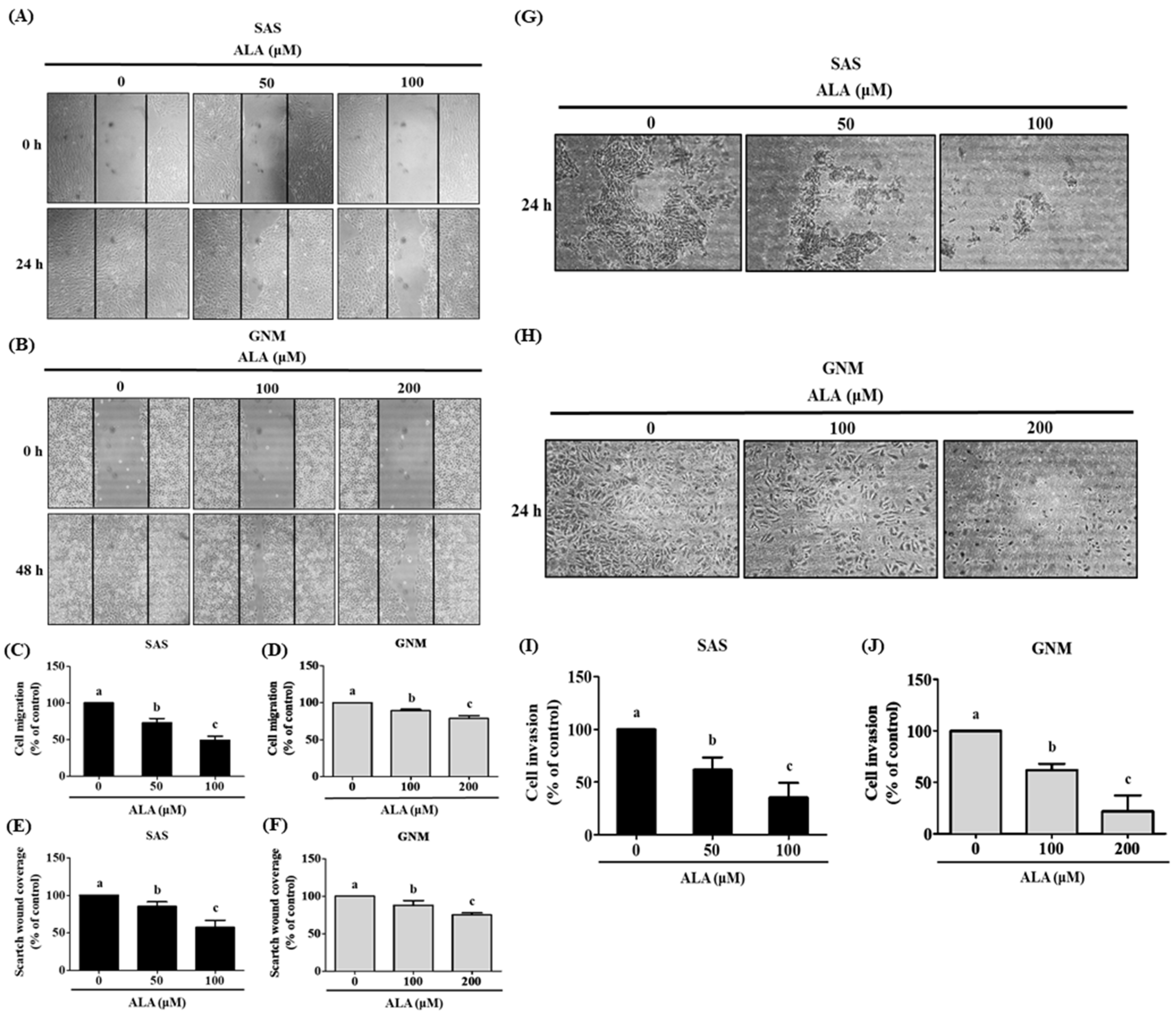
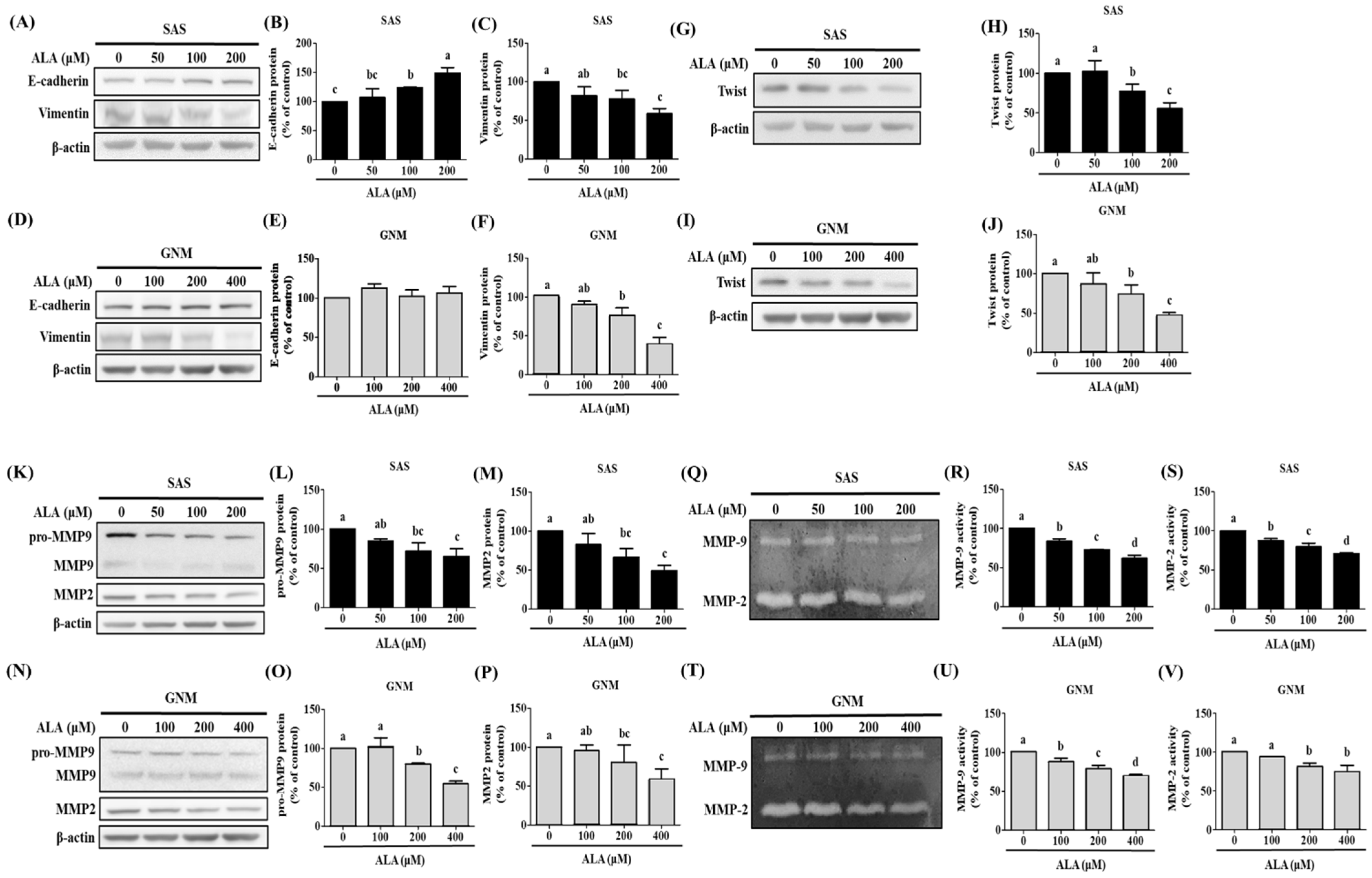
3.2. Effects of ALA on Cell Migration, Invasion, and Related Protein Expression in SAS and GNM Cells
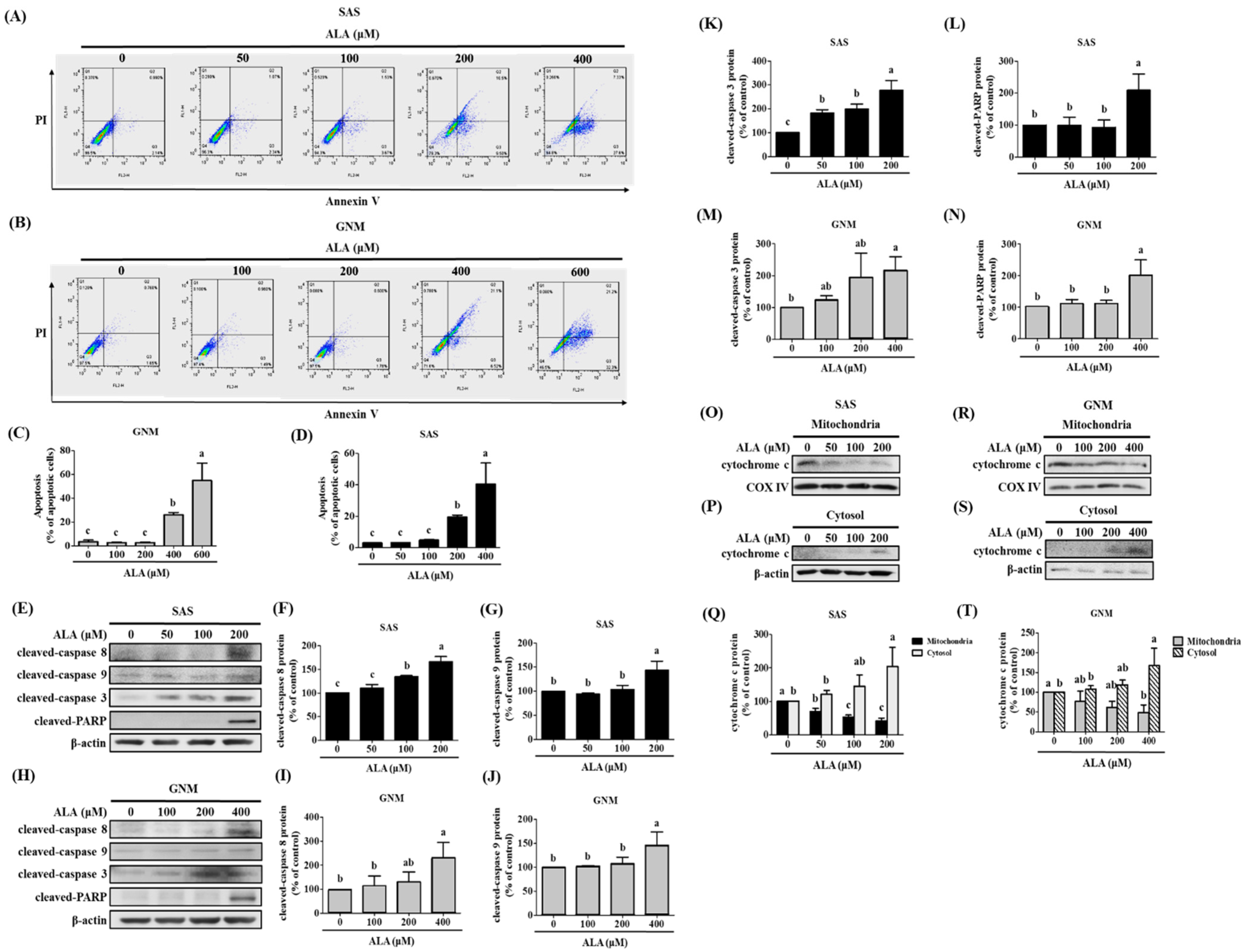
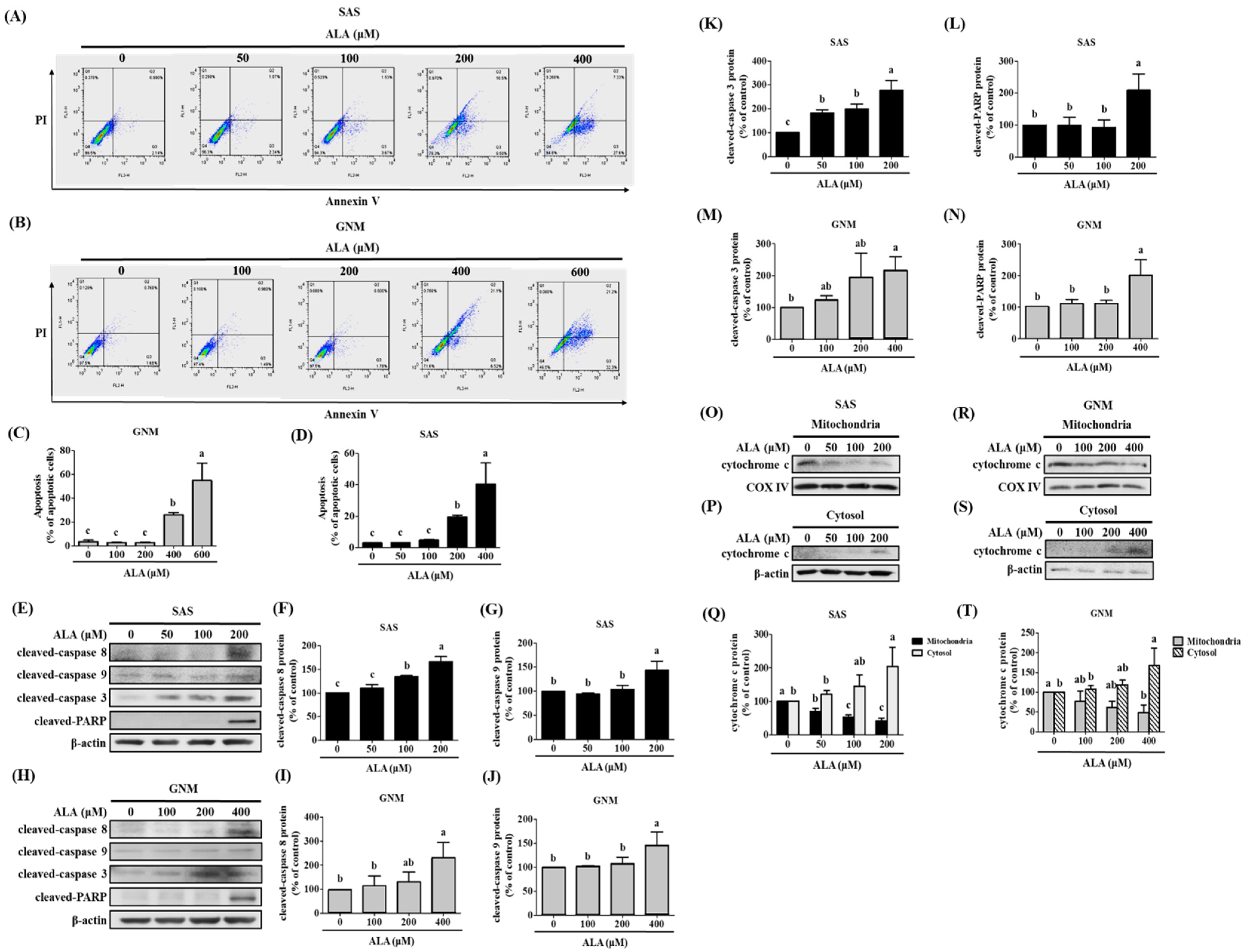
3.3. Effects of ALA on Cell Apoptosis and Related Protein Expression in SAS and GNM Cells
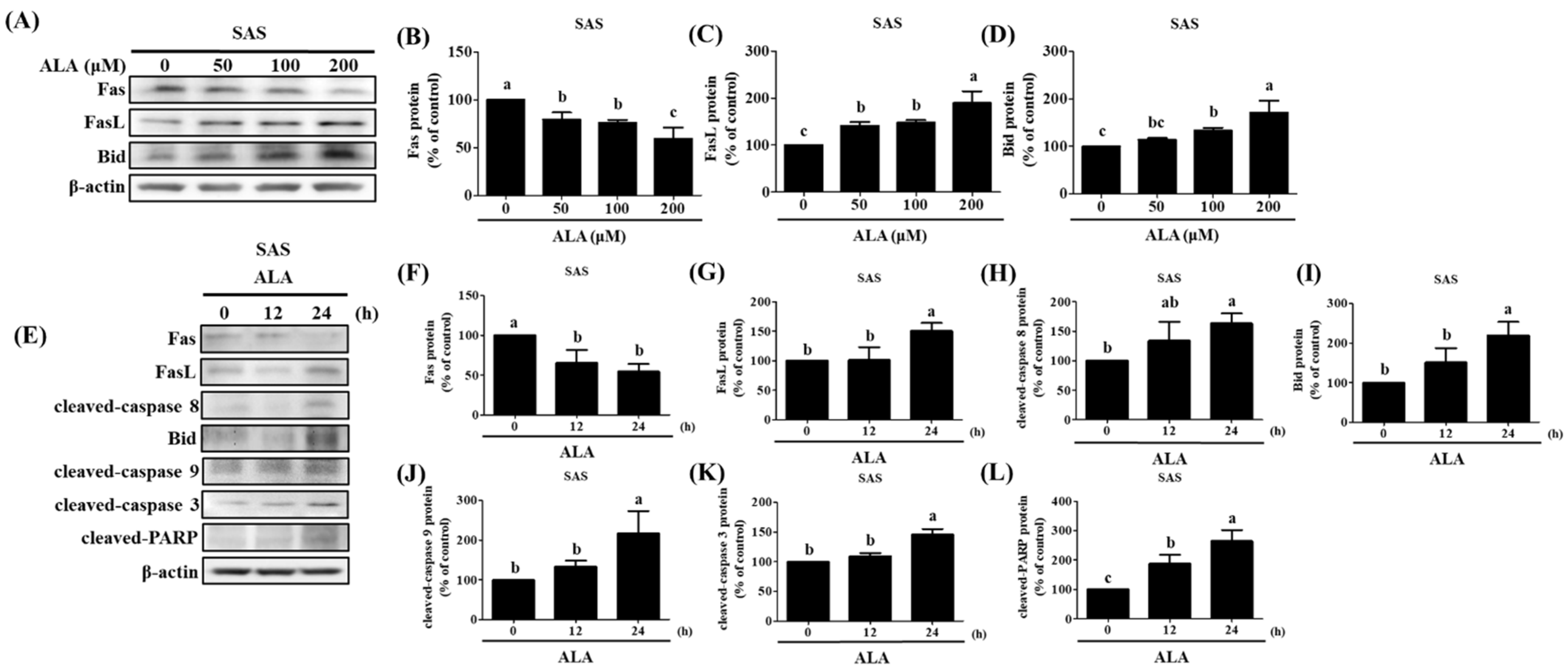
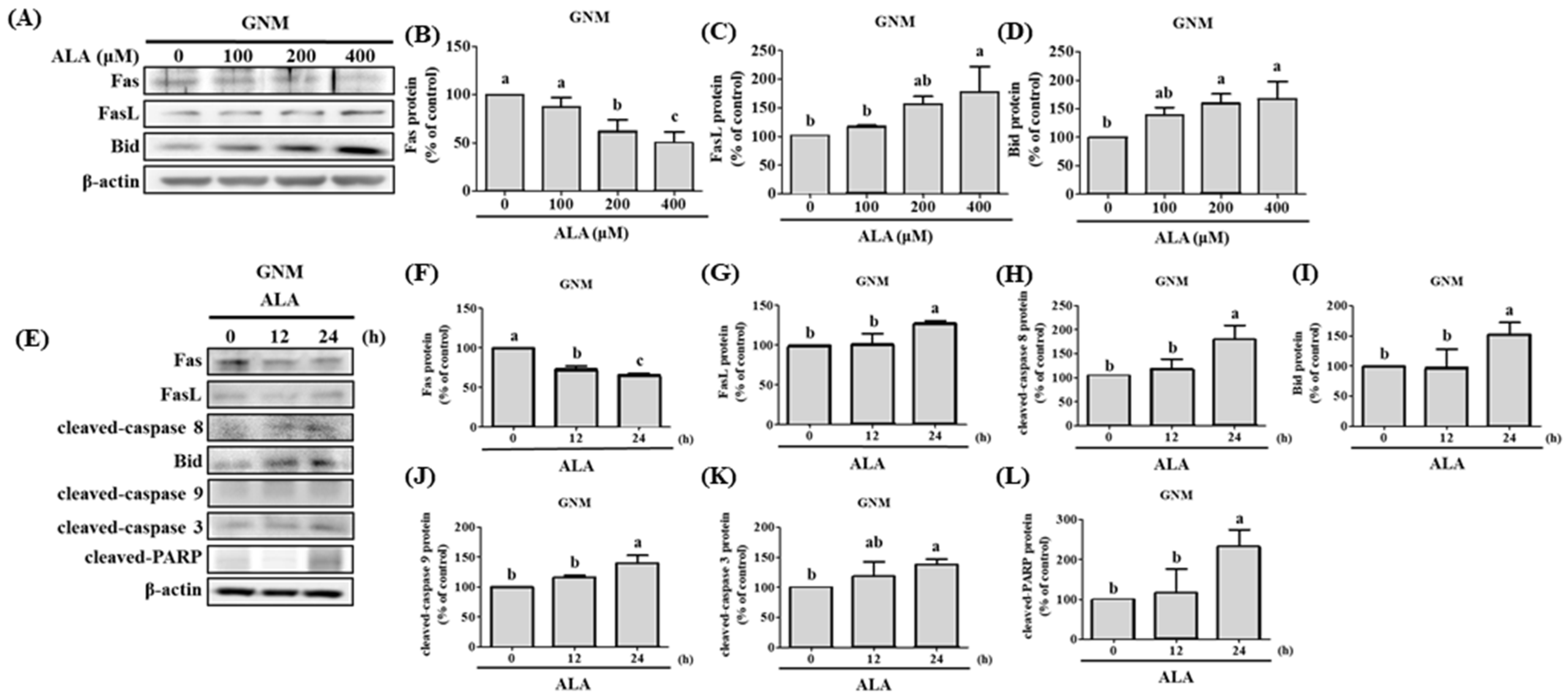
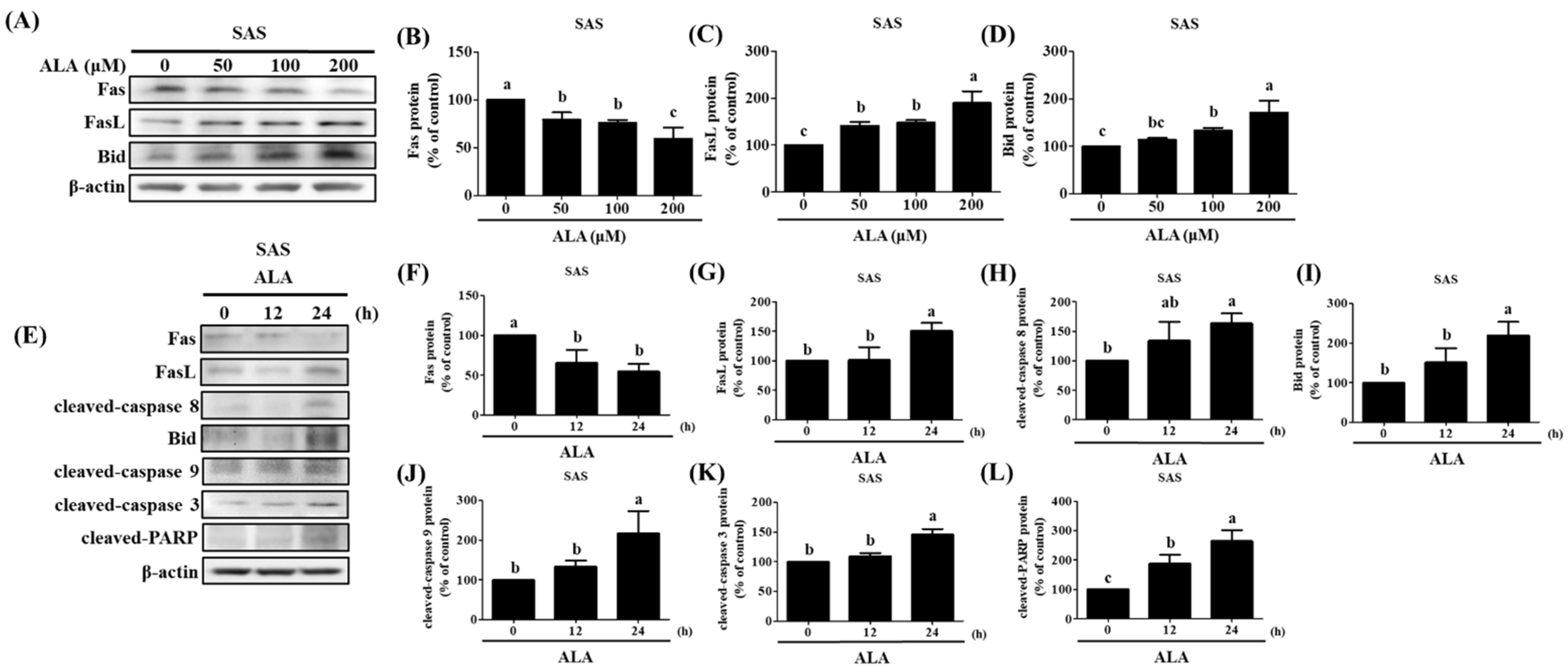
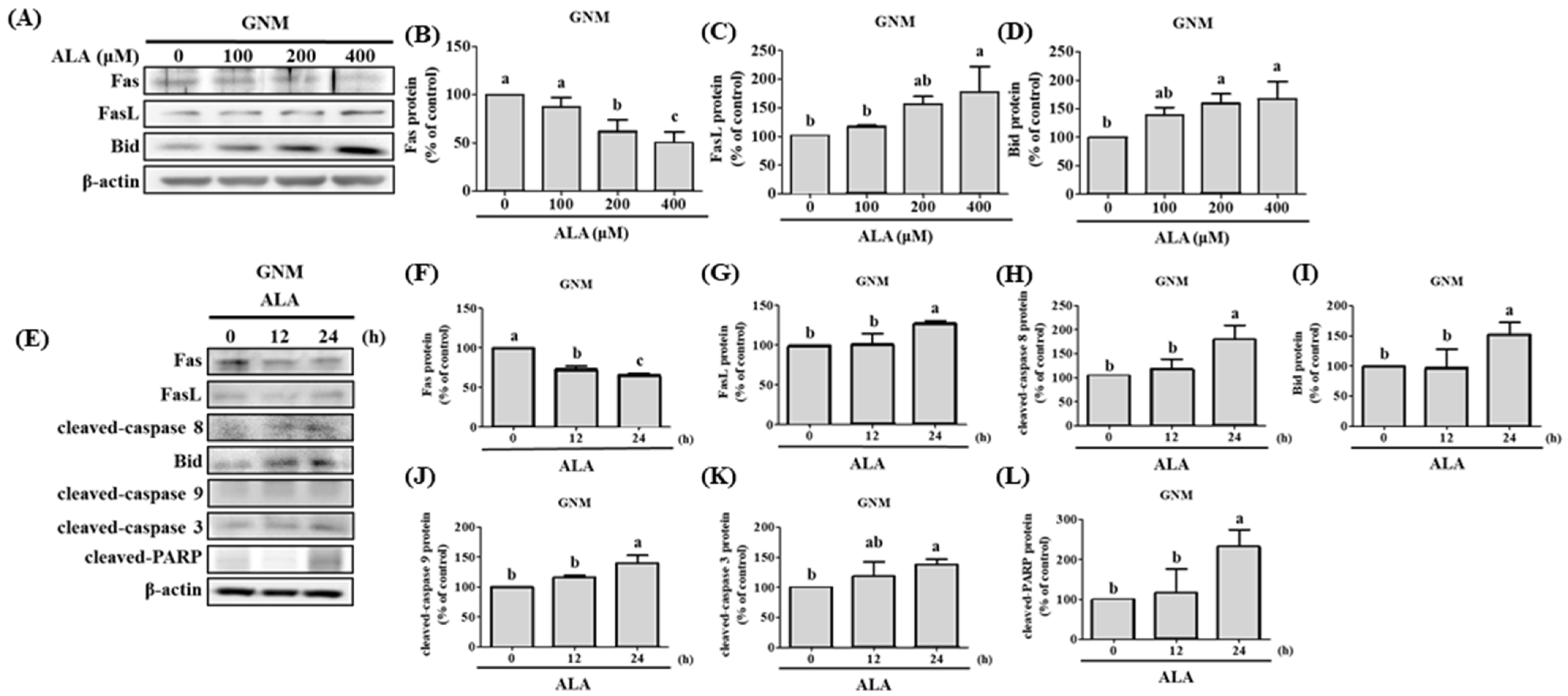
3.4. ALA’s Ability to Regulate the Expression of Fas, FasL, Bid, and Apoptosis-Related Proteins in SAS and GNM Cells
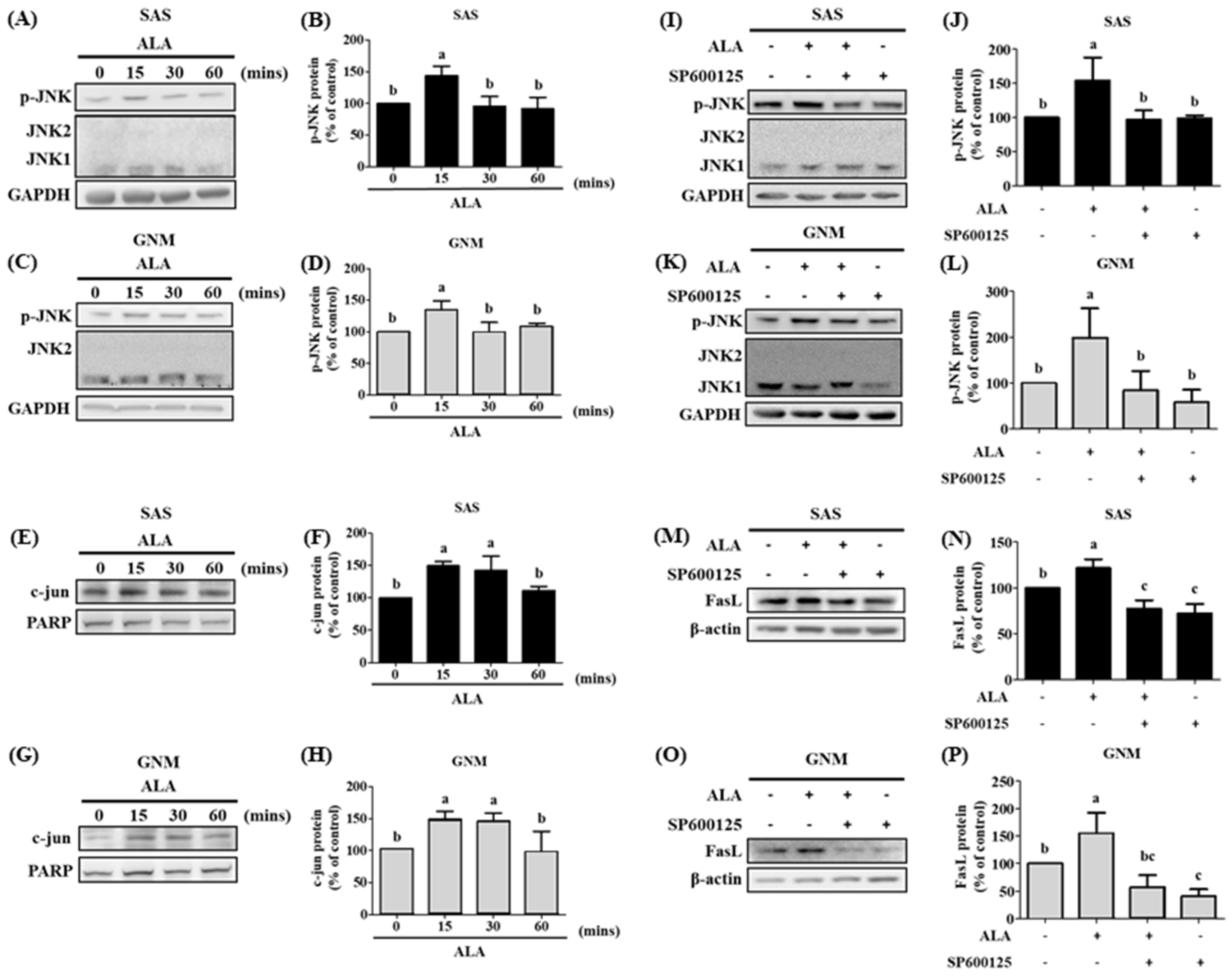
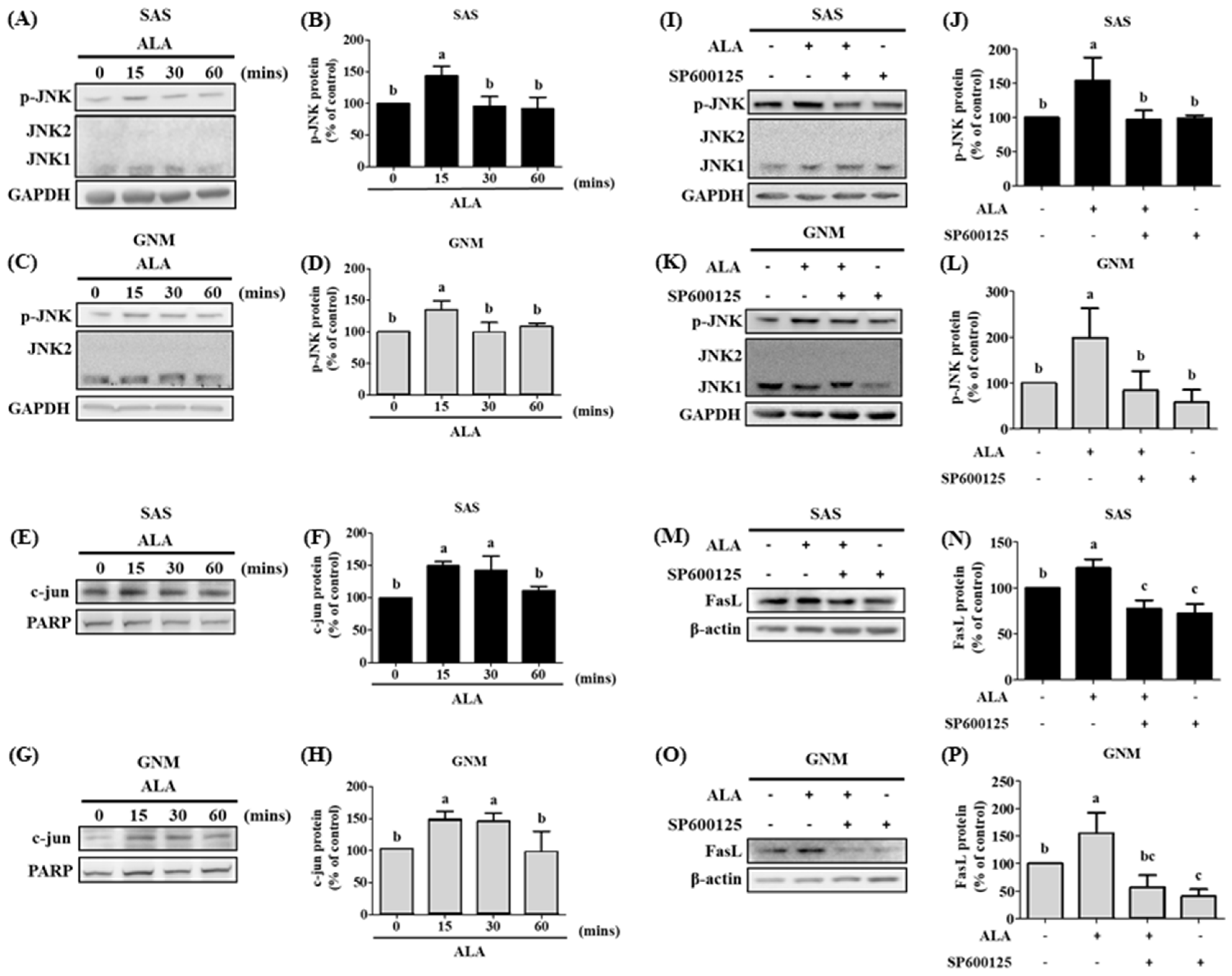
3.5. The Effects of Different Treatment Times of ALA on the Expression of Phosphorylated JNK Protein, Nuclear c-jun Protein Accumulation, and Their Association with FasL in SAS and GNM Cells
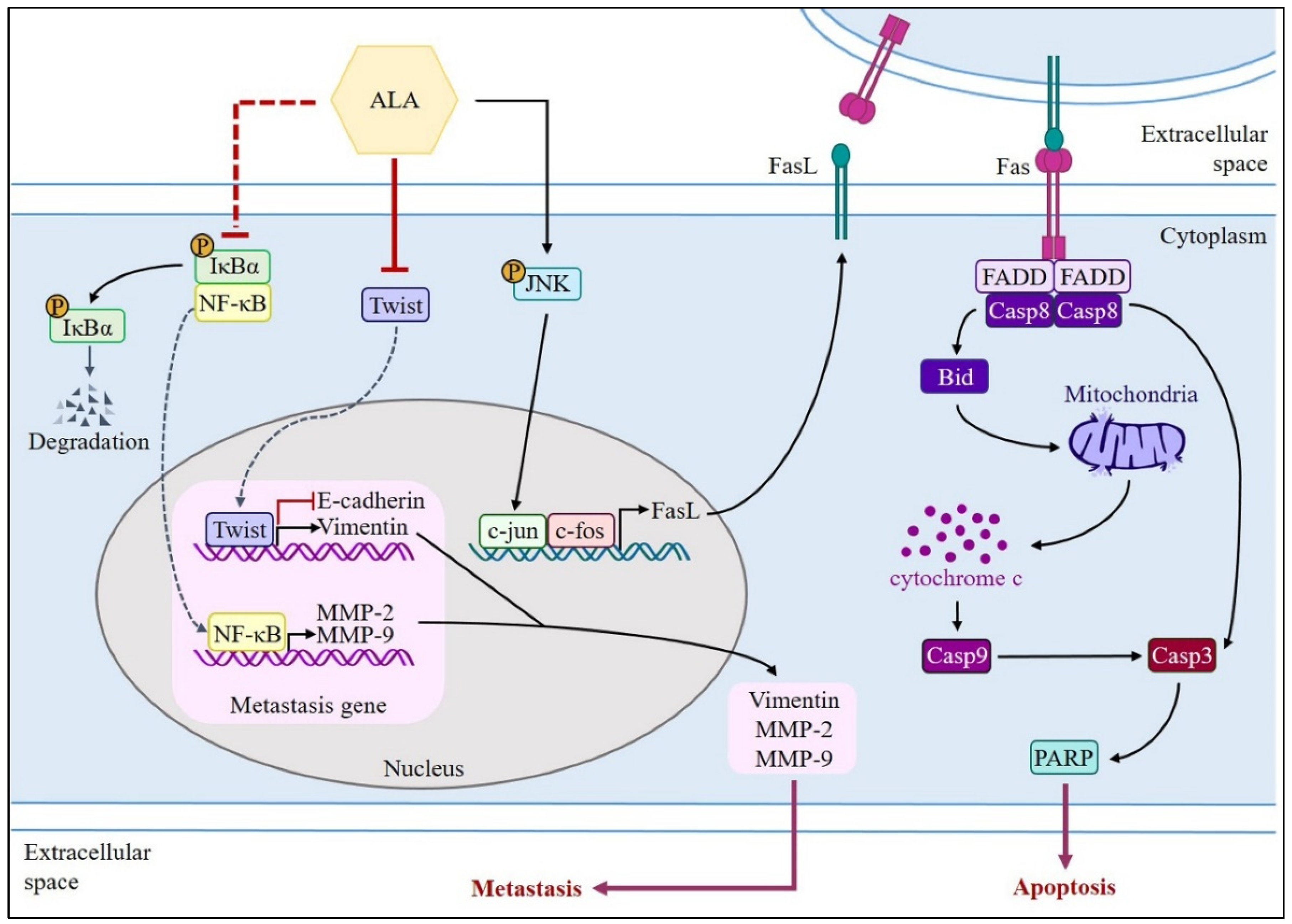
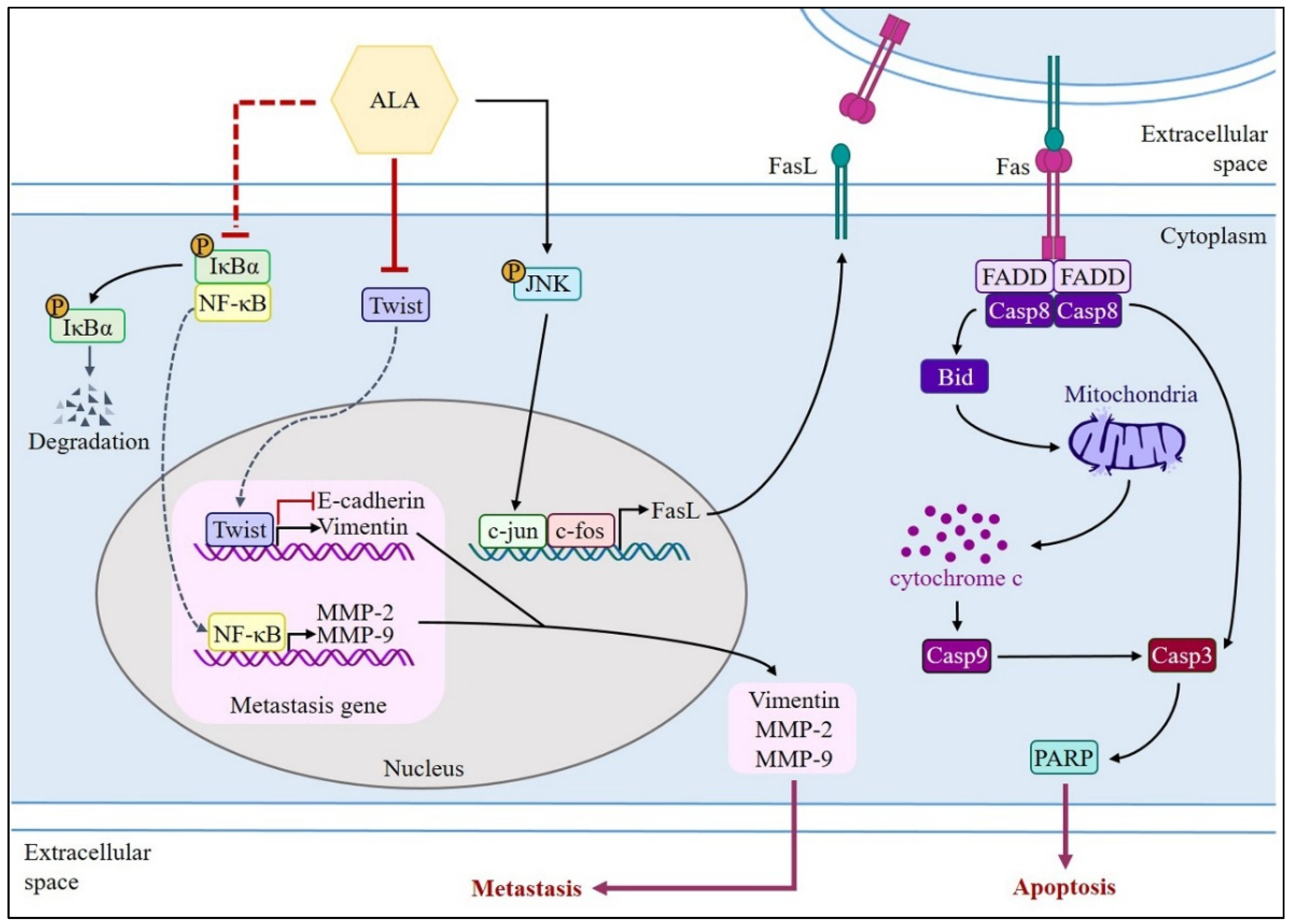
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. Diagnostic and prognostic value of chromosome studies in cancer. Ann. Clin. Lab. Sci. 1974, 4, 234–240. [Google Scholar] [PubMed]
- Bajaj, J.; Diaz, E. Stem cells in cancer initiation and progression. J. Cell Biol. 2019, 219, e201911053. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Neagu, M. Proteoglycans and immunobiology of cancer—Therapeutic implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef] [PubMed]
- Tzanakakis, G.; Giatagana, E.M. Proteoglycans in the pathogenesis of hormone-dependent cancers: Mediators and effectors. Cancers 2020, 12, 2401. [Google Scholar] [CrossRef]
- Giatagana, E.M.; Berdiaki, A. Lumican in carcinogenesis—Revisited. Biomolecules 2021, 11, 1319. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Jiang, R.S. Smoking, alcohol, and betel quid and oral cancer: A prospective cohort study. J. Oncol. 2011, 2011, 525976. [Google Scholar] [CrossRef]
- Dhanuthai, K.; Rojanawatsirivej, S. Oral cancer: A multicenter study. Med. Oral Patol. Oral Cir. Bucal. 2018, 23, e23. [Google Scholar] [CrossRef]
- Grewal, P.; Viswanathen, V.A. Liver cancer and alcohol. Clin. Liver Dis. 2012, 16, 839–850. [Google Scholar] [CrossRef]
- Stornetta, A.; Guidolin, V. Alcohol-derived acetaldehyde exposure in the oral cavity. Cancers 2018, 10, 20. [Google Scholar] [CrossRef]
- Curado, M.P.; Hashibe, M. Recent changes in the epidemiology of head and neck cancer. Curr. Opin. Oncol. 2009, 21, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Kumar, A. Assessment of relationship of ABO blood groups in oral cancer patients-a retrospective study. Ann. Maxillofac. Surg. 2021, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Lu, Q. Oral microbiota alteration associated with oral cancer and areca chewing. Oral Dis. 2021, 27, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surg. Oncol. Clin. 2015, 24, 491–508. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar] [PubMed]
- Kademani, D. Oral cancer. Mayo Clin. Proc. 2007, 82, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.T.; Barnes, E.L. The extracapsular spread of tumors in cervical node metastasis. Arch. Otolaryngol. 1981, 107, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Shin, D.Y. Prognostic significance of epithelial-mesenchymal transition of extracapsular spread tumors in lymph node metastases of head and neck cancer. Ann. Surg. Oncol. 2014, 21, 1904–1911. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Merindol, N.; Riquet, A. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014, 4, e206. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Sun, L. SET8 promotes epithelial–mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Adrain, C. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Swanson, D.; Block, R. Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 2012, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Welch, A.A.; Shakya-Shrestha, S. Dietary intake and status of n–3 polyunsaturated fatty acids in a population of fish-eating and non-fish-eating meat-eaters, vegetarians, and vegans and the precursor-product ratio of α-linolenic acid to long-chain n–3 polyunsaturated fatty acids: Results from the EPIC-Norfolk cohort. Am. J. Clin. Nutr. 2010, 92, 1040–1051. [Google Scholar] [PubMed]
- Roy, S.; Rawat, A.K. Alpha-linolenic acid stabilizes HIF-1 α and downregulates FASN to promote mitochondrial apoptosis for mammary gland chemoprevention. Oncotarget 2017, 8, 70049. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Nam, Y.A. α-Linolenic acid: Nutraceutical, pharmacological and toxicological evaluation. Food Chem. Toxicol. 2014, 70, 163–178. [Google Scholar] [CrossRef]
- Yuan, Q.; Xie, F. The review of alpha-linolenic acid: Sources, metabolism, and pharmacology. Phytother. Res. 2022, 36, 164–188. [Google Scholar] [CrossRef]
- Yang, L.; Yuan, J. α-linolenic acid inhibits human renal cell carcinoma cell proliferation through PPAR-γ activation and COX-2 inhibition. Oncol. Lett. 2013, 6, 197–202. [Google Scholar] [CrossRef]
- Deshpande, R.; Mansara, P. Alpha-linolenic acid regulates Cox2/VEGF/MAP kinase pathway and decreases the expression of HPV oncoproteins E6/E7 through restoration of p53 and Rb expression in human cervical cancer cell lines. Tumor Biol. 2016, 37, 3295–3305. [Google Scholar] [CrossRef]
- Li, J.; Gu, Z. Dietary supplementation of α-linolenic acid induced conversion of n-3 LCPUFAs and reduced prostate cancer growth in a mouse model. Lipids Health Dis. 2017, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Kang, B.H. Vimentin is a potential prognostic factor for tongue squamous cell carcinoma among five epithelial–mesenchymal transition-related proteins. PLoS ONE 2017, 12, e0178581. [Google Scholar] [CrossRef] [PubMed]
- Seyedmajidi, M.; Seifi, S. Immunohistochemical expression of TWIST in oral squamous cell carcinoma and its correlation with clinicopathologic factors. J. Cancer Res. Ther. 2018, 14, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.W.; Hsu, S.C. Quercetin inhibits migration and invasion of SAS human oral cancer cells through inhibition of NF-κB and matrix metalloproteinase-2/-9 signaling pathways. Anticancer Res. 2013, 33, 1941–1950. [Google Scholar] [PubMed]
- Lee, C.C.; Hsiao, C.Y. Suppression of oral cancer by induction of cell cycle arrest and apoptosis using Juniperus communis extract. Biosci. Rep. 2020, 40, BSR20202083. [Google Scholar] [CrossRef] [PubMed]
- Won, D.H.; Kim, L.H. In vitro and in vivo anti-cancer activity of silymarin on oral cancer. Tumor Biol. 2018, 40, 1010428318776170. [Google Scholar] [CrossRef]
- Suhara, T.; Kim, H.S. Suppression of Akt signaling induces Fas ligand expression: Involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol. Cell. Biol. 2002, 22, 680–691. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Park, S.J.; Yoon, B.H. GENT2: An updated gene expression database for normal and tumor tissues. BMC Med. Genom. 2019, 12, 101. [Google Scholar] [CrossRef]
- Das, U.N. “Cell membrane theory of senescence” and the role of bioactive lipids in aging, and aging associated diseases and their therapeutic implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Greene, E.R. EET signaling in cancer. Cancer Metastasis Rev. 2011, 30, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Wijendran, V.; Hayes, K.C. Dietary n-6 and n-3 fatty acid balance and cardiovascular health. Annu. Rev. Nutr. 2004, 24, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Ambigaipalan, P. Omega-3 polyunsaturated fatty acids and their health benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Kumlin, M. Dietary long-chain n− 3 fatty acids for the prevention of cancer: A review of potential mechanisms. Am. J. Clin. Nutr. 2004, 79, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Corsetto, P.A.; Cremona, A. Chemical–physical changes in cell membrane microdomains of breast cancer cells after omega-3 PUFA incorporation. Cell Biochem. Biophys. 2012, 64, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liu, Q. Docosahexaenoic acid ameliorates the Toll-Like Receptor 22-triggered inflammation in fish by disrupting lipid raft formation. J. Nutr. 2022, 152, 1991–2002. [Google Scholar] [CrossRef]
- Basiouni, S.; Stöckel, K. Polyunsaturated fatty acid supplements modulate mast cell membrane microdomain composition. Cell. Immunol. 2021, 275, 42–46. [Google Scholar] [CrossRef]
- Burns, R.N.; Moniri, N.H. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem. Biophys. Res. Commun. 2010, 396, 1030–1035. [Google Scholar] [CrossRef]
- Senatorov, I.S.; Moniri, N.H. The role of free-fatty acid receptor-4 (FFA4) in human cancers and cancer cell lines. Biochem. Pharmacol. 2018, 150, 170–180. [Google Scholar] [CrossRef]
- Vecchini, A.; Ceccarelli, V. Dietary α-linolenic acid reduces COX-2 expression and induces apoptosis of hepatoma cells. J. Lipid Res. 2004, 45, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Truan, J.S.; Chen, J.M. Flaxseed oil reduces the growth of human breast tumors (MCF-7) at high levels of circulating estrogen. Mol. Nutr. Food Res. 2010, 54, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.; Mansara, P. Alpha-linolenic acid regulates the growth of breast and cervical cancer cell lines through regulation of NO release and induction of lipid peroxidation. J. Mol. Biochem. 2013, 2, 6–17. [Google Scholar]
- Fan, H.; Huang, W. α-Linolenic Acid Suppresses Proliferation and Invasion in Osteosarcoma Cells via Inhibiting Fatty Acid Synthase. Molecules 2022, 27, 2741. [Google Scholar] [CrossRef] [PubMed]
- Mason-Ennis, J.K.; LeMay-Nedjelski, L.P. Exploration of mechanisms of α-linolenic acid in reducing the growth of oestrogen receptor positive breast cancer cells (MCF-7). J. Funct. Foods 2016, 24, 513–519. [Google Scholar] [CrossRef]
- Wiggins, A.K.; Kharotia, S. α-Linolenic acid reduces growth of both triple negative and luminal breast cancer cells in high and low estrogen environments. Nutr. Cancer 2015, 67, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.S.; Batirel, S. Alpha linolenic acid and oleic acid additively down-regulate malignant potential and positively cross-regulate AMPK/S6 axis in OE19 and OE33 esophageal cancer cells. Metabolism 2014, 63, 1447–1454. [Google Scholar] [CrossRef]
- Fan, C.C.; Wang, T.Y. Expression of E-cadherin, Twist, and p53 and their prognostic value in patients with oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1735–1744. [Google Scholar] [CrossRef]
- Wang, S.C.; Sun, H.L. α-Linolenic acid inhibits the migration of human triple-negative breast cancer cells by attenuating Twist1 expression and suppressing Twist1-mediated epithelial-mesenchymal transition. Biochem. Pharmacol. 2020, 180, 114152. [Google Scholar] [CrossRef]
- Chen, H.W.; Chao, C.Y. Inhibition of matrix metalloproteinase-9 expression by docosahexaenoic acid mediated by heme oxygenase 1 in 12-O-tetradecanoylphorbol-13-acetate-induced MCF-7 human breast cancer cells. Arch. Toxicol. 2013, 87, 857–869. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.H.; Chang, W.M. A Fas ligand (FasL)-fused humanized antibody against tumor-associated glycoprotein 72 selectively exhibits the cytotoxic effect against oral cancer cells with a low FasL/Fas ratio. Mol. Cancer Ther. 2017, 16, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.; Khachigian, L. Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ. 2003, 10, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, A.; Rodriguez-Leyva, D. Bioavailability of α-linolenic acid from flaxseed diets as a function of the age of the subject. Eur. J. Clin. Nutr. 2009, 63, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Schwab, U.S.; Callaway, J.C. Effects of hempseed and flaxseed oils on the profile of serum lipids, serum total and lipoprotein lipid concentrations and haemostatic factors. Eur. J. Nutr. 2006, 45, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Martinchik, A.; Baturin, A. Nutritional value and functional properties of flaxseed. Vopr. Pitan. 2012, 81, 4–10. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.-C.; Yu, C.-C.; Shih, Y.-W.; Liu, K.-L.; Chen, H.-W.; Wu, C.-C.; Yang, Y.-C.; Yeh, E.-L.; Li, C.-C. Protective Effect of Alpha-Linolenic Acid on Human Oral Squamous Cell Carcinoma Metastasis and Apoptotic Cell Death. Nutrients 2023, 15, 4992. https://doi.org/10.3390/nu15234992
Su C-C, Yu C-C, Shih Y-W, Liu K-L, Chen H-W, Wu C-C, Yang Y-C, Yeh E-L, Li C-C. Protective Effect of Alpha-Linolenic Acid on Human Oral Squamous Cell Carcinoma Metastasis and Apoptotic Cell Death. Nutrients. 2023; 15(23):4992. https://doi.org/10.3390/nu15234992
Chicago/Turabian StyleSu, Ching-Chyuan, Cheng-Chia Yu, Yi-Wen Shih, Kai-Li Liu, Haw-Wen Chen, Chih-Chung Wu, Ya-Chen Yang, En-Ling Yeh, and Chien-Chun Li. 2023. "Protective Effect of Alpha-Linolenic Acid on Human Oral Squamous Cell Carcinoma Metastasis and Apoptotic Cell Death" Nutrients 15, no. 23: 4992. https://doi.org/10.3390/nu15234992
APA StyleSu, C.-C., Yu, C.-C., Shih, Y.-W., Liu, K.-L., Chen, H.-W., Wu, C.-C., Yang, Y.-C., Yeh, E.-L., & Li, C.-C. (2023). Protective Effect of Alpha-Linolenic Acid on Human Oral Squamous Cell Carcinoma Metastasis and Apoptotic Cell Death. Nutrients, 15(23), 4992. https://doi.org/10.3390/nu15234992