Thermoneutral Housing Enables Studies of Vertical Transmission of Obesogenic Diet-Driven Metabolic Diseases

 , , , , and
, , , , and
Abstract
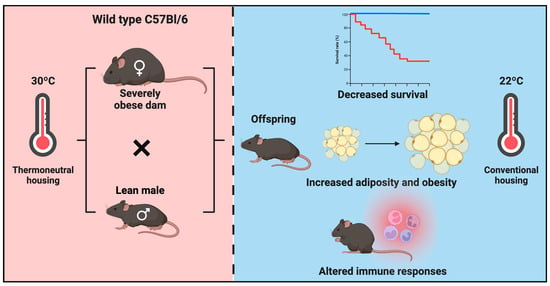
1. Introduction
2. Materials and Methods
2.1. Mice
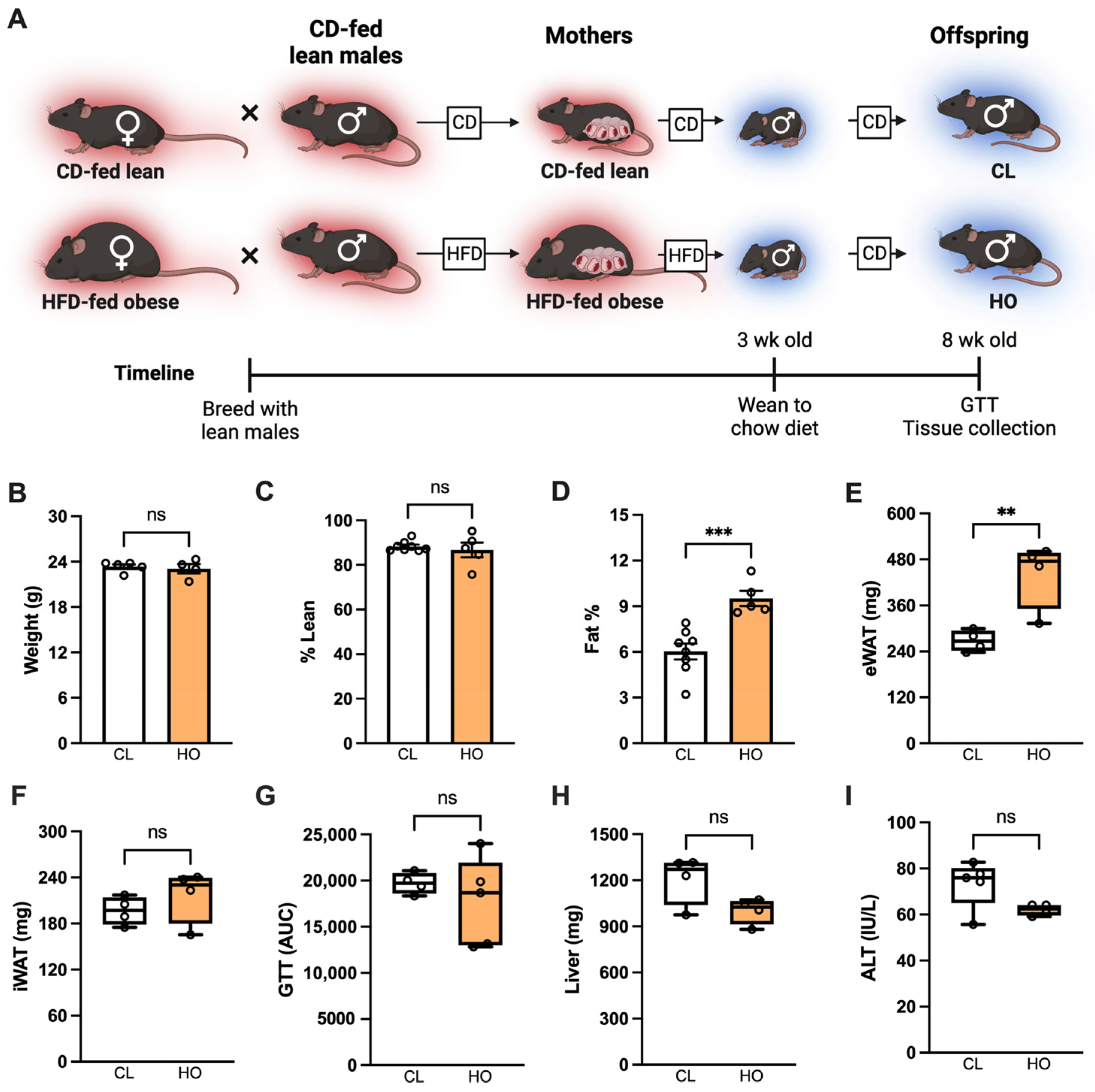
2.2. Maternal Obesity Model and Mouse Study Design
2.2.1. Experimental Design: Pregnancy and Neonatal Offspring Health
2.2.2. Experimental Design: Offspring Health at Baseline
2.2.3. Experimental Design: Offspring Health during HFD Challenge
2.2.4. Experimental Design: Microbiome Manipulation
2.3. In Vivo and Serum Assays (GTT, EchoMRI, Hemavet, ALT)
2.4. Cytokine and Endotoxin Quantification
2.5. Mouse Embryonic Fibroblast (MEF) Isolation and Culture
2.6. Flow Cytometry
2.7. Shotgun Metagenome Sequencing and Data Analysis
2.8. Proteomics
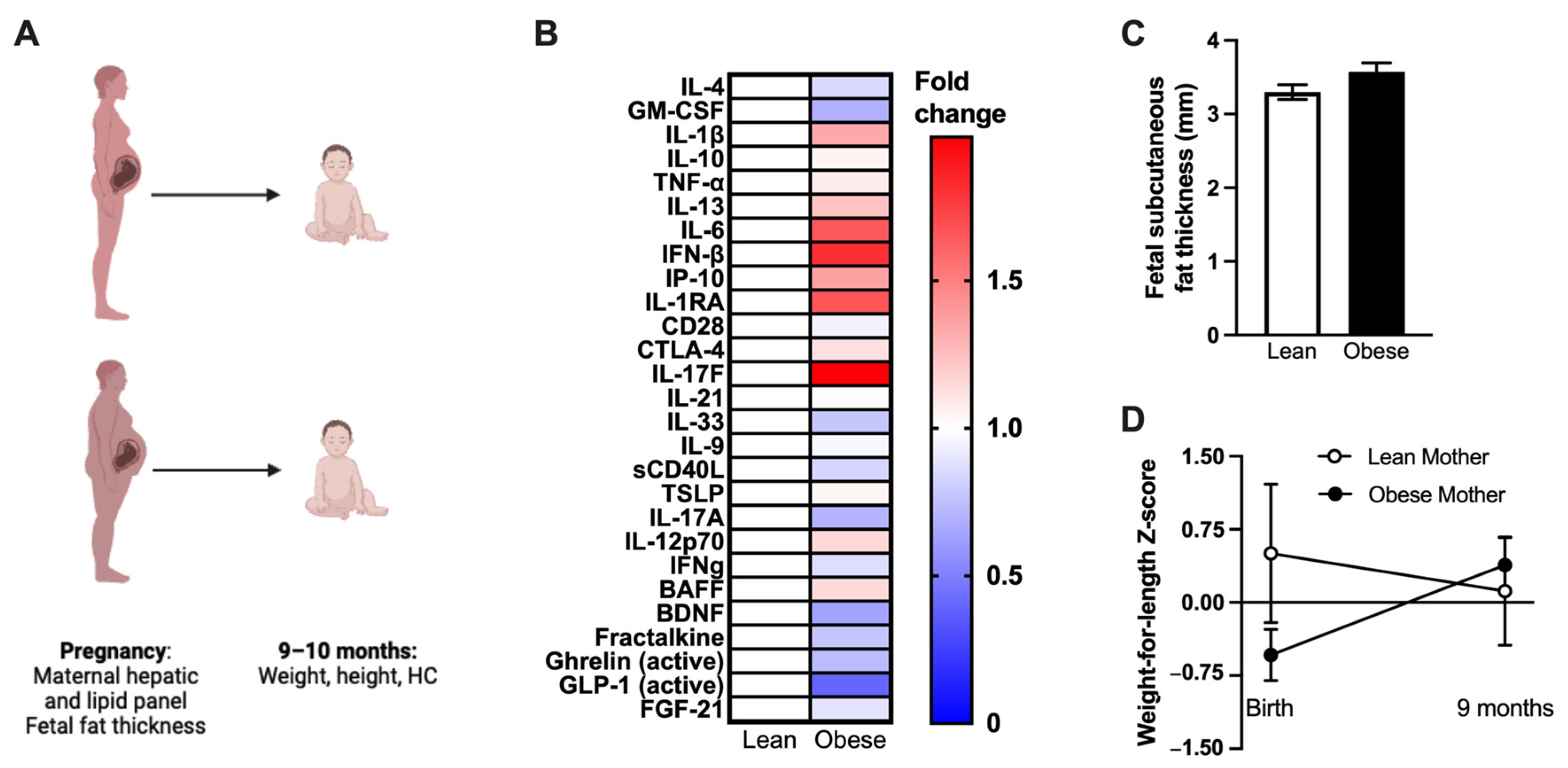
2.9. Human Study Design
2.10. Statistical Analysis
3. Results
3.1. Maternal Obesity Impacts Neonatal Offspring Health in Mice
3.2. Absence of Metabolic Dysfunction at Baseline in Offspring of HFD-Fed Obese Dams
3.3. Development of Metabolic Dysfunction in Offspring of HFD-Fed Obese Dams after HFD Feeding
3.4. Maternal HFD and Antibiotic Treatment Impact the Intestinal Microbiome in Mice
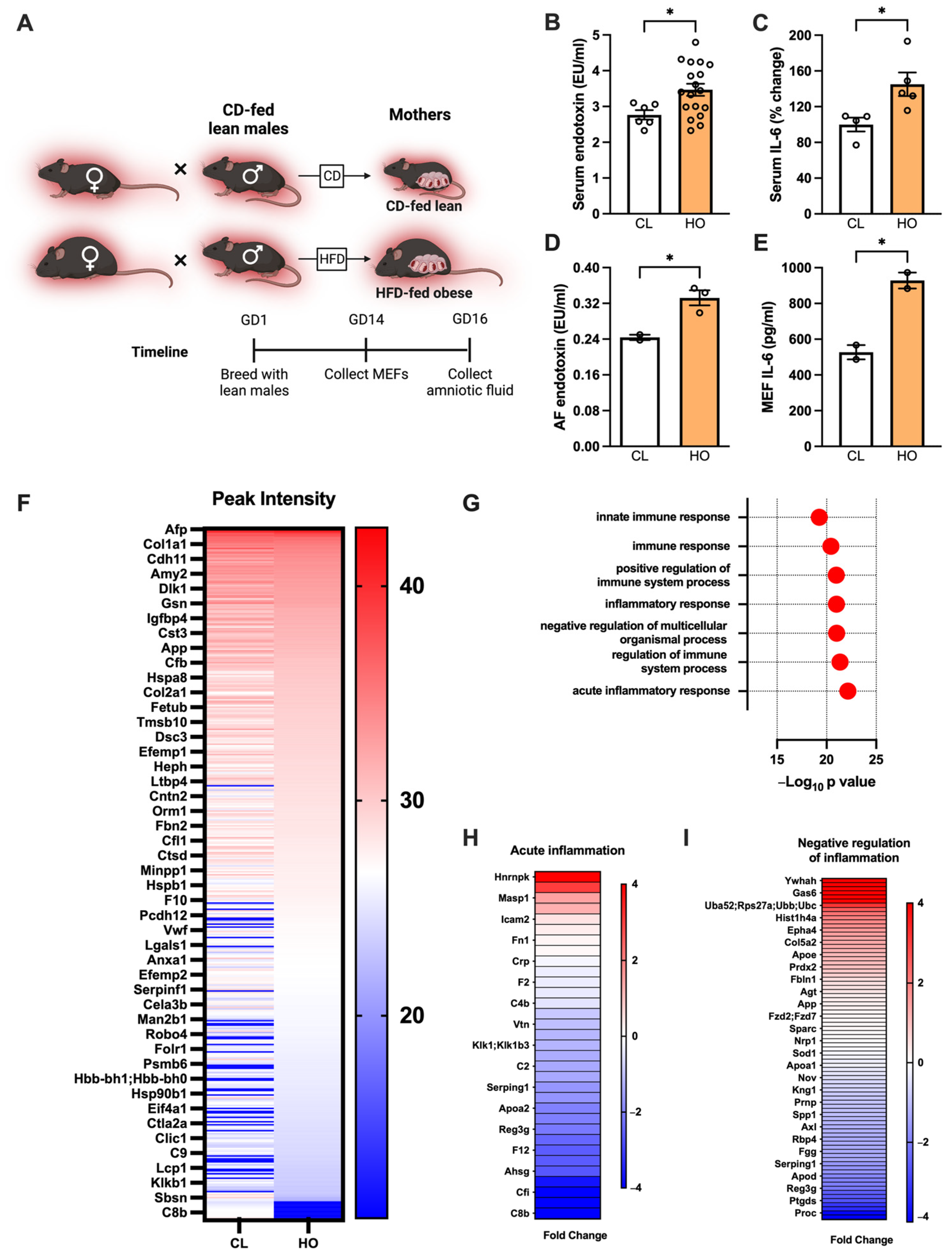
3.5. Maternal Obesity-Associated Inflammation Transfers to the Fetus in Utero in Mice
3.6. Human Mothers with Obesity Have Altered Inflammation, and Their Infants Experience Differential Weight Gain after Birth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–17. [Google Scholar]
- Lappas, M. Effect of pre-existing maternal obesity, gestational diabetes and adipokines on the expression of genes involved in lipid metabolism in adipose tissue. Metab. Clin. Exp. 2014, 63, 250–262. [Google Scholar] [CrossRef]
- Suzuki, A.; Diehl, A.M. Nonalcoholic Steatohepatitis. Annu. Rev. Med. 2017, 68, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.; Ehrenberg, H. Review article: The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG Int. J. Obstet. Gynaecol. 2006, 113, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Smith, G.D.; O’Callaghan, M.; Alati, R.; Mamun, A.A.; Williams, G.M.; Najman, J.M. Epidemiologic evidence for the fetal overnutrition hypothesis: Findings from the mater-university study of pregnancy and its outcomes. Am. J. Epidemiol. 2007, 165, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Comas-Armangue, G.; Makharadze, L.; Gomez-Velazquez, M.; Teperino, R. The Legacy of Parental Obesity: Mechanisms of Non-Genetic Transmission and Reversibility. Biomedicines 2022, 10, 2461. [Google Scholar] [CrossRef] [PubMed]
- Bellatorre, A.; Scherzinger, A.; Stamm, E.; Martinez, M.; Ringham, B.; Dabelea, D. Fetal Overnutrition and Adolescent Hepatic Fat Fraction: The Exploring Perinatal Outcomes in Children Study. J. Pediatr. 2018, 192, 165–170.e161. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef]
- Thorn, S.R.; Baquero, K.C.; Newsom, S.A.; El Kasmi, K.C.; Bergman, B.C.; Shulman, G.I.; Grove, K.L.; Friedman, J.E. Early Life Exposure to Maternal Insulin Resistance Has Persistent Effects on Hepatic NAFLD in Juvenile Nonhuman Primates. Diabetes 2014, 63, 2702–2713. [Google Scholar] [CrossRef]
- Thompson, M.D.; Hinrichs, H.; Faerber, A.; Tarr, P.I.; Davidson, N.O. Maternal obesogenic diet enhances cholestatic liver disease in offspring. J. Lipid Res. 2022, 63, 100205. [Google Scholar] [CrossRef]
- Tajaddini, A.; Kendig, M.D.; Prates, K.V.; Westbrook, R.F.; Morris, M.J. Male Rat Offspring Are More Impacted by Maternal Obesity Induced by Cafeteria Diet than Females-Additive Effect of Postweaning Diet. Int. J. Mol. Sci. 2022, 23, 1442. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.L.; Johnson, J.; Vemulapalli, T.H.; Crisler, J.R.; Shepherd, R. Commonly Used Animal Models. Princ. Anim. Res. Grad. Undergrad. Stud. 2017, 117–175. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.D. The blessings and curses of C57BL/6 substrains in mouse genetic studies. Ann. N. Y. Acad. Sci. 2011, 1245, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.C.; Harley, I.T.W.; Pfluger, P.T.; Trompette, A.; Stankiewicz, T.E.; Allen, J.L.; Moreno-Fernandez, M.E.; Damen, M.S.M.A.; Oates, J.R.; Alarcon, P.C.; et al. A BAFF/APRIL axis regulates obesogenic diet-driven weight gain. Nat. Commun. 2021, 12, 2911. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Sharma, V.; Stankiewicz, T.E.; Oates, J.R.; Doll, J.R.; Damen, M.S.M.A.; Almanan, M.A.T.A.; Chougnet, C.A.; Hildeman, D.A.; Divanovic, S. Aging mitigates the severity of obesity-associated metabolic sequelae in a gender independent manner. Nutr. Diabetes 2021, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Giles, D.A.; Oates, J.R.; Chan, C.C.; Damen, M.S.M.A.; Doll, J.R.; Stankiewicz, T.E.; Chen, X.; Chetal, K.; Karns, R.; et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. 2021, 33, 1187–1204.e1189. [Google Scholar] [CrossRef]
- Singer, K.; Maley, N.; Mergian, T.; DelProposto, J.; Cho, K.W.; Zamarron, B.F.; Martinez-Santibanez, G.; Geletka, L.; Muir, L.; Wachowiak, P.; et al. Differences in Hematopoietic Stem Cells Contribute to Sexually Dimorphic Inflammatory Responses to High Fat Diet-induced Obesity. J. Biol. Chem. 2015, 290, 13250–13262. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-fernandez, M.E.; Stankiewicz, T.E.; Graspeuntner, S.; Cappelletti, M.; Wu, D.; Mukherjee, R.; Chan, C.C.; Lawson, M.J.; Klarquist, J.; et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat. Med. 2017, 23, 829–838. [Google Scholar] [CrossRef]
- Giles, D.A.; Ramkhelawon, B.; Donelan, E.M.; Stankiewicz, T.E.; Hutchison, S.B.; Mukherjee, R.; Cappelletti, M.; Karns, R.; Karp, C.L.; Moore, K.J.; et al. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol. Metab. 2016, 5, 1121–1130. [Google Scholar] [CrossRef]
- Pettersson, U.S.; Waldén, T.B.; Carlsson, P.-O.; Jansson, L.; Phillipson, M. Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue. PLoS ONE 2012, 7, e46057. [Google Scholar] [CrossRef]
- Yang, Y.; Smith, D.L., Jr.; Keating, K.D.; Allison, D.B.; Nagy, T.R. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity 2014, 22, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Omar, B.; Pacini, G.; Ahrén, B. Differential development of glucose intolerance and pancreatic islet adaptation in multiple diet induced obesity models. Nutrients 2012, 4, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Hwang, L.-L.; Wang, C.-H.; Li, T.-L.; Chang, S.-D.; Lin, L.-C.; Chen, C.-P.; Chen, C.-T.; Liang, K.-C.; Ho, I.-K.; Yang, W.-S.; et al. Sex Differences in High-fat Diet-induced Obesity, Metabolic Alterations and Learning, and Synaptic Plasticity Deficits in Mice. Obesity 2010, 18, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wang, C.; Jin, Y.; Meng, Q.; Wu, J.; Sun, H. Kaempferol-induced GPER upregulation attenuates atherosclerosis via the PI3K/AKT/Nrf2 pathway. Pharm. Biol. 2021, 59, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Maric, I.; Krieger, J.P.; van der Velden, P.; Börchers, S.; Asker, M.; Vujicic, M.; Wernstedt Asterholm, I.; Skibicka, K.P. Sex and Species Differences in the Development of Diet-Induced Obesity and Metabolic Disturbances in Rodents. Front. Nutr. 2022, 9, 828522. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Gascoin, G.; Musial, B.; Carr, S.; Duque-Guimaraes, D.; Blackmore, H.L.; Alfaradhi, M.Z.; Loche, E.; Sferruzzi-Perri, A.N.; Fowden, A.L.; et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci. Rep. 2017, 7, 44650. [Google Scholar] [CrossRef] [PubMed]
- Blackler, G.; Akingbasote, J.; Cairns, E.; Howlett, C.; Kiser, P.; Barra, L. The effect of HLA-DRB1*04:01 on a mouse model of atherosclerosis. J. Transl. Autoimmun. 2023, 7, 100203. [Google Scholar] [CrossRef] [PubMed]
- De Souza, F.B.; Novaes, R.D.; Santos, C.F.F.; de Deus, F.A.; Santos, F.C.; Ladeira, L.C.M.; Gonçalves, R.V.; Bastos, D.S.S.; Souza, A.C.F.; Machado-Neves, M.; et al. High-fat diet and caffeine interact to modulate bone microstructure and biomechanics in mice. Life Sci. 2021, 276, 119450. [Google Scholar] [CrossRef]
- Li, H.; Lelliott, C.; Håkansson, P.; Ploj, K.; Tuneld, A.; Verolin-Johansson, M.; Benthem, L.; Carlsson, B.; Storlien, L.; Michaëlsson, E. Intestinal, adipose, and liver inflammation in diet-induced obese mice. Metabolism 2008, 57, 1704–1710. [Google Scholar] [CrossRef]
- King, V.; Dakin, R.S.; Liu, L.; Hadoke, P.W.; Walker, B.R.; Seckl, J.R.; Norman, J.E.; Drake, A.J. Maternal obesity has little effect on the immediate offspring but impacts on the next generation. Endocrinology 2013, 154, 2514–2524. [Google Scholar] [CrossRef]
- Marin, V.; Rosso, N.; Dal Ben, M.; Raseni, A.; Boschelle, M.; Degrassi, C.; Nemeckova, I.; Nachtigal, P.; Avellini, C.; Tiribelli, C.; et al. An Animal Model for the Juvenile Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. PLoS ONE 2016, 11, e0158817. [Google Scholar] [CrossRef] [PubMed]
- Saben, J.L.; Boudoures, A.L.; Asghar, Z.; Thompson, A.; Drury, A.; Zhang, W.; Chi, M.; Cusumano, A.; Scheaffer, S.; Moley, K. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Griffin, C.; Abrishami, S.; Eter, L.; Lanzetta, N.; Hak, L.; Clemente, J.; Agarwal, D.; Lerner, A.; Westerhoff, M.; et al. Sex hormones regulate metainflammation in diet-induced obesity in mice. J. Biol. Chem. 2021, 297, 101229. [Google Scholar] [CrossRef]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Matthaei, S. Regulation of energy balance by leptin. Exp. Clin. Endocrinol. Diabetes 1996, 104, 293–300. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Batt, R.A.L.; Harrison, G.A. The reproductive system of the adipose mouse. J. Hered. 1963, 54, 135–138. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; De Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immunol. 2021, 11, 622468. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.R.; Jenkins, T.G.; Carrell, D.T.; Hotaling, J.M. Obesity, male infertility, and the sperm epigenome. Fertil. Steril. 2017, 107, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Sass, S.; Wu, M.; Dyckhoff, D.; Tschöp, M.; Theis, F.; Marschall, S.; de Angelis, M.H.; Beckers, J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 2016, 48, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 2014, 35, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.H.; Smith, J.; McLea, S.A.; Heizer, A.B.; Richardson, J.L.; Myatt, L. Effect of increasing maternal body mass index on oxidative and nitrative stress in the human placenta. Placenta 2009, 30, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Couch, S.C.; Glanz, K.; Zhou, C.; Sallis, J.F.; Saelens, B.E. Home Food Environment in Relation to Children’s Diet Quality and Weight Status. J. Acad. Nutr. Diet. 2014, 114, 1569–1579.e1561. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Sakwinska, O.; Soh, S.E.; Ngom-Bru, C.; Brück, W.M.; Berger, B.; Brüssow, H.; Lee, Y.S.; Yap, F.; Chong, Y.S.; et al. Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. mBio 2015, 6, e02419-14. [Google Scholar] [CrossRef]
- Xie, R.; Sun, Y.; Wu, J.; Huang, S.; Jin, G.; Guo, Z.; Zhang, Y.; Liu, T.; Liu, X.; Cao, X.; et al. Maternal High Fat Diet Alters Gut Microbiota of Offspring and Exacerbates DSS-Induced Colitis in Adulthood. Front. Immunol. 2018, 9, 2608. [Google Scholar] [CrossRef]
- Ma, J.; Prince, A.L.; Bader, D.; Hu, M.; Ganu, R.; Baquero, K.; Blundell, P.; Alan Harris, R.; Frias, A.E.; Grove, K.L.; et al. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat. Commun. 2014, 5, 3889. [Google Scholar] [CrossRef]
- Myles, I.A.; Fontecilla, N.M.; Janelsins, B.M.; Vithayathil, P.J.; Segre, J.A.; Datta, S.K. Parental dietary fat intake alters offspring microbiome and immunity. J. Immunol. 2013, 191, 3200–3209. [Google Scholar] [CrossRef]
- Paul, H.A.; Bomhof, M.R.; Vogel, H.J.; Reimer, R.A. Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci. Rep. 2016, 6, 20683. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.N.; Dunn-Meynell, A.A.; Hartman, T.G.; Levin, B.E. Postnatal environment overrides genetic and prenatal factors influencing offspring obesity and insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R768–R778. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Messaoudi, I. The impact of maternal obesity during pregnancy on offspring immunity. Mol. Cell. Endocrinol. 2015, 418, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Haghiac, M.; Surace, P.; Challier, J.C.; Guerre-Millo, M.; Singh, K.; Waters, T.; Minium, J.; Presley, L.; Catalano, P.M.; et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity 2011, 19, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Maguire, R.L.; House, J.S.; Lloyd, D.T.; Skinner, H.G.; Allen, T.K.; Raffi, A.M.; Skaar, D.A.; Park, S.S.; McCullough, L.E.; Kollins, S.H.; et al. Associations between maternal obesity, gestational cytokine levels and child obesity in the NEST cohort. Pediatr. Obes. 2021, 16, e12763. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.M.; Dunkel Schetter, C.; Carroll, J.E.; Mancuso, R.A.; Breen, E.C.; Okun, M.L.; Hobel, C.; Coussons-Read, M. Inflammatory and immune marker trajectories from pregnancy to one-year post-birth. Cytokine 2022, 149, 155758. [Google Scholar] [CrossRef]
- Leddy, M.A.; Power, M.L.; Schulkin, J. The impact of maternal obesity on maternal and fetal health. Rev. Obstet. Gynecol. 2008, 1, 170–178. [Google Scholar]
- Sureshchandra, S.; Marshall, N.E.; Mendoza, N.; Jankeel, A.; Zulu, M.Z.; Messaoudi, I. Functional and genomic adaptations of blood monocytes to pregravid obesity during pregnancy. iScience 2021, 24, 102690. [Google Scholar] [CrossRef]
- Sureshchandra, S.; Wilson, R.M.; Rais, M.; Marshall, N.E.; Purnell, J.Q.; Thornburg, K.L.; Messaoudi, I. Maternal Pregravid Obesity Remodels the DNA Methylation Landscape of Cord Blood Monocytes Disrupting Their Inflammatory Program. J. Immunol. 2017, 199, 2729–2744. [Google Scholar] [CrossRef]
- Castellana, B.; Perdu, S.; Kim, Y.; Chan, K.; Atif, J.; Marziali, M.; Beristain, A.G. Maternal obesity alters uterine NK activity through a functional KIR2DL1/S1 imbalance. Immunol. Cell Biol. 2018, 96, 805–819. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Karp, C.L. Unstressing intemperate models: How cold stress undermines mouse modeling. J. Exp. Med. 2012, 209, 1069–1074. [Google Scholar] [CrossRef]
- James, C.M.; Olejniczak, S.H.; Repasky, E.A. How murine models of human disease and immunity are influenced by housing temperature and mild thermal stress. Temperature 2023, 10, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Swoap, S.J.; Overton, J.M.; Garber, G. Effect of ambient temperature on cardiovascular parameters in rats and mice: A comparative approach. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R391–R396. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.D.; Chambers, J.B.; Henderson, R.P.; Rashotte, M.E.; Overton, J.M. Cardiovascular responses to caloric restriction and thermoneutrality in C57BL/6J mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R1459–R1467. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, K.; Chawla, A. Warming the mouse to model human diseases. Nat. Rev. Endocrinol. 2017, 13, 458–465. [Google Scholar] [CrossRef]
- Ginting, R.P.; Lee, J.M.; Lee, M.W. The Influence of Ambient Temperature on Adipose Tissue Homeostasis, Metabolic Diseases and Cancers. Cells 2023, 12, 881. [Google Scholar] [CrossRef]
- De Jong, J.M.A.; Sun, W.; Pires, N.D.; Frontini, A.; Balaz, M.; Jespersen, N.Z.; Feizi, A.; Petrovic, K.; Fischer, A.W.; Bokhari, M.H.; et al. Human brown adipose tissue is phenocopied by classical brown adipose tissue in physiologically humanized mice. Nat. Metab. 2019, 1, 830–843. [Google Scholar] [CrossRef]
- Eng, J.W.; Reed, C.B.; Kokolus, K.M.; Pitoniak, R.; Utley, A.; Bucsek, M.J.; Ma, W.W.; Repasky, E.A.; Hylander, B.L. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat. Commun. 2015, 6, 6426. [Google Scholar] [CrossRef]
- Leigh, N.D.; Kokolus, K.M.; O’Neill, R.E.; Du, W.; Eng, J.W.L.; Qiu, J.; Chen, G.L.; McCarthy, P.L.; Farrar, J.D.; Cao, X.; et al. Housing Temperature–Induced Stress Is Suppressing Murine Graft-versus-Host Disease through β2-Adrenergic Receptor Signaling. J. Immunol. 2015, 195, 5045–5054. [Google Scholar] [CrossRef]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylander, B.L. Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front. Immunol. 2018, 9, 164. [Google Scholar] [CrossRef]
- Povinelli, B.J.; Kokolus, K.M.; Eng, J.W.; Dougher, C.W.; Curtin, L.; Capitano, M.L.; Sailsbury-Ruf, C.T.; Repasky, E.A.; Nemeth, M.J. Standard sub-thermoneutral caging temperature influences radiosensitivity of hematopoietic stem and progenitor cells. PLoS ONE 2015, 10, e0120078. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ganeshan, K.; Hong, C.; Nguyen, K.D.; Qiu, Y.; Kim, J.; Tangirala, R.K.; Tontonoz, P.; Chawla, A. Thermoneutral Housing Accelerates Metabolic Inflammation to Potentiate Atherosclerosis but Not Insulin Resistance. Cell Metab. 2016, 23, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Zhou, L.; Zhao, X.; Song, L.; Lu, Y.; Zhong, N.; Yang, P.; Sun, B.; Zhang, X. Thermoneutral housing temperature regulates T-regulatory cell function and inhibits ovabumin-induced asthma development in mice. Sci. Rep. 2017, 7, 7123. [Google Scholar] [CrossRef] [PubMed]
- Noah, T.K.; Lee, J.B.; Brown, C.A.; Yamani, A.; Tomar, S.; Ganesan, V.; Newberry, R.D.; Huffnagle, G.B.; Divanovic, S.; Hogan, S.P. Thermoneutrality Alters Gastrointestinal Antigen Passage Patterning and Predisposes to Oral Antigen Sensitization in Mice. Front. Immunol. 2021, 12, 636198. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.R.; Sawada, K.; Giles, D.A.; Alarcon, P.C.; Damen, M.; Szabo, S.; Stankiewicz, T.E.; Moreno-Fernandez, M.E.; Divanovic, S. Thermoneutral housing shapes hepatic inflammation and damage in mouse models of non-alcoholic fatty liver disease. Front. Immunol. 2023, 14, 1095132. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, P.C.; Damen, M.; Ulanowicz, C.J.; Sawada, K.; Oates, J.R.; Toth, A.; Wayland, J.L.; Chung, H.; Stankiewicz, T.E.; Moreno-Fernandez, M.E.; et al. Obesity amplifies influenza virus-driven disease severity in male and female mice. Mucosal Immunol. 2023; in press. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, F.; Morris, S.; Orekhova, T.; Sehy, D. The in vivo cytokine capture assay for measurement of cytokine production in the mouse. Curr. Protoc. Immunol. 2003, 54, 6–28. [Google Scholar] [CrossRef]
- Stemmer, K.; Kotzbeck, P.; Zani, F.; Bauer, M.; Neff, C.; Müller, T.D.; Pfluger, P.T.; Seeley, R.J.; Divanovic, S. Thermoneutral housing is a critical factor for immune function and diet-induced obesity in C57BL/6 nude mice. Int. J. Obes. 2015, 39, 791–797. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, e0149783. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ files (Version 1.33). 2011. Available online: https://github.com/najoshi/sickle (accessed on 7 April 2023).
- Wood, D.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, G.F.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.M.; Stevens, H.H.; Wagner, H. Vegan: Community Ecology Package. 2015. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 7 April 2023).
- Xia, Y.; Sun, J. Hypothesis testing and statistical analysis of microbiome. Genes Dis. 2017, 4, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Marshall, N.E.; Messaoudi, I. Impact of pregravid obesity on maternal and fetal immunity: Fertile grounds for reprogramming. J. Leukoc. Biol. 2019, 106, 1035–1050. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- WHO Multicentre Growth Reference Study Group; de Onis, M. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr. Suppl. 2006, 450, 76–85. [Google Scholar] [CrossRef]
- Hylander, B.L.; Gordon, C.J.; Repasky, E.A. Manipulation of Ambient Housing Temperature To Study the Impact of Chronic Stress on Immunity and Cancer in Mice. J. Immunol. 2019, 202, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Gilley, S.P.; Ruebel, M.L.; Sims, C.; Zhong, Y.; Turner, D.; Lan, R.S.; Pack, L.M.; Piccolo, B.D.; Chintapalli, S.V.; Abraham, A.; et al. Associations between maternal obesity and offspring gut microbiome in the first year of life. Pediatr. Obes. 2022, 17, e12921. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Z.; Chen, L.; Tang, L.; Wen, S.; Liu, Y.; Yuan, J. Diet induced maternal obesity affects offspring gut microbiota and persists into young adulthood. Food Funct. 2018, 9, 4317–4327. [Google Scholar] [CrossRef]
- Goldstein, J.A.; Gallagher, K.; Beck, C.; Kumar, R.; Gernand, A.D. Maternal-Fetal Inflammation in the Placenta and the Developmental Origins of Health and Disease. Front. Immunol. 2020, 11, 531543. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.S.; Barlow, S.E.; Rao, G.; Inge, T.H.; Hayman, L.L.; Steinberger, J.; Urbina, E.M.; Ewing, L.J.; Daniels, S.R. Severe obesity in children and adolescents: Identification, associated health risks, and treatment approaches: A scientific statement from the American Heart Association. Circulation 2013, 128, 1689–1712. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Mehendale, A.M. Increased Incidence of Obesity in Children and Adolescents Post-COVID-19 Pandemic: A Review Article. Cureus 2022, 14, e29348. [Google Scholar] [CrossRef] [PubMed]
- Ayonrinde, O.T.; Oddy, W.H.; Adams, L.A.; Mori, T.A.; Beilin, L.J.; de Klerk, N.; Olynyk, J.K. Infant nutrition and maternal obesity influence the risk of non-alcoholic fatty liver disease in adolescents. J. Hepatol. 2017, 67, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Denizli, M.; Capitano, M.L.; Kua, K.L. Maternal obesity and the impact of associated early-life inflammation on long-term health of offspring. Front. Cell Infect. Microbiol. 2022, 12, 940937. [Google Scholar] [CrossRef] [PubMed]
- Poston, L. Maternal obesity, gestational weight gain and diet as determinants of offspring long term health. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, A.; Sferruzzi-Perri, A.N. Developmental programming of offspring adipose tissue biology and obesity risk. Int. J. Obesity 2021, 45, 1170–1192. [Google Scholar] [CrossRef]
- Wei, W.; Zhang, X.; Zhou, B.; Ge, B.; Tian, J.; Chen, J. Effects of female obesity on conception, pregnancy and the health of offspring. Front. Endocrinol. 2022, 13, 949228. [Google Scholar] [CrossRef]
- Schoonejans, J.M.; Ozanne, S.E. Developmental programming by maternal obesity: Lessons from animal models. Diabet. Med. 2021, 38, e14694. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef]
- Pettitt, D.J.; Jovanovic, L. Birth weight as a predictor of type 2 diabetes mellitus: The U-shaped curve. Curr. Diabetes Rep. 2001, 1, 78–81. [Google Scholar] [CrossRef]
- Wang, J.J.; Yu, X.W.; Wu, R.Y.; Sun, X.F.; Cheng, S.F.; Ge, W.; Liu, J.C.; Li, Y.P.; Liu, J.; Zou, S.H.; et al. Starvation during pregnancy impairs fetal oogenesis and folliculogenesis in offspring in the mouse. Cell Death Dis. 2018, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Sutton, G.M.; Centanni, A.V.; Butler, A.A. Protein malnutrition during pregnancy in C57BL/6J mice results in offspring with altered circadian physiology before obesity. Endocrinology 2010, 151, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S.; et al. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014, 345, 1255903. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Derse, A.; Ferey, J.L.; Reid, M.; Xie, Y.; Christ, M.; Chatterjee, D.; Nguyen, C.; Harasymowicz, N.; Guilak, F.; et al. Transgenerational impact of maternal obesogenic diet on offspring bile acid homeostasis and nonalcoholic fatty liver disease. Am. J. Physiol.-Endocrinol. Metab. 2019, 316, E674–E686. [Google Scholar] [CrossRef]
- Bibi, S.; Kang, Y.; Du, M.; Zhu, M.J. Maternal high-fat diet consumption enhances offspring susceptibility to DSS-induced colitis in mice. Obesity 2017, 25, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.-M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.J.M.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Pedroni, S.M.A.; Turban, S.; Kipari, T.; Dunbar, D.R.; McInnes, K.; Saunders, P.T.K.; Morton, N.M.; Norman, J.E. Pregnancy in obese mice protects selectively against visceral adiposity and is associated with increased adipocyte estrogen signalling. PLoS ONE 2014, 9, e94680. [Google Scholar] [CrossRef]
- Perez, P.A.; DiPatrizio, N.V. Impact of maternal western diet-induced obesity on offspring mortality and peripheral endocannabinoid system in mice. PLoS ONE 2018, 13, e0205021. [Google Scholar] [CrossRef]
- Monaco-Brown, M.; Lawrence, D.A. Obesity and Maternal-Placental-Fetal Immunology and Health. Front. Pediatr. 2022, 10, 859885. [Google Scholar] [CrossRef]
- Lean, S.C.; Candia, A.A.; Gulacsi, E.; Lee, G.C.L.; Sferruzzi-Perri, A.N. Obesogenic diet in mice compromises maternal metabolic physiology and lactation ability leading to reductions in neonatal viability. Acta Physiol. 2022, 236, e13861. [Google Scholar] [CrossRef]
- John, R.M. Prenatal Adversity Modulates the Quality of Maternal Care Via the Exposed Offspring. BioEssays News Rev. Mol. Cell. Dev. Biol. 2019, 41, e1900025. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-P.; Chen, Y.-H.; Gao, L.; Wang, P.; Wang, X.-L.; Luo, B.; Li, J.; Xu, D.-X. Differential effects of high-fat diets before pregnancy and/or during pregnancy on fetal growth development. Life Sci. 2018, 212, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Sasson, I.E.; Vitins, A.P.; Mainigi, M.A.; Moley, K.H.; Simmons, R.A. Pre-gestational vs gestational exposure to maternal obesity differentially programs the offspring in mice. Diabetologia 2015, 58, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.; Ahmed, M.L.; Emmett, P.M.; Preece, M.A.; Dunger, D.B. Association between postnatal catch-up growth and obesity in childhood: Prospective cohort study. BMJ 2000, 320, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Elahi, M.M.; Cagampang, F.R.; Mukhtar, D.; Anthony, F.W.; Ohri, S.K.; Hanson, M.A. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br. J. Nutr. 2009, 102, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Nivoit, P.; Morens, C.; Van Assche, F.A.; Jansen, E.; Poston, L.; Remacle, C.; Reusens, B. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009, 52, 1133–1142. [Google Scholar] [CrossRef]
- Odaka, Y.; Nakano, M.; Tanaka, T.; Kaburagi, T.; Yoshino, H.; Sato-Mito, N.; Sato, K. The Influence of a High-Fat Dietary Environment in the Fetal Period on Postnatal Metabolic and Immune Function. Obesity 2010, 18, 1688–1694. [Google Scholar] [CrossRef]
- Kumar, S.; Raj, V.S.; Ahmad, A.; Saini, V. Amoxicillin modulates gut microbiota to improve short-term high-fat diet induced pathophysiology in mice. Gut Pathog. 2022, 14, 40. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y. Additive effects of maternal high fat diet during lactation on mouse offspring. PLoS ONE 2014, 9, e92805. [Google Scholar] [CrossRef]
- Snowden, J.M.; Mission, J.F.; Marshall, N.E.; Quigley, B.; Main, E.; Gilbert, W.M.; Chung, J.H.; Caughey, A.B. The Impact of maternal obesity and race/ethnicity on perinatal outcomes: Independent and joint effects. Obesity 2016, 24, 1590–1598. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HFD (Ts) | HF-HC/HF-HS | Cafeteria | HFD or WD (Tn) |
|---|---|---|---|---|
| Weight gain | 30–50% increase [20,22,23] | 50% increase [27,28,29] | 30–60 + % increase [30,31,32] | 100 + % increase [20] |
| GTT | ~50% increase [22,23] | 50–100% increase [27,28] | Studies needed, glucose high [31] | 100% increase [20] |
| Liver weight | No change [20] | Increased [33] | Increased [31] | Increased [20] |
| Liver triglyceride | No change [20] | Studies needed | Increased [31] | Increased [20] |
| ALT | No change [20] | Increased [33] | Increased [31] | Increased [20] |
| Serum triglyceride | Increased [22]/ not increased [25] | Increased or decreased [33,34] | Increased [30,32] | Studies needed * |
| Serum LDL | Mildly increased [26] | No change or increased [29,33] | No change [23,25] | Studies needed * |
| Atherosclerotic lesions | None [26] | None [29] | Studies needed | Studies needed * |
| Parameter | Lean (n = 11) | Obese (n = 30) |
|---|---|---|
| Age (years) | 30 ± 4 | 30 ± 6 |
| White race | 11 (100%) | 22 (73%) |
| Family history of NAFLD | 1 (9%) | 3 (10%) |
| Pre-pregnancy NAFLD | 0 (0%) | 5 (17%) |
| Pre-pregnancy T2D | 0 (0%) | 5 (17%) |
| Gestational diabetes | 2 (18%) | 9 (30%) |
| Bariatric surgery history | 0 (0%) | 2 (7%) |
| Insulin use in pregnancy | 0 (0%) | 7 (23%) |
| Multivitamin use in pregnancy | 8 (73%) | 7 (23%) * |
| Pre-pregnancy BMI (kg/m2) | 22.2 ± 2.9 | 39.2 ± 8.8 * |
| Pregnancy BMI (kg/m2) | 25.2 ± 2.7 | 41.2 ± 7.9 * |
| Pregnancy number | 3 ± 2 | 3 ± 3 |
| Living children | 1 ± 1 | 1 ± 1 |
| C-section delivery | 5 (45%) | 12 (40%) |
| BMI at 9 months (kg/m2) | 20.2 ± 2.4 | 38.1 ± 8.8 * |
| Parameter | Lean (n = 10) | Obese (n = 28) |
|---|---|---|
| HDL (mg/dL) | 67 ± 15 | 61 ± 12 |
| TG (mg/dL) | 199 ± 39 | 210 ± 71 |
| LDL (mg/dL) | 153 ± 46 | 122 ± 34 * |
| TC (mg/dL) | 237 ± 49 | 225 ± 44 |
| Total bilirubin (mg/dL) | 0.34 ± 0.21 | 0.32 ± 0.12 |
| Albumin (g/dL) | 2.9 ± 0.2 | 2.8 ± 0.2 |
| AST (units/L) | 21 ± 4 | 24 ± 29 |
| ALT (units/L) | 13 ± 7 | 28 ± 56 |
| ALP (units/L) | 108 ± 35 | 125 ± 36 |
| Protein (g/dL) | 6.3 ± 0.3 | 6.4 ± 0.4 |
| One-hour GCT (mg/dL) 1 | 109 ± 16 | 122 ± 26 |
| Parameter | Lean (n = 10) | Obese (n = 29) |
|---|---|---|
| White race 1 | 8 (89%) | 21 (75%) |
| Male sex | 5 (50%) | 13 (45%) |
| Gestational age at birth (weeks) | 39 ± 1 | 38 ± 1 |
| Birth weight within AGA range 2 | 9 (82%) | 25 (83%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wayland, J.L.; Doll, J.R.; Lawson, M.J.; Stankiewicz, T.E.; Oates, J.R.; Sawada, K.; Damen, M.S.M.A.; Alarcon, P.C.; Haslam, D.B.; Trout, A.T.; et al. Thermoneutral Housing Enables Studies of Vertical Transmission of Obesogenic Diet-Driven Metabolic Diseases. Nutrients 2023, 15, 4958. https://doi.org/10.3390/nu15234958
Wayland JL, Doll JR, Lawson MJ, Stankiewicz TE, Oates JR, Sawada K, Damen MSMA, Alarcon PC, Haslam DB, Trout AT, et al. Thermoneutral Housing Enables Studies of Vertical Transmission of Obesogenic Diet-Driven Metabolic Diseases. Nutrients. 2023; 15(23):4958. https://doi.org/10.3390/nu15234958
Chicago/Turabian StyleWayland, Jennifer L., Jessica R. Doll, Matthew J. Lawson, Traci E. Stankiewicz, Jarren R. Oates, Keisuke Sawada, Michelle S. M. A. Damen, Pablo C. Alarcon, David B. Haslam, Andrew T. Trout, and et al. 2023. "Thermoneutral Housing Enables Studies of Vertical Transmission of Obesogenic Diet-Driven Metabolic Diseases" Nutrients 15, no. 23: 4958. https://doi.org/10.3390/nu15234958
APA StyleWayland, J. L., Doll, J. R., Lawson, M. J., Stankiewicz, T. E., Oates, J. R., Sawada, K., Damen, M. S. M. A., Alarcon, P. C., Haslam, D. B., Trout, A. T., DeFranco, E. A., Klepper, C. M., Woo, J. G., Moreno-Fernandez, M. E., Mouzaki, M., & Divanovic, S. (2023). Thermoneutral Housing Enables Studies of Vertical Transmission of Obesogenic Diet-Driven Metabolic Diseases. Nutrients, 15(23), 4958. https://doi.org/10.3390/nu15234958