
Reducing Dietary Polyunsaturated to Saturated Fatty Acids Ratio Improves Lipid and Glucose Metabolism in Obese Zucker Rats
 , ,
, ,  ,
,  , ,
, , 

Abstract
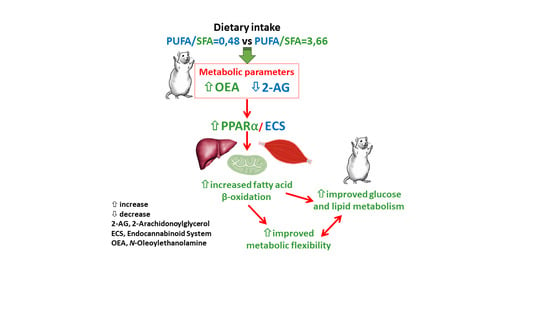
:
1. Introduction
2. Materials and Methods
2.1. Experimental Diets
2.2. Animals and Experimental Procedure
2.3. Lipid Analyses
2.3.1. Reagents
2.3.2. FA Analysis
2.3.3. N-Acylethanolamines (NAE) and Endocannabinoids (EC) Analysis
2.4. Plasma and Hepatic Parameters
2.5. Mitochondria Isolation and Function
Oxidative Stress
2.6. Statistical Analysis
3. Results
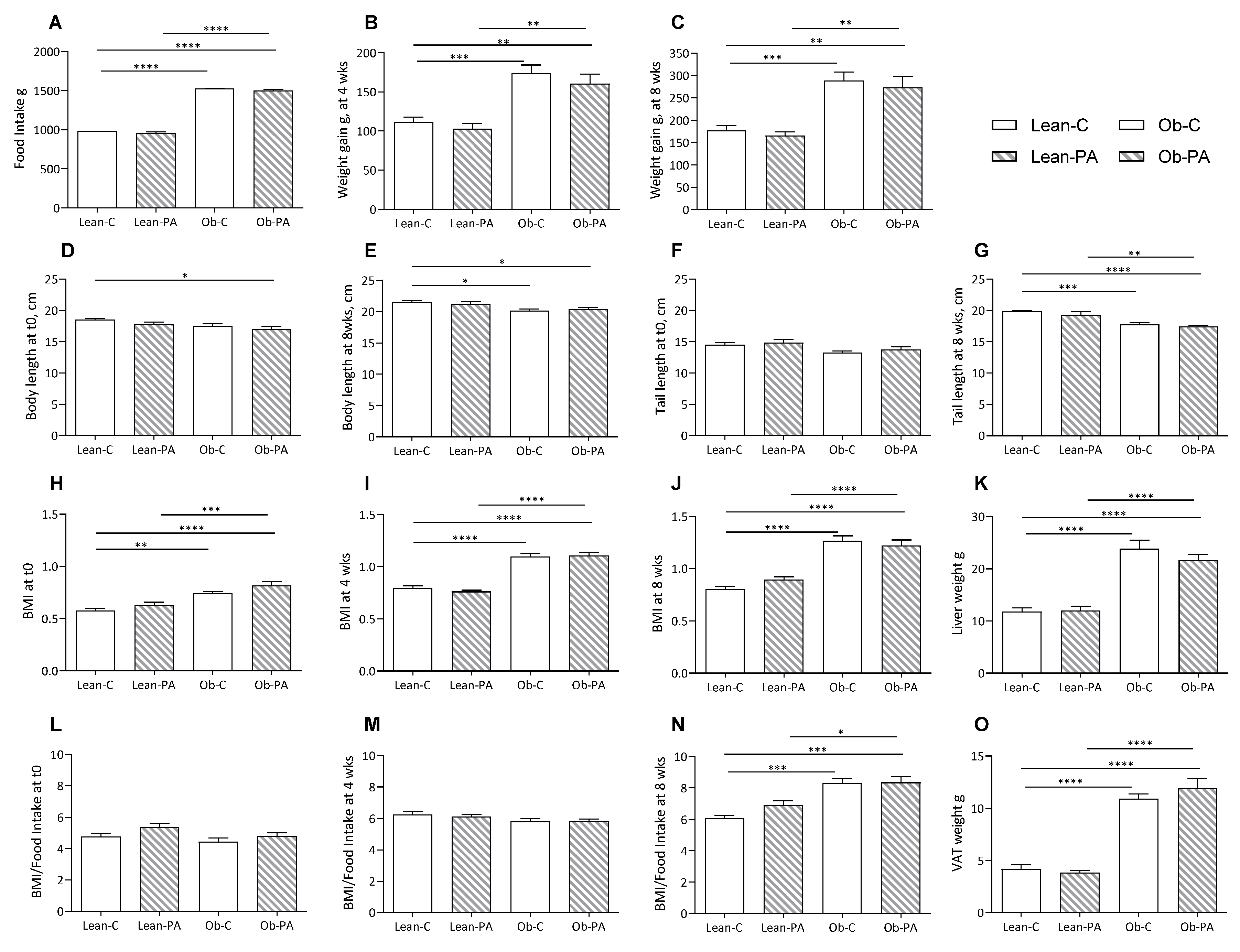
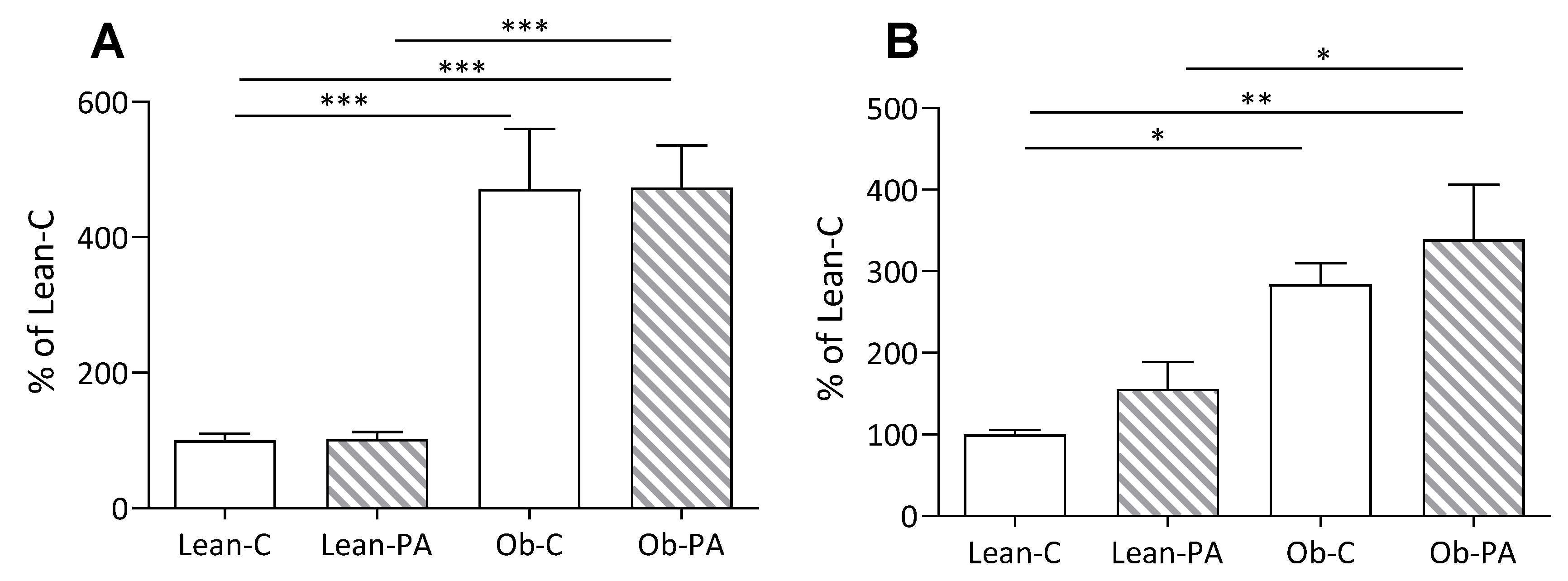
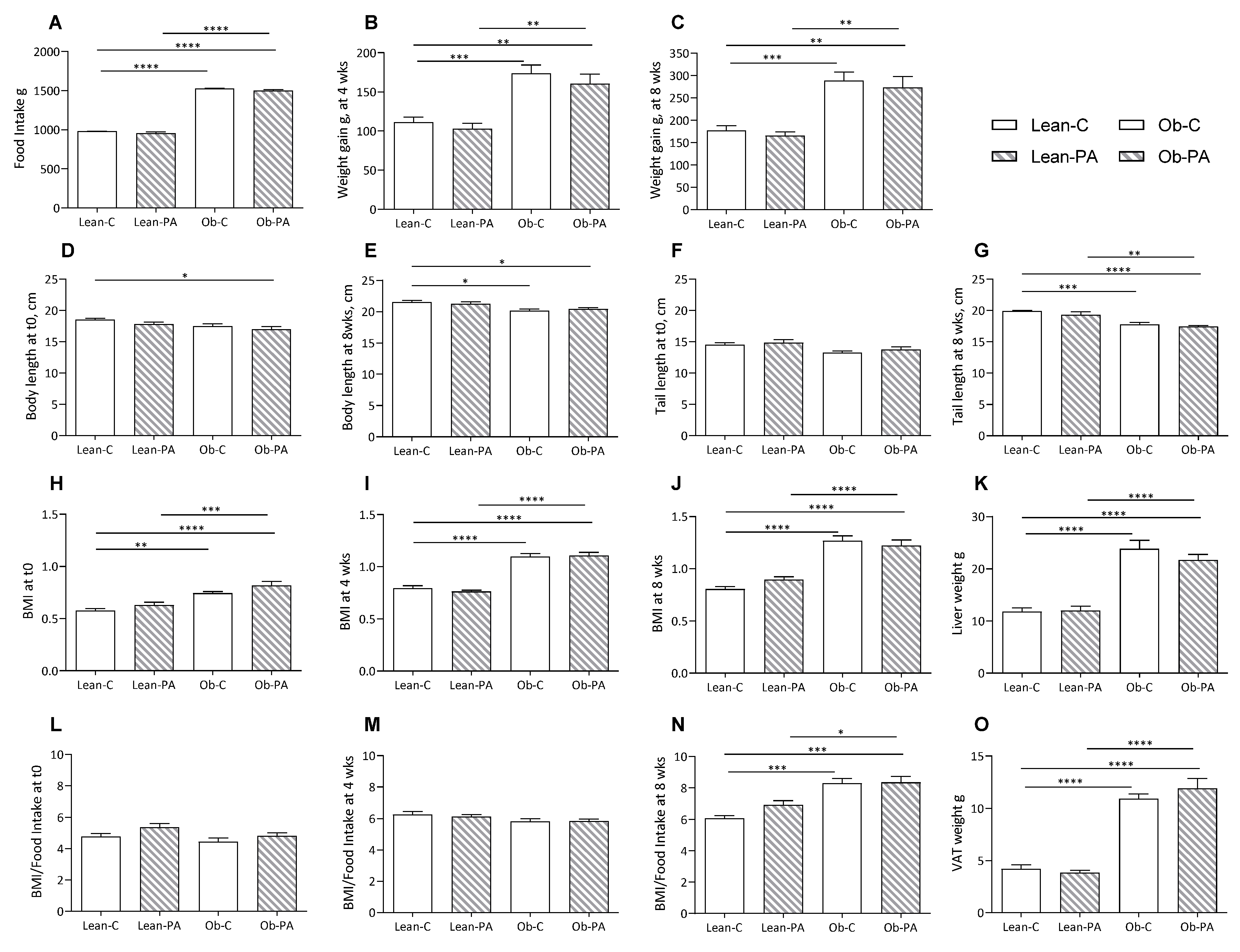
3.1. Body Composition, Food Intake, and Tissues Total Lipids
3.2. Tissue Fatty Acid Profiles
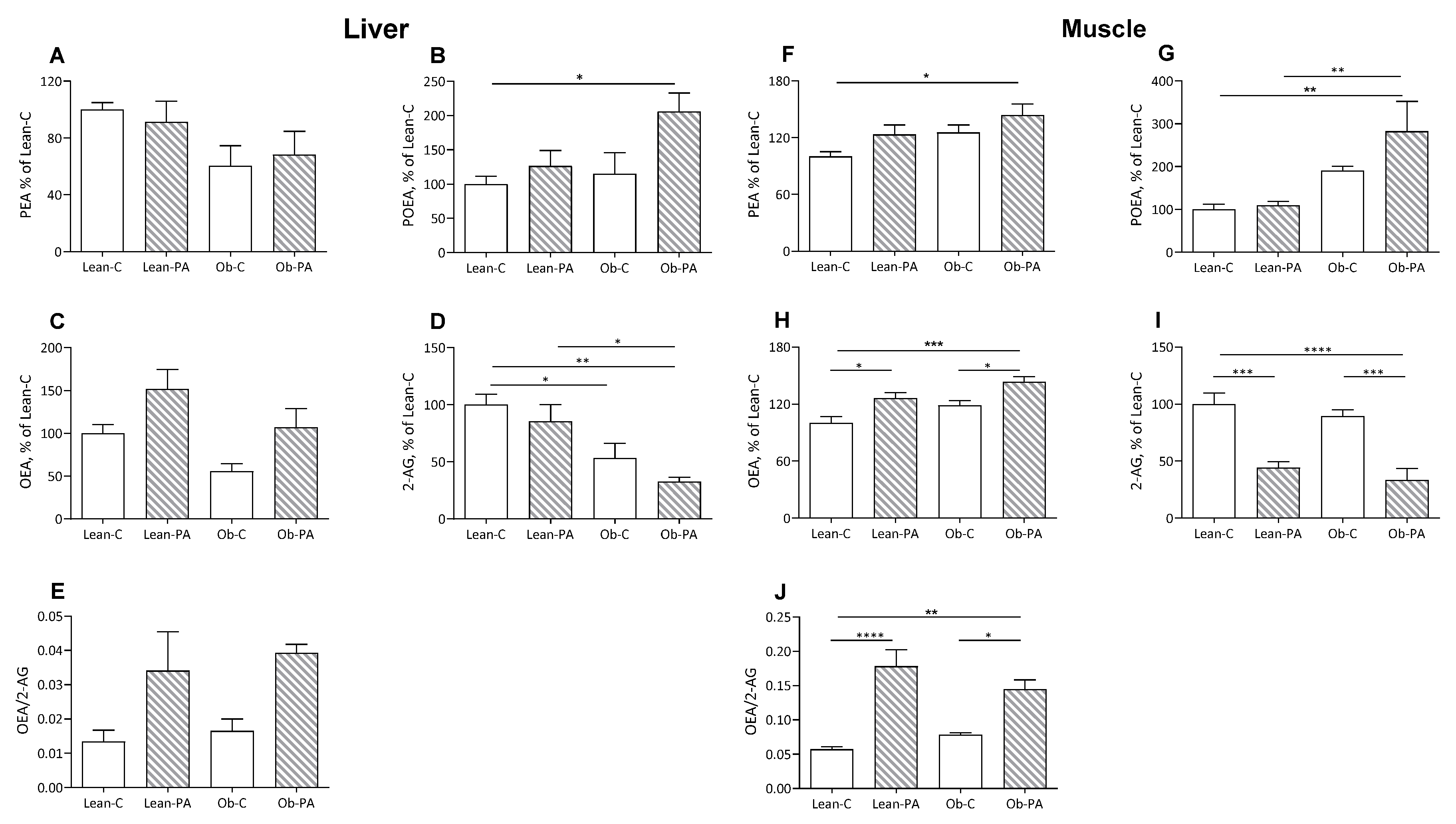
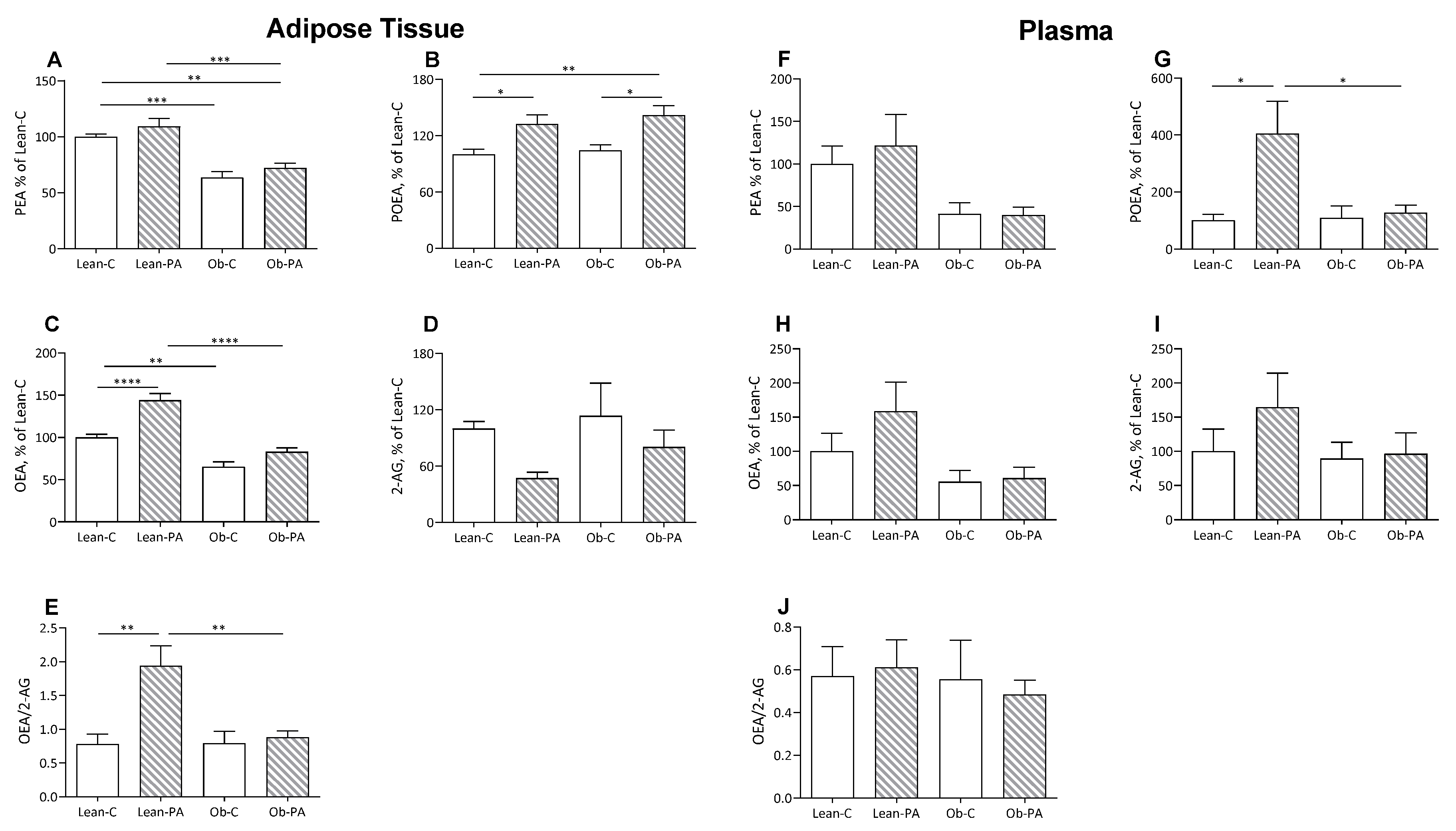
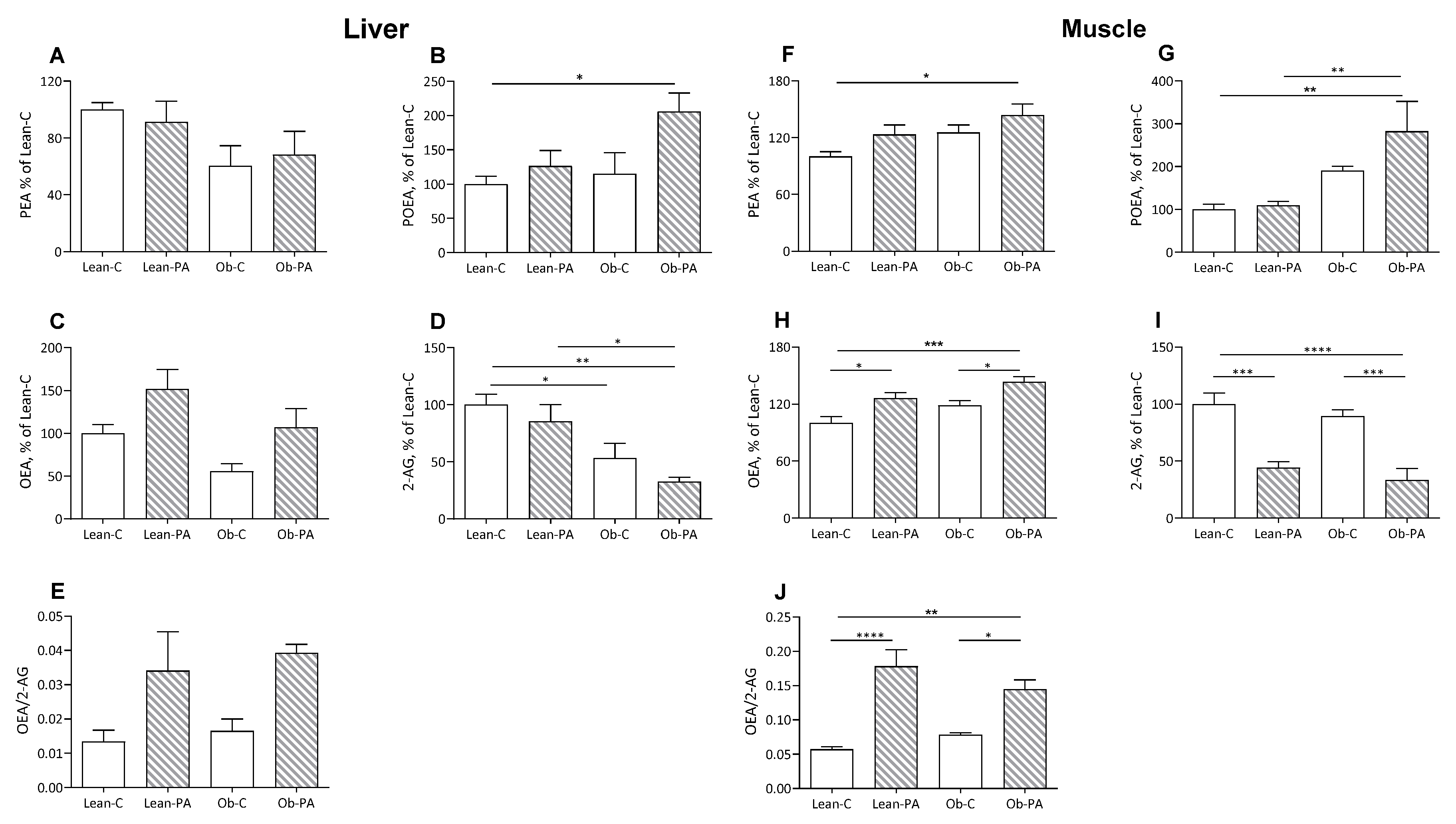
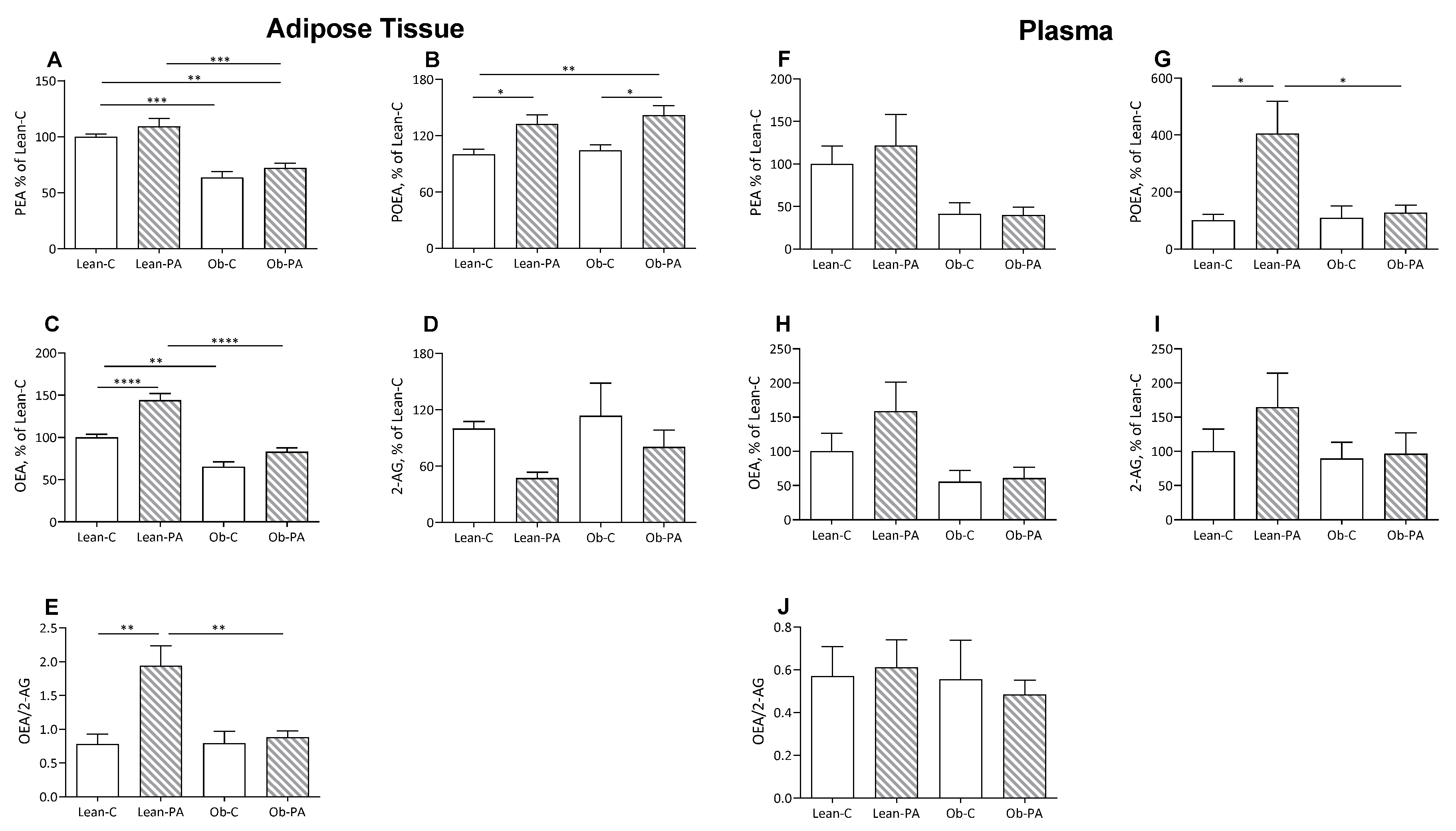
3.3. NAE and EC Tissue Profiles
3.4. FA Correlations with NAE and EC Tissue Levels
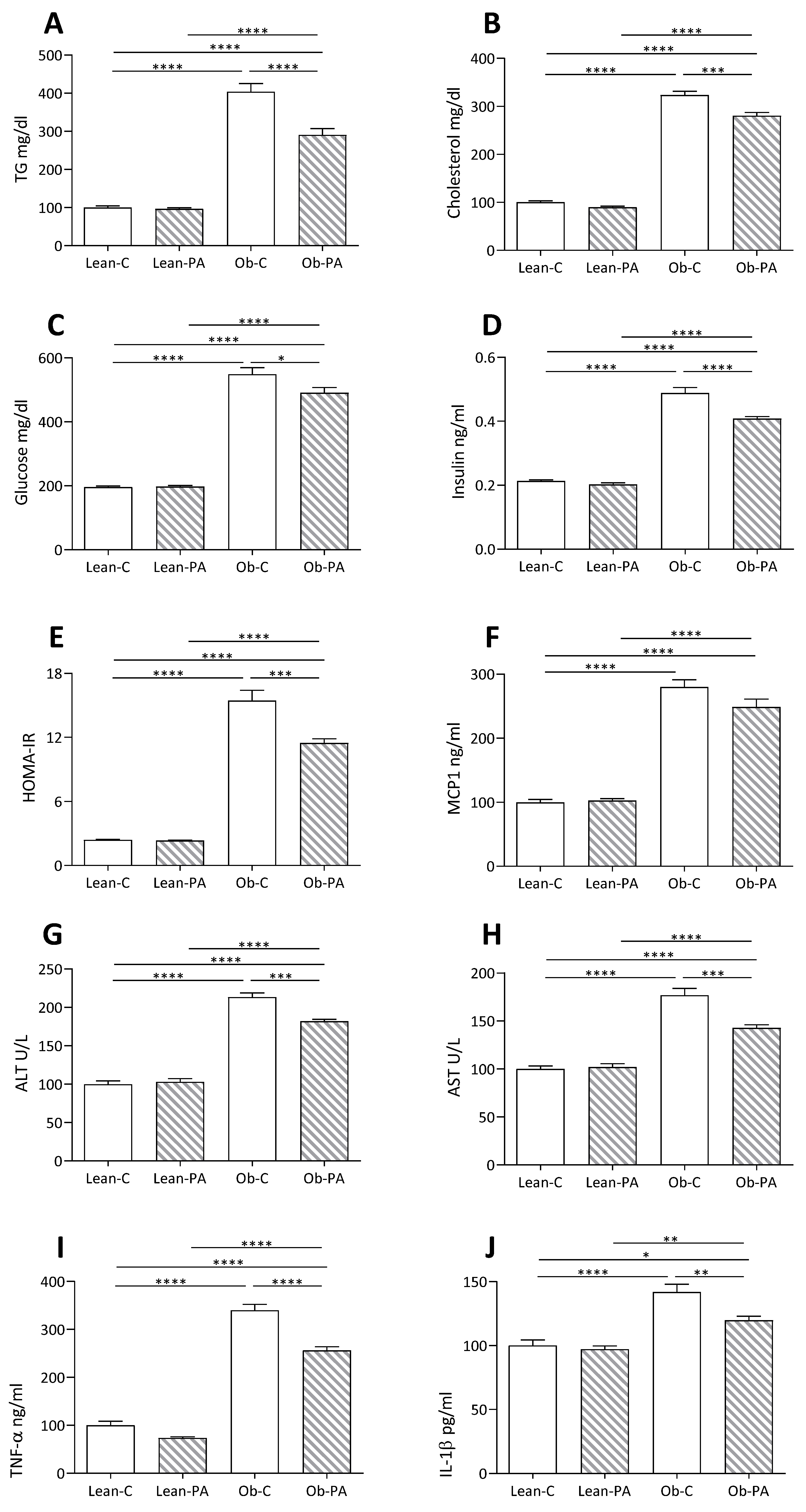
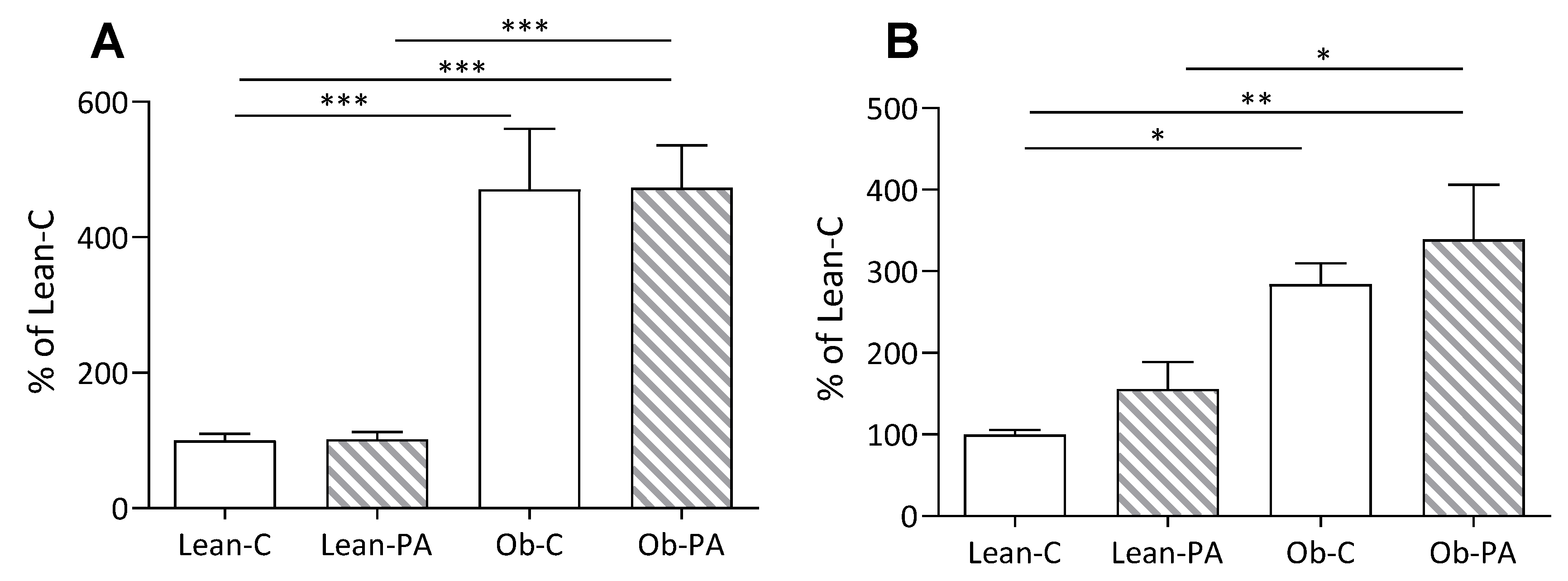
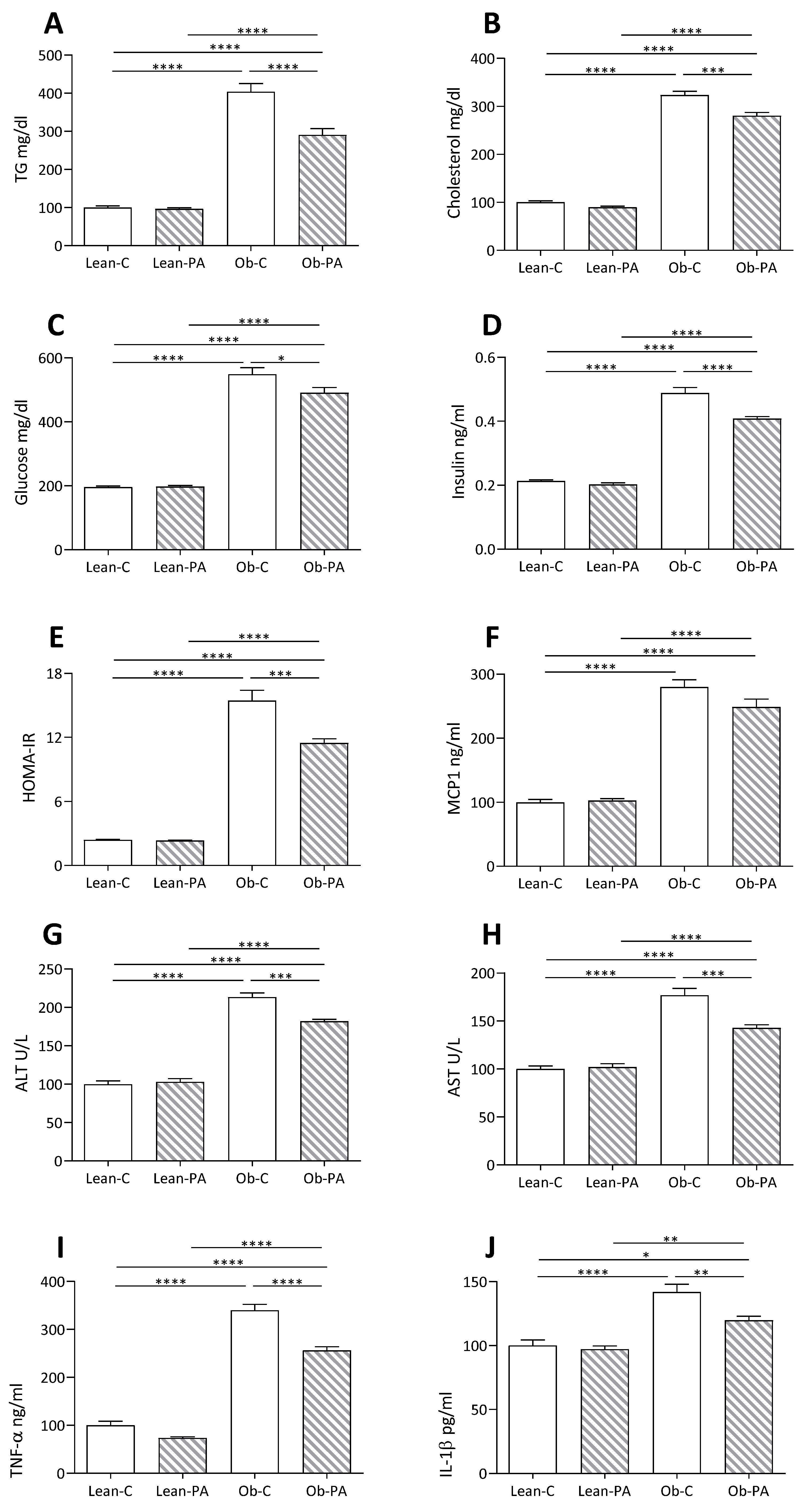
3.5. Plasma and Hepatic Parameters
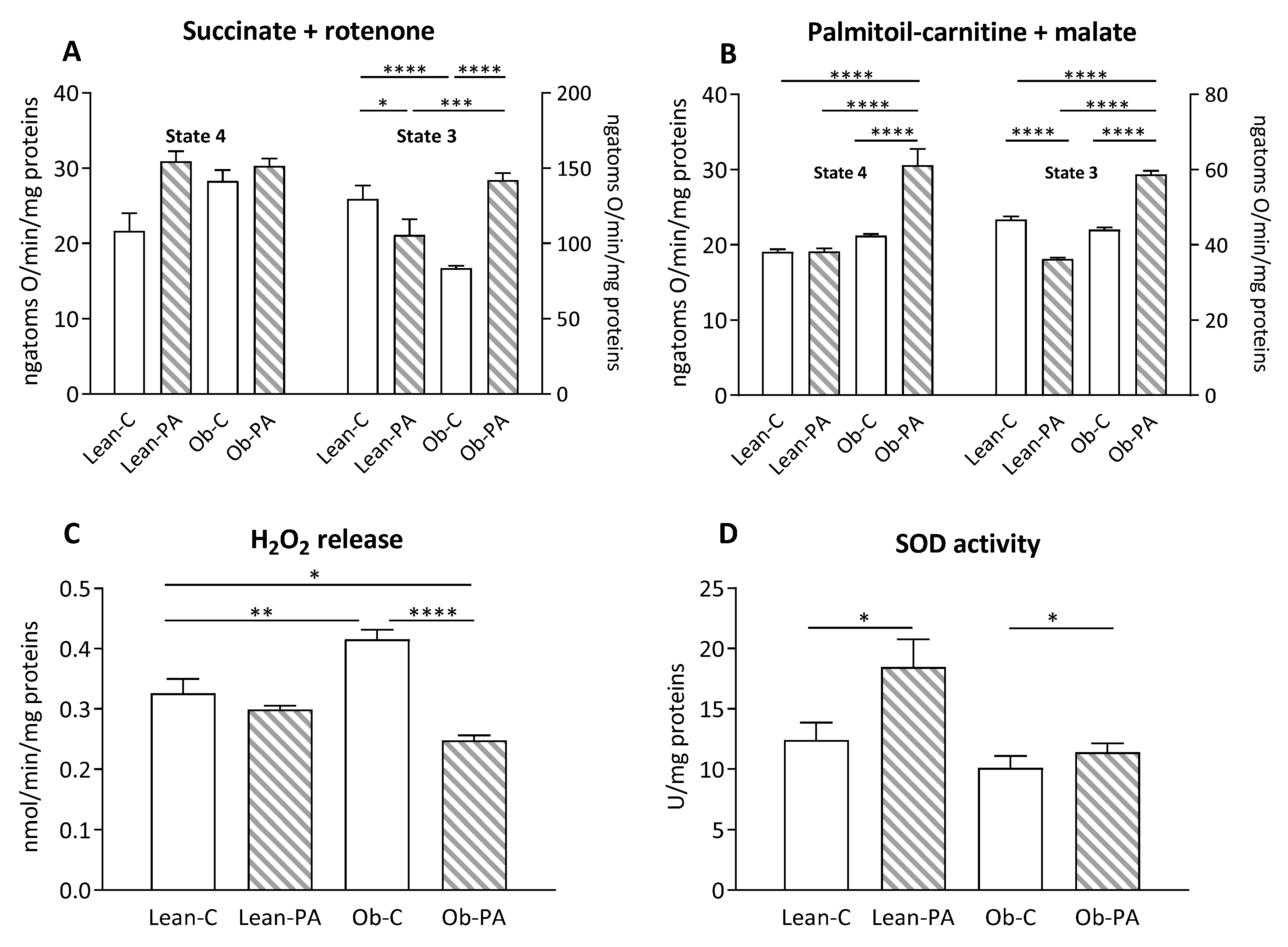
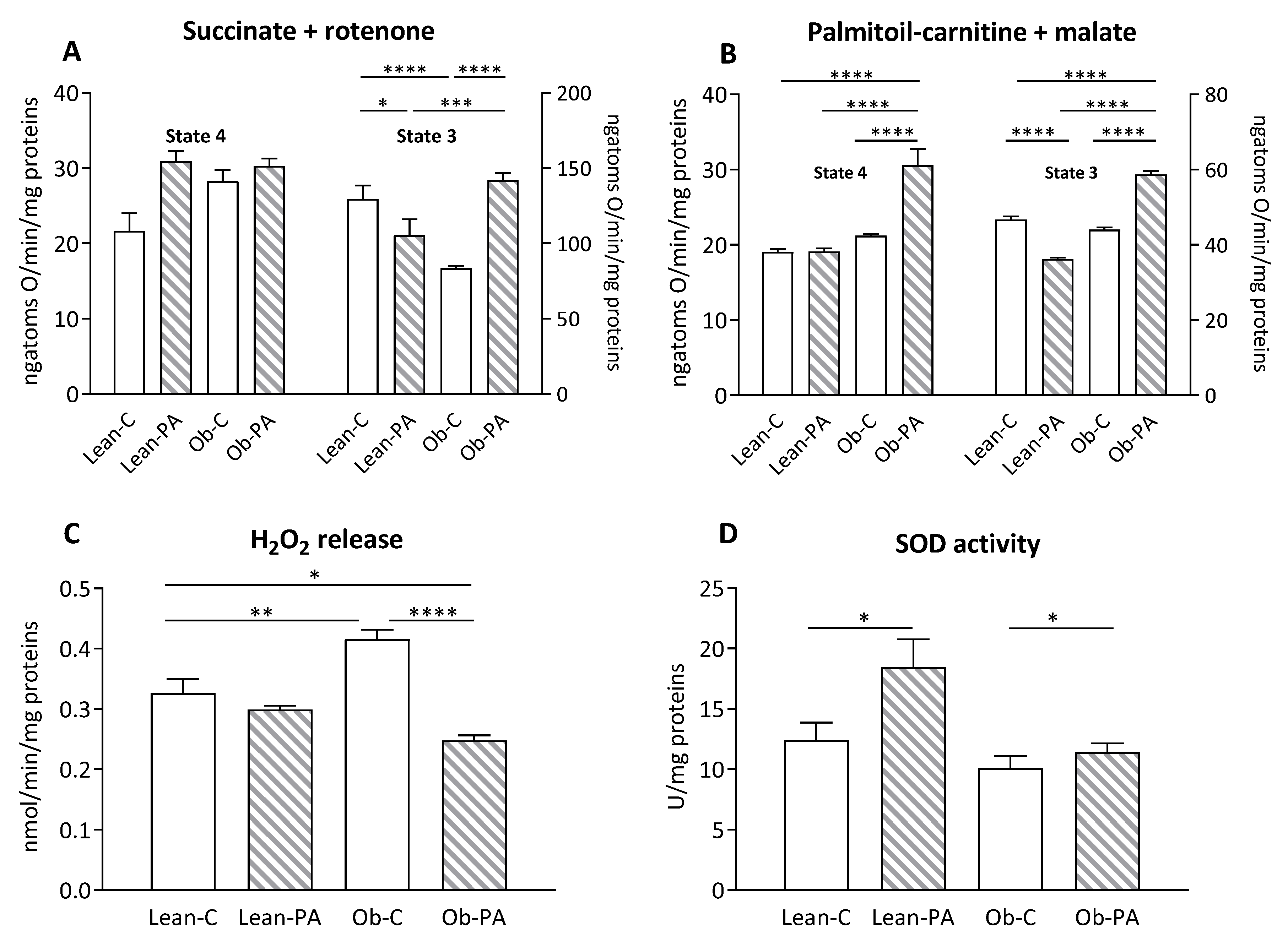
3.6. Modulation of Hepatic Mitochondrial Function and Efficiency and Oxidative Stress
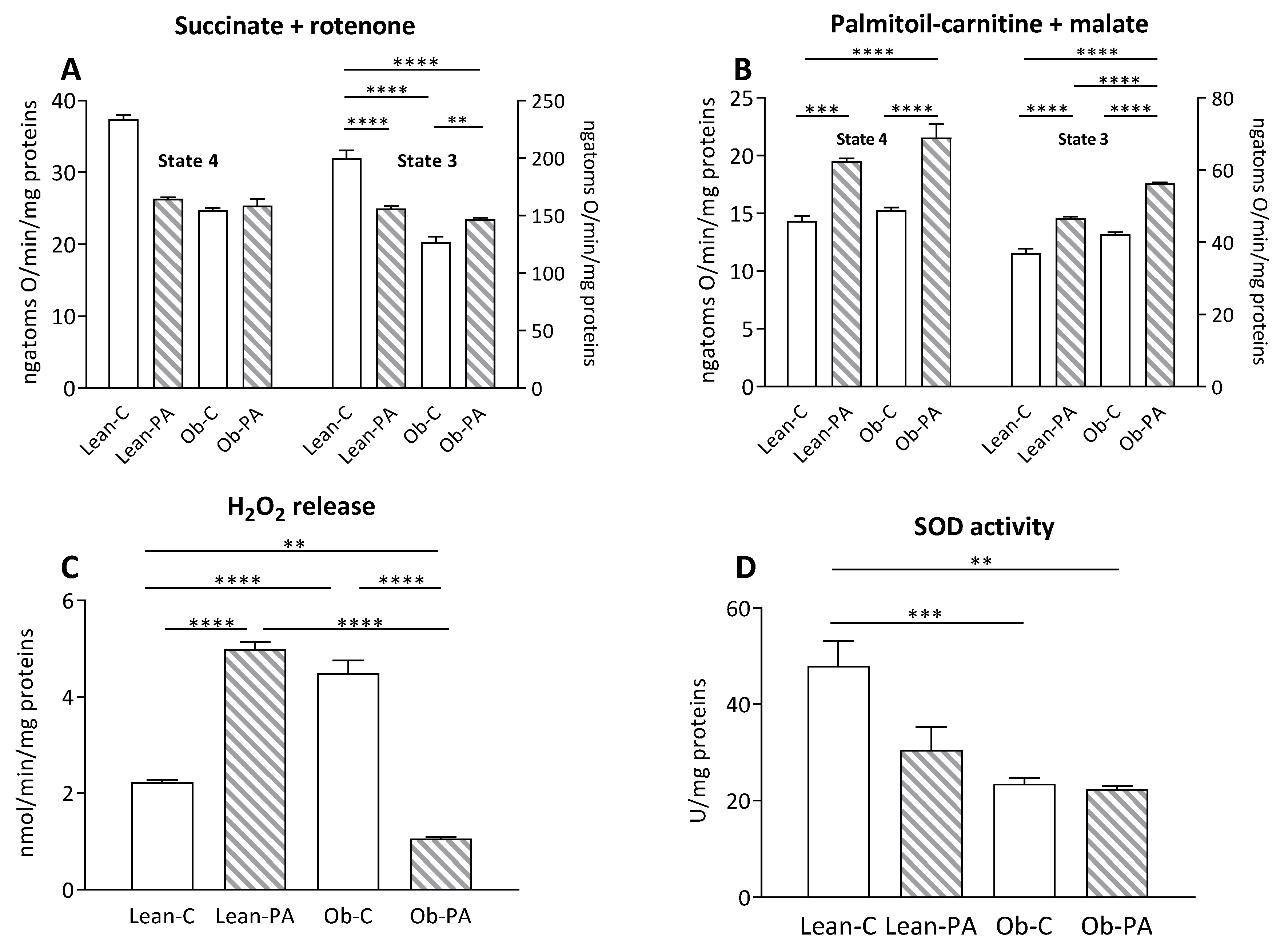
3.7. Modulation of Skeletal Muscle Mitochondrial Function and Efficiency and Oxidative Stress
4. Discussion
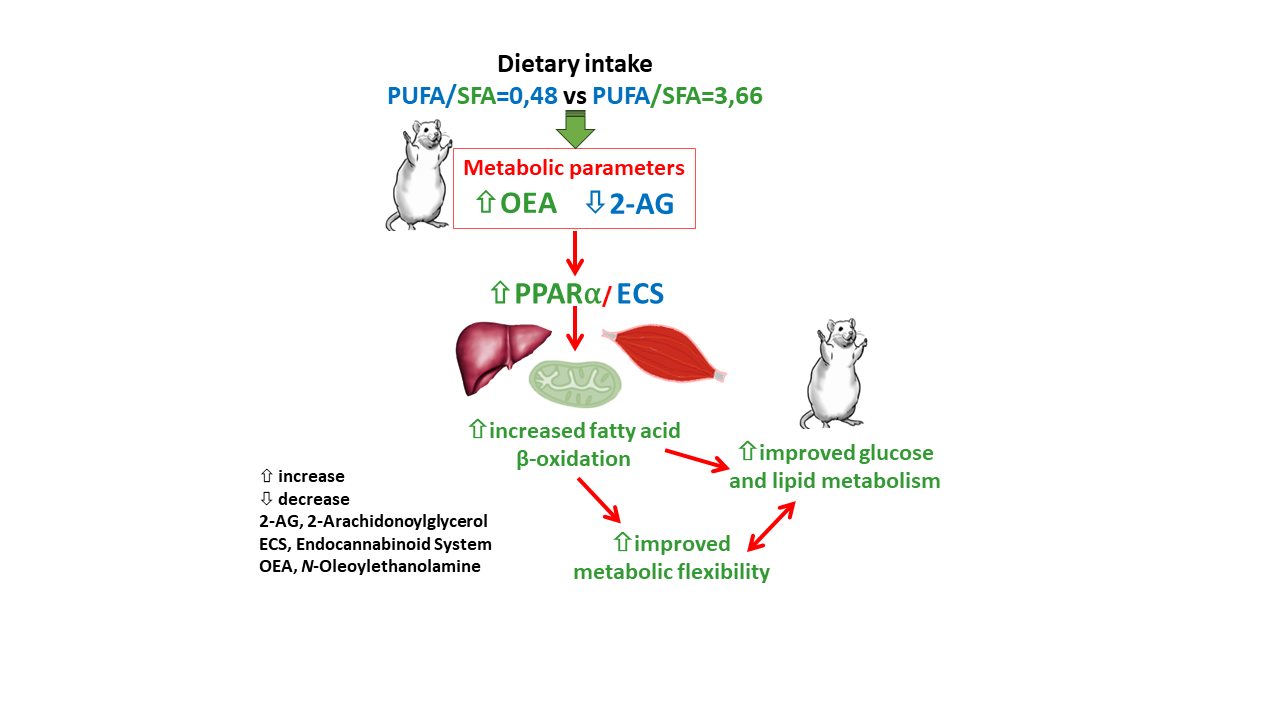
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, G.A.; Popkin, B.M. Dietary Fat Intake Does Affect Obesity! Am. J. Clin. Nutr. 1998, 68, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Use of High-Fat Diets to Study Rodent Obesity as a Model of Human Obesity. Int. J. Obes. 2019, 43, 1491–1492. [Google Scholar] [CrossRef]
- Hooper, L.; Abdelhamid, A.S.; Jimoh, O.F.; Bunn, D.; Skeaff, C.M. Effects of Total Fat Intake on Body Fatness in Adults. Cochrane Database Syst. Rev. 2020, 6, CD013636. [Google Scholar] [CrossRef] [PubMed]
- Heini, A.F.; Weinsier, R.L. Divergent Trends in Obesity and Fat Intake Patterns: The American Paradox. Am. J. Med. 1997, 102, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Duster, M.; Roberts, T.; Devinsky, O. United States Dietary Trends Since 1800: Lack of Association Between Saturated Fatty Acid Consumption and Non-Communicable Diseases. Front. Nutr. 2022, 8, 748847. [Google Scholar] [CrossRef]
- Monsanto, S.P.; Hintze, K.J.; Ward, R.E.; Larson, D.P.; Lefevre, M.; Benninghoff, A.D. The New Total Western Diet for Rodents Does Not Induce an Overweight Phenotype or Alter Parameters of Metabolic Syndrome in Mice. Nutr. Res. 2016, 36, 1031–1044. [Google Scholar] [CrossRef]
- Hintze, K.J.; Benninghoff, A.D.; Cho, C.E.; Ward, R.E. Modeling the Western Diet for Preclinical Investigations. Adv. Nutr. 2018, 9, 263–271. [Google Scholar] [CrossRef]
- Hariri, N.; Thibault, L. High-Fat Diet-Induced Obesity in Animal Models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef]
- Lavau, M.; Susini, C. [U-14C]Glucose Metabolism in Vivo in Rats Rendered Obese by a High Fat Diet. J. Lipid Res. 1975, 16, 134–142. [Google Scholar] [CrossRef]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef]
- Lemonnier, D.; Tremolieres, J. [Experimental obesity induced by high-fat diets]. Probl. Actuels Endocrinol. Nutr. 1963, 7, 77–85. [Google Scholar] [PubMed]
- Reeves, P.G. Components of the AIN-93 Diets as Improvements in the AIN-76A Diet. J. Nutr. 1997, 127, S838–S841. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in Db/Db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Murakami, T.; Ishida, K.; Mizuno, A.; Kuwajima, M.; Shima, K. Phenotype-Linked Amino Acid Alteration in Leptin Receptor cDNA from Zucker Fatty (Fa/Fa) Rat. Biochem. Biophys. Res. Commun. 1996, 222, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- DeLany, J.P.; Windhauser, M.M.; Champagne, C.M.; Bray, G.A. Differential Oxidation of Individual Dietary Fatty Acids in Humans. Am. J. Clin. Nutr. 2000, 72, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Banni, S.; Di Marzo, V. Effect of Dietary Fat on Endocannabinoids and Related Mediators: Consequences on Energy Homeostasis, Inflammation and Mood. Mol. Nutr. Food Res. 2010, 54, 82–92. [Google Scholar] [CrossRef]
- Naughton, S.S.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Fatty Acid Modulation of the Endocannabinoid System and the Effect on Food Intake and Metabolism. Int. J. Endocrinol. 2013, 2013, 361895. [Google Scholar] [CrossRef]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and Related N-Acylethanolamines: Biological Activities and Metabolism. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Kim, H.-J.; Kaburagi, Y.; Yasuda, K.; Ezaki, O. A Low Fish Oil Inhibits SREBP-1 Proteolytic Cascade, While a High-Fish-Oil Feeding Decreases SREBP-1 mRNA in Mice Liver: Relationship to Anti-Obesity. J. Lipid Res. 2003, 44, 369–379. [Google Scholar] [CrossRef]
- Roomp, K.; Kristinsson, H.; Schvartz, D.; Ubhayasekera, K.; Sargsyan, E.; Manukyan, L.; Chowdhury, A.; Manell, H.; Satagopam, V.; Groebe, K.; et al. Combined Lipidomic and Proteomic Analysis of Isolated Human Islets Exposed to Palmitate Reveals Time-Dependent Changes in Insulin Secretion and Lipid Metabolism. PLoS ONE 2017, 12, e0176391. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T. Role of Fatty Acid Elongase Elovl6 in the Regulation of Energy Metabolism and Pathophysiological Significance in Diabetes. Diabetol. Int. 2021, 12, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Sun, Q.; Hu, F.B.; Krauss, R.M. Saturated Fatty Acids and Risk of Coronary Heart Disease: Modulation by Replacement Nutrients. Curr. Atheroscler. Rep. 2010, 12, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Bertram, H.C.; Bonjour, J.-P.; de Groot, L.C.; de Oliveira Otto, M.C.; Feeney, E.L.; Garg, M.L.; Givens, I.; Kok, F.J.; Krauss, R.M.; et al. WHO Draft Guidelines on Dietary Saturated and Trans Fatty Acids: Time for a New Approach? BMJ 2019, 366, l4137. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Martin, N.; Abdelhamid, A.; Davey Smith, G. Reduction in Saturated Fat Intake for Cardiovascular Disease. Cochrane Database Syst. Rev. 2015, 5, CD011737. [Google Scholar] [CrossRef] [PubMed]
- Brontestewart, B. Effects of Feeding Different Fats on Serum-Cholesterol Level. Lancet 1956, 267, 521–527. [Google Scholar] [CrossRef]
- Lawrence, G.D. Perspective: The Saturated Fat–Unsaturated Oil Dilemma: Relations of Dietary Fatty Acids and Serum Cholesterol, Atherosclerosis, Inflammation, Cancer, and All-Cause Mortality. Adv. Nutr. 2021, 12, 647–656. [Google Scholar] [CrossRef]
- Cook, S.L.; Konrad, S.D.; Goh, Y.K.; French, M.A.; Clandinin, M.T. Palmitic Acid Effect on Lipoprotein Profiles and Endogenous Cholesterol Synthesis or Clearance in Humans. Asia Pac. J. Clin. Nutr. 1997, 6, 6–11. [Google Scholar] [PubMed]
- Cavaliere, G.; Trinchese, G.; Bergamo, P.; De Filippo, C.; Mattace Raso, G.; Gifuni, G.; Putti, R.; Moni, B.H.; Canani, R.B.; Meli, R.; et al. Polyunsaturated Fatty Acids Attenuate Diet Induced Obesity and Insulin Resistance, Modulating Mitochondrial Respiratory Uncoupling in Rat Skeletal Muscle. PLoS ONE 2016, 11, e0149033. [Google Scholar] [CrossRef]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C. AIN-93 Purified Diets for Laboratory Rodents: Final Report of the American Institute of Nutrition Ad Hoc Writing Committee on the Reformulation of the AIN-76A Rodent Diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef]
- Klurfeld, D.M.; Gregory, J.F.; Fiorotto, M.L. Should the AIN-93 Rodent Diet Formulas Be Revised? J. Nutr. 2021, 151, 1380–1382. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Murru, E.; Manca, C.; Carta, G.; Banni, S. Impact of Dietary Palmitic Acid on Lipid Metabolism. Front. Nutr. 2022, 9, 861664. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.J.; Kelly, M.A.; Abbott, S.K. Polyunsaturated Fats, Membrane Lipids and Animal Longevity. J. Comp. Physiol. B 2014, 184, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Sette, S.; Le Donne, C.; Piccinelli, R.; Arcella, D.; Turrini, A.; Leclercq, C. The Third Italian National Food Consumption Survey, INRAN-SCAI 2005–06—Part 1: Nutrient Intakes in Italy. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 922–932. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Chiang, S.P.; Gessert, C.F.; Lowry, O.H. Colorimetric Determination of Extracted Lipids. An Adaptation for Microgram Amounts of Lipids Obtained from Cerumen. Curr. List Med. Lit. Res. Rep. 1957, 33, 56–113. [Google Scholar]
- Batetta, B.; Griinari, M.; Carta, G.; Murru, E.; Ligresti, A.; Cordeddu, L.; Giordano, E.; Sanna, F.; Bisogno, T.; Uda, S.; et al. Endocannabinoids May Mediate the Ability of (n − 3) Fatty Acids to Reduce Ectopic Fat and Inflammatory Mediators in Obese Zucker Rats. J. Nutr. 2009, 139, 1495–1501. [Google Scholar] [CrossRef]
- Stark, K.D. The Percentage of N-3 Highly Unsaturated Fatty Acids in Total HUFA as a Biomarker for Omega-3 Fatty Acid Status in Tissues. Lipids 2008, 43, 45–53. [Google Scholar] [CrossRef]
- Manca, C.; Carta, G.; Murru, E.; Abolghasemi, A.; Ansar, H.; Errigo, A.; Cani, P.D.; Banni, S.; Pes, G.M. Circulating Fatty Acids and Endocannabinoidome-Related Mediator Profiles Associated to Human Longevity. GeroScience 2021, 43, 1783–1798. [Google Scholar] [CrossRef]
- Cacho, J.; Sevillano, J.; De Castro, J.; Herrera, E.; Ramos, M.P. Validation of Simple Indexes to Assess Insulin Sensitivity during Pregnancy in Wistar and Sprague-Dawley Rats. Am. J. Physiol.-Endocrinol. Metab. 2008, 295, E1269–E1276. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Catapano, A.; Trinchese, G.; Cavaliere, G.; Culurciello, R.; Fogliano, C.; Penna, E.; Lucci, V.; Crispino, M.; Avallone, B.; et al. Dietary Micronutrient Management to Treat Mitochondrial Dysfunction in Diet-Induced Obese Mice. Int. J. Mol. Sci. 2021, 22, 2862. [Google Scholar] [CrossRef]
- Trinchese, G.; Cavaliere, G.; Cimmino, F.; Catapano, A.; Carta, G.; Pirozzi, C.; Murru, E.; Lama, A.; Meli, R.; Bergamo, P.; et al. Decreased Metabolic Flexibility in Skeletal Muscle of Rat Fed with a High-Fat Diet Is Recovered by Individual CLA Isomer Supplementation via Converging Protective Mechanisms. Cells 2020, 9, 823. [Google Scholar] [CrossRef] [PubMed]
- Hartree, E.F. Determination of Protein: A Modification of the Lowry Method That Gives a Linear Photometric Response. Anal. Biochem. 1972, 48, 422–427. [Google Scholar] [CrossRef]
- Estabrook, R.W. [7] Mitochondrial Respiratory Control and the Polarographic Measurement of ADP : O Ratios. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1967; Volume 10, pp. 41–47. ISBN 978-0-12-181850-0. [Google Scholar]
- Barja, G. Mitochondrial Free Radical Production and Aging in Mammals and Birdsa. Ann. N. Y. Acad. Sci. 1998, 854, 224–238. [Google Scholar] [CrossRef]
- Flohé, L.; ötting, F. [10] Superoxide Dismutase Assays. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1984; Volume 105, pp. 93–104. ISBN 978-0-12-182005-3. [Google Scholar]
- Jeffery, N.M.; Cortina, M.; Newsholme, E.A.; Calder, P.C. Effects of Variations in the Proportions of Saturated, Monounsaturated and Polyunsaturated Fatty Acids in the Rat Diet on Spleen Lymphocyte Functions. Br. J. Nutr. 1997, 77, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Johansson, H.-E.; Haenni, A.; Zethelius, B. Platelet Counts and Liver Enzymes after Bariatric Surgery. J. Obes. 2013, 2013, 567984. [Google Scholar] [CrossRef]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated Fatty Acids Promote Endoplasmic Reticulum Stress and Liver Injury in Rats with Hepatic Steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef]
- Abbott, S.K.; Else, P.L.; Atkins, T.A.; Hulbert, A.J. Fatty Acid Composition of Membrane Bilayers: Importance of Diet Polyunsaturated Fat Balance. Biochim. Biophys. Acta BBA—Biomembr. 2012, 1818, 1309–1317. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Lisai, S.; Sirigu, A.; Piras, A.; Collu, M.; Batetta, B.; Gambelli, L.; Banni, S. Dietary Triacylglycerols with Palmitic Acid in the Sn-2 Position Modulate Levels of N-Acylethanolamides in Rat Tissues. PLoS ONE 2015, 10, e0120424. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.T.; Ntambi, J.M. Stearoyl-CoA Desaturase and Its Relation to High-Carbohydrate Diets and Obesity. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2009, 1791, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Claiborn, K.C.; Hotamisligil, G.S. De Novo Lipogenesis Products and Endogenous Lipokines. Diabetes 2016, 65, 1800–1807. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Invest. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Flowers, M.T.; Ntambi, J.M. Role of Stearoyl-Coenzyme A Desaturase in Regulating Lipid Metabolism. Curr. Opin. Lipidol. 2008, 19, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, A.; Ntambi, J.M. Stearoyl-CoA Desaturase as a New Drug Target for Obesity Treatment. Obes. Rev. 2005, 6, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez de Fonseca, F.; Navarro, M.; Gómez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodríguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An Anorexic Lipid Mediator Regulated by Feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodríguez de Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide Regulates Feeding and Body Weight through Activation of the Nuclear Receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Suardíaz, M.; Estivill-Torrús, G.; Goicoechea, C.; Bilbao, A.; de Fonseca, F.R. Analgesic Properties of Oleoylethanolamide (OEA) in Visceral and Inflammatory Pain. Pain 2007, 133, 99–110. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Howey, D.C.; McErlean, E.; Wang, M.-D.; Gomez, E.V.; Russo, J.M. Effects of a Potent and Selective PPAR-α Agonist in Patients With Atherogenic Dyslipidemia or Hypercholesterolemia: Two Randomized Controlled Trials. JAMA 2007, 297, 1362. [Google Scholar] [CrossRef]
- Tovar, R.; Gavito, A.L.; Vargas, A.; Soverchia, L.; Hernandez-Folgado, L.; Jagerovic, N.; Baixeras, E.; Ciccocioppo, R.; Rodríguez de Fonseca, F.; Decara, J. Palmitoleoylethanolamide Is an Efficient Anti-Obesity Endogenous Compound: Comparison with Oleylethanolamide in Diet-Induced Obesity. Nutrients 2021, 13, 2589. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.K.; Bui, H.H.; Beavers, L.S.; Farb, T.B.; Ficorilli, J.; Chesterfield, A.K.; Kuo, M.-S.; Bokvist, K.; Barrett, D.G.; Efanov, A.M. Regulation of GPR119 Receptor Activity with Endocannabinoid-like Lipids. Am. J. Physiol.-Endocrinol. Metab. 2012, 303, E1469–E1478. [Google Scholar] [CrossRef] [PubMed]
- de Souza, C.O.; Vannice, G.K.; Rosa Neto, J.C.; Calder, P.C. Is Palmitoleic Acid a Plausible Nonpharmacological Strategy to Prevent or Control Chronic Metabolic and Inflammatory Disorders? Mol. Nutr. Food Res. 2018, 62, 1700504. [Google Scholar] [CrossRef]
- Frigolet, M.E.; Gutiérrez-Aguilar, R. The Role of the Novel Lipokine Palmitoleic Acid in Health and Disease. Adv. Nutr. 2017, 8, 173S–181S. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a Lipokine, a Lipid Hormone Linking Adipose Tissue to Systemic Metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef]
- Blüher, M.; Engeli, S.; Klöting, N.; Berndt, J.; Fasshauer, M.; Bátkai, S.; Pacher, P.; Schön, M.R.; Jordan, J.; Stumvoll, M. Dysregulation of the Peripheral and Adipose Tissue Endocannabinoid System in Human Abdominal Obesity. Diabetes 2006, 55, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Matias, I.; Lemieux, I.; Petrosino, S.; Alméras, N.; Després, J.-P.; Di Marzo, V. Circulating Endocannabinoid Levels, Abdominal Adiposity and Related Cardiometabolic Risk Factors in Obese Men. Int. J. Obes. 2007, 31, 692–699. [Google Scholar] [CrossRef]
- Alvheim, A.R.; Torstensen, B.E.; Lin, Y.H.; Lillefosse, H.H.; Lock, E.-J.; Madsen, L.; Frøyland, L.; Hibbeln, J.R.; Malde, M.K. Dietary Linoleic Acid Elevates the Endocannabinoids 2-AG and Anandamide and Promotes Weight Gain in Mice Fed a Low Fat Diet. Lipids 2014, 49, 59–69. [Google Scholar] [CrossRef]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A Proposed Autacoid Mechanism Controlling Mastocyte Behaviour. Agents Actions 1993, 39, C145–C147. [Google Scholar] [CrossRef]
- Barbier, O.; Torra, I.P.; Duguay, Y.; Blanquart, C.; Fruchart, J.-C.; Glineur, C.; Staels, B. Pleiotropic Actions of Peroxisome Proliferator–Activated Receptors in Lipid Metabolism and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A. Peroxisome Proliferator-Activated Receptors: From Genes to Physiology. Recent Prog. Horm. Res. 2001, 56, 239–265. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.; Lama, A.; Pirozzi, C.; Cavaliere, G.; Trinchese, G.; Di Guida, F.; Nitrato Izzo, A.; Cimmino, F.; Paciello, O.; De Biase, D.; et al. Palmitoylethanolamide Counteracts Hepatic Metabolic Inflexibility Modulating Mitochondrial Function and Efficiency in Diet-induced Obese Mice. FASEB J. 2020, 34, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)Function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Banni, S.; Carta, G.; Murru, E.; Cordeddu, L.; Giordano, E.; Sirigu, A.; Berge, K.; Vik, H.; Maki, K.C.; Di Marzo, V.; et al. Krill Oil Significantly Decreases 2-Arachidonoylglycerol Plasma Levels in Obese Subjects. Nutr. Metab. 2011, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, L.; Mollica, M.P.; Sica, R.; Donizzetti, I.; Gifuni, G.; Pignalosa, A.; Cavaliere, G.; Putti, R. Differential Effects of High-Fish Oil and High-Lard Diets on Cells and Cytokines Involved in the Inflammatory Process in Rat Insulin-Sensitive Tissues. Int. J. Mol. Sci. 2014, 15, 3040–3063. [Google Scholar] [CrossRef]
- Román, G.C.; Jackson, R.E.; Gadhia, R.; Román, A.N.; Reis, J. Mediterranean Diet: The Role of Long-Chain ω-3 Fatty Acids in Fish; Polyphenols in Fruits, Vegetables, Cereals, Coffee, Tea, Cacao and Wine; Probiotics and Vitamins in Prevention of Stroke, Age-Related Cognitive Decline, and Alzheimer Disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Brainard, J.; Song, F.; Wang, X.; Abdelhamid, A.; Hooper, L.; PUFAH Group. Omega-3, Omega-6, and Total Dietary Polyunsaturated Fat for Prevention and Treatment of Type 2 Diabetes Mellitus: Systematic Review and Meta-Analysis of Randomised Controlled Trials. BMJ 2019, 366, l4697. [Google Scholar] [CrossRef]
- Carlson, S.E.; DeVoe, P.W.; Barness, L.A. Effect of Infant Diets with Different Polyunsaturated to Saturated Fat Ratios on Circulating High-Density Lipoproteins. J. Pediatr. Gastroenterol. Nutr. 1982, 1, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Trinchese, G.; Cavaliere, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Chun, J.T.; Penna, E.; Negri, R.; Muredda, L.; Demurtas, A.; et al. Human Milk and Donkey Milk, Compared to Cow Milk, Reduce Inflammatory Mediators and Modulate Glucose and Lipid Metabolism, Acting on Mitochondrial Function and Oleylethanolamide Levels in Rat Skeletal Muscle. Front. Physiol. 2018, 9, 32. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FA | Diet-C | Diet-PA | (Diet-PA)-(Diet-C) |
|---|---|---|---|
| % of the diet | |||
| 12:0 | 0.003 | 0.43 | +0.427 |
| 14:0 | 0.02 | 0.19 | +0.17 |
| 16:0 | 0.84 | 1.91 | +1.07 |
| 18:0 | 0.26 | 0.26 | +0.00 |
| 18:1n9 | 1.78 | 2.88 | +1.10 |
| 18:2n6 | 3.69 | 1.23 | −2.46 |
| 18:3n3 | 0.41 | 0.10 | −0.31 |
| SFA | 1.12 | 2.79 | +1.67 |
| MUFA | 1.78 | 2.88 | +1.10 |
| PUFA | 4.10 | 1.33 | −2.77 |
| PUFA/SFA | 3.66 | 0.48 | −3.18 |
| MUFA/SFA | 1.59 | 1.03 | −0.56 |
| n3/n6 | 0.41 | 0.08 | −0.33 |
| total FA | 7.00 | 7.00 | 0.00 |
| Lean-C | Lean-PA | Ob-C | Ob-PA | |||||
|---|---|---|---|---|---|---|---|---|
| FA in Liver | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM |
| 12:0 | 100 | 16.18 | 161.21 | 28.28 | 103.20 | 16.35 | 297.37 | 48.18 *** $ ££ |
| 14:0 | 100 | 19.32 | 110.54 | 15.70 | 314.52 | 36.33 *** | 331.37 | 22.69 *** $$$ |
| 16:0 | 100 | 6.53 | 108.85 | 4.10 | 162.83 | 10.47 *** | 167.89 | 5.12 *** $$$ |
| 18:0 | 100 | 7.55 | 104.71 | 5.60 | 36.32 | 4.13 *** | 42.42 | 2.71 *** $$$ |
| 14:1n5 | 100 | 21.31 | 87.55 | 23.51 | 215.45 | 35.57 | 271.84 | 40.78 ** $$ |
| 16:1n7 | 100 | 13.80 | 121.45 | 15.98 | 462.09 | 35.65 *** | 465.15 | 14.86 *** $$$ |
| 18:1n9 | 100 | 11.23 | 175.04 | 22.19 * | 299.65 | 16.34 *** | 326.24 | 13.38 *** $$$ |
| 18:3n3 | 100 | 23.63 | 23.04 | 2.48 ** | 50.47 | 14.73 | 14.51 | 1.72 ** |
| 20:5n3 | 100 | 24.61 | 33.51 | 0.91 | 58.66 | 23.25 | 14.64 | 1.98 * |
| 22:6n3 | 100 | 6.36 | 64.54 | 5.88 *** | 23.44 | 5.35 *** | 16.73 | 1.23 *** $$$ |
| 18:2n6 | 100 | 7.44 | 49.25 | 3.49 *** | 43.06 | 4.95 *** | 18.44 | 1.72 *** $$ ££ |
| 18:3n6 | 100 | 11.71 | 55.76 | 4.51 | 101.36 | 19.13 | 44.55 | 5.98 * £ |
| 20:4n6 | 100 | 5.87 | 98.49 | 7.67 | 25.25 | 4.96 *** | 25.41 | 1.49 *** $$$ |
| 22:4n6 | 100 | 13.17 | 93.81 | 12.02 | 51.09 | 13.50 | 35.71 | 3.60 * $ |
| 20:3n9 | 100 | 41.28 | 139.71 | 18.60 | 39.42 | 7.46 | 64.98 | 6.98 $ |
| SFA | 100 | 6.58 | 106.36 | 5.62 | 110.77 | 6.46 | 116.32 | 3.75 |
| MUFA | 100 | 12.60 | 166.51 | 20.96 * | 337.97 | 19.35 *** | 361.31 | 10.75 *** $$$ |
| PUFA | 100 | 5.24 | 79.22 | 5.71 * | 33.99 | 5.56 *** | 24.39 | 1.73 *** $$$ |
| PUFAn3 | 100 | 6.91 | 59.96 | 4.96 *** | 27.02 | 6.11 *** | 16.98 | 1.10 *** $$$ |
| PUFAn6 | 100 | 5.24 | 82.02 | 6.13 * | 34.96 | 5.34 *** | 24.81 | 1.76 *** $$$ |
| PUFAn9 | 100 | 10.79 | 175.87 | 21.30 * | 293.85 | 15.75 *** | 320.73 | 13.01 *** $$$ |
| n3HUFA score | 100 | 4.31 | 65.77 | 1.68 *** | 82.51 | 3.45 ** | 60.39 | 2.52 *** £££ |
| Lean-C | Lean-PA | Ob-C | Ob-PA | |||||
|---|---|---|---|---|---|---|---|---|
| FA in AT | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM |
| 12:0 | 100 | 9.50 | 751.58 | 158.74 *** | 61.35 | 10.96 | 335.97 | 72.98 $$ |
| 14:0 | 100 | 4.96 | 157.02 | 2.74 *** | 118.75 | 2.76 ** | 138.43 | 3.27 *** $$ ££ |
| 16:0 | 100 | 1.73 | 118.24 | 1.57 *** | 126.44 | 2.81 *** | 129.94 | 2.13 *** $$ |
| 18:0 | 100 | 3.84 | 82.18 | 4.13 | 92.81 | 9.17 | 80.06 | 6.31 |
| 14:1n5 | 100 | 7.38 | 126.4 | 36.99 | 282.3 | 72.82 | 318.97 | 102.03 |
| 16:1n7 | 100 | 5.99 | 141.60 | 18.42 | 253.89 | 38.60 ** | 291.07 | 40.62 *** $$ |
| 18:1n9 | 100 | 2.15 | 163.63 | 1.23 *** | 116.27 | 1.61 *** | 152.64 | 1.29 ***$$ £££ |
| 18:3n3 | 100 | 3.15 | 17.78 | 1.80 *** | 54.35 | 1.73 *** | 12.27 | 0.60 *** £££ |
| 20:5n3 | 100 | 8.15 | 36.89 | 8.93 *** | 176.71 | 4.67 *** | 51.47 | 7.20 *** £££ |
| 22:6n3 | 100 | 6.80 | 26.36 | 3.90 *** | 182.64 | 18.40 *** | 39.99 | 6.14 *** £££ |
| 18:2n6 | 100 | 2.52 | 33.29 | 1.67 *** | 50.75 | 0.59 *** | 20.02 | 0.68 *** $$$ £££ |
| 18:3n6 | 100 | 2.84 | 24.22 | 3.23 *** | 72.08 | 5.17 *** | 24.79 | 1.91 *** £££ |
| 20:4n6 | 100 | 6.23 | 52.06 | 9.14 *** | 128.17 | 3.10 * | 58.04 | 2.50 *** £££ |
| 22:5n6 | 100 | 5.78 | 91.49 | 13.55 | 179.19 | 9.53 *** | 125.86 | 10.60 $$ £££ |
| 22:4n6 | 100 | 8.36 | 45.00 | 15.50 *** | 163.81 | 11.10 *** | 61.63 | 5.91 * £££ |
| 20:3n9 | 100 | 7.25 | 185.01 | 14.87 ** | 199.51 | 9.22 *** | 322.49 | 23.94 *** $$$ £££ |
| SFAs | 100 | 2.12 | 123.17 | 0.72 *** | 120.06 | 3.17 *** | 125.53 | 1.52 *** |
| MUFA | 100 | 1.94 | 158.39 | 2.77 *** | 134.41 | 4.30 *** | 170.70 | 3.75 *** £££ |
| PUFA | 100 | 2.43 | 33.36 | 1.68 *** | 55.25 | 0.67 *** | 21.98 | 0.69 *** $$$ £££ |
| PUFAn3 | 100 | 3.25 | 18.19 | 1.80 *** | 61.30 | 1.93 *** | 13.95 | 0.78 *** £££ |
| PUFAn6 | 100 | 2.44 | 34.06 | 1.72 *** | 53.41 | 0.60 *** | 21.58 | 0.73 *** $$$ £££ |
| PUFAn9 | 100 | 2.15 | 163.67 | 1.26 *** | 116.49 | 1.60 *** | 153.09 | 1.28 *** $$$ £££ |
| n3HUFA score | 100 | 5.42 | 47.10 | 2.95 *** | 108.96 | 6.76 | 51.96 | 6.47 *** £££ |
| Lean-C | Lean-PA | Ob-C | Ob-PA | |||||
|---|---|---|---|---|---|---|---|---|
| FA in Muscle | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM |
| 12:0 | 100 | 14.60 | 938.73 | 150.72 *** | 108.02 | 11.98 | 639.02 | 28.29 *** £££ |
| 14:0 | 100 | 9.38 | 190.96 | 23.22 *** | 160.17 | 9.73 * | 227.50 | 7.27 *** £ |
| 16:0 | 100 | 2.76 | 98.80 | 8.03 | 100.41 | 6.72 | 113.71 | 3.08 |
| 18:0 | 100 | 10.21 | 65.67 | 8.23 * | 38.81 | 4.95 *** | 39.56 | 3.77 *** |
| 14:1n5 | 100 | 45.35 | 83.64 | 34.14 | 81.89 | 10.89 | 53.15 | 14.26 |
| 16:1n7 | 100 | 16.55 | 148.51 | 33.59 | 480.03 | 48.16 *** | 517.92 | 46.40 *** $$$ |
| 18:1n9 | 100 | 12.12 | 187.65 | 29.75 ** | 186.26 | 6.75 ** | 218.53 | 6.22 *** |
| 18:3n3 | 100 | 14.62 | 29.71 | 5.25 *** | 117.41 | 13.27 | 28.84 | 2.80 *** £££ |
| 20:5n3 | 100 | 16.21 | 51.19 | 11.35 * | 134.08 | 9.55 | 66.17 | 7.20 ££ |
| 22:6n3 | 100 | 10.36 | 44.86 | 7.64 *** | 41.18 | 3.67 *** | 26.81 | 2.77 *** |
| 18:2n6 | 100 | 6.89 | 55.94 | 4.61 *** | 80.81 | 3.18 * | 40.73 | 2.13 *** £££ |
| 18:3n6 | 100 | 11.08 | 53.51 | 5.93 * | 131.46 | 12.96 | 57.55 | 1.82 * £££ |
| 20:4n6 | 100 | 9.67 | 74.06 | 12.34 | 51.89 | 3.43 ** | 43.89 | 3.53 *** |
| 22:5n6 | 100 | 8.47 | 166.04 | 18.96 ** | 88.45 | 10.09 | 113.29 | 13.08 |
| 22:4n6 | 100 | 6.31 | 88.34 | 12.76 | 99.77 | 6.40 | 76.50 | 6.99 |
| 20:3n9 | 100 | 40.50 | 275.93 | 37.52 * | 169.98 | 9.11 | 434.43 | 27.57 *** $ |
| SFA | 100 | 4.74 | 107.02 | 9.26 | 81.73 | 5.14 | 95.50 | 2.20 |
| MUFA | 100 | 12.52 | 180.92 | 29.86 * | 234.65 | 12.79 *** | 267.22 | 11.59 *** $ |
| PUFA | 100 | 4.49 | 64.65 | 7.57 *** | 74.52 | 2.38 ** | 44.07 | 2.82 *** $ ££ |
| PUFAn3 | 100 | 7.84 | 44.13 | 6.48 *** | 54.86 | 2.52 *** | 29.57 | 2.21 *** £ |
| PUFAn6 | 100 | 5.91 | 65.24 | 6.29 *** | 74.24 | 2.47 ** | 44.28 | 3.06 *** $ ££ |
| PUFAn9 | 100 | 12.24 | 189.50 | 29.45 ** | 186.95 | 6.85 ** | 221.65 | 6.08 *** |
| n3HUFA score | 100 | 5 | 65.64 | 2.75 *** | 77.77 | 1.81 ** | 62.77 | 3.72 *** £ |
| Lean-C | Lean-PA | Ob-C | Ob-PA | |||||
|---|---|---|---|---|---|---|---|---|
| FA In Plasma | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM |
| 12:0 | 100 | 18.78 | 209.46 | 34.44 * | 38.44 | 4.61 | 105.08 | 3.43 $ |
| 14:0 | 100 | 7.14 | 137.55 | 13.40 | 109.90 | 23.35 | 140.18 | 12.29 |
| 16:0 | 100 | 7.78 | 123.45 | 8.61 | 111.39 | 19.95 | 124.35 | 6.88 |
| 18:0 | 100 | 5.74 | 107.32 | 9.12 | 91.96 | 19.56 | 111.69 | 9.52 |
| 14:1n5 | 100 | 27.11 | 54.79 | 13.70 | 33.51 | 6.42 * | 49.57 | 3.60 |
| 16:1n7 | 100 | 10.68 | 162.83 | 21.17 | 381.26 | 57.99 ** | 395.88 | 41.91 *** $$ |
| 18:1n9 | 100 | 11.87 | 178.24 | 19.68 | 165.94 | 26.62 | 188.75 | 14.83 * |
| 18:3n3 | 100 | 8.68 | 22.27 | 1.57 *** | 59.53 | 13.42 * | 15.26 | 1.95 *** ££ |
| 20:5n3 | 100 | 10.36 | 36.26 | 2.44 | 154.81 | 24.57 | 57.19 | 4.29 ££ |
| 22:6n3 | 100 | 8.39 | 56.26 | 6.59 ** | 84.50 | 9.15 | 60.55 | 7.46 * |
| 18:2n6 | 100 | 6.08 | 46.45 | 2.41 *** | 53.58 | 8.14 *** | 24.21 | 1.71 *** $ ££ |
| 18:3n6 | 100 | 7.74 | 81.66 | 7.74 | 111.12 | 4.14 | 72.39 | 5.17 * £ |
| 20:4n6 | 100 | 12 | 80.11 | 9.06 | 73.88 | 18.99 | 71.51 | 12.83 |
| 22:4n6 | 100 | 14.21 | 84.01 | 10.36 | 162.83 | 36.15 | 106.72 | 10.96 |
| 22:5n6 | 100 | 7.94 | 345.81 | 21.10 *** | 232.90 | 35.34 * | 436.17 | 21.48 *** £££ |
| 20:3n9 | 100 | 6.14 | 441.52 | 88.73 ** | 270.42 | 88.73 | 710.60 | 47.49 *** $ £££ |
| SFA | 100 | 6.12 | 119.03 | 8.04 | 120.82 | 17.15 | 119.25 | 4.38 |
| MUFA | 100 | 7.70 | 149.22 | 18.45 | 337.44 | 50.38 ** | 352.24 | 36.99 ** $$ |
| PUFA | 100 | 5.45 | 67.55 | 5.27 * | 72.67 | 7.55 * | 57.62 | 5.59 ** |
| PUFAn3 | 100 | 4.80 | 49.71 | 4.69 *** | 87.07 | 5.51 | 53.10 | 4.63 *** £££ |
| PUFAn6 | 100 | 5.77 | 65.22 | 5.41 * | 68.09 | 8.00 * | 51.76 | 5.89 *** |
| PUFAn9 | 100 | 11.72 | 180.56 | 19.41 | 167.23 | 26.64 | 193.36 | 14.62 * |
| n3HUFA score | 100 | 2.89 | 62.55 | 0. 96 ** | 109.14 | 9.90 | 69.45 | 1.84 * ££ |
| Liver | Muscle | Plasma | AT | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NAE | Lean | Obese | Lean | Obese | Lean | Obese | Lean | Obese | |||||||||
| EC | r | p | r | p | r | p | r | p | r | p | r | p | r | p | r | p | |
| 16:0 | PEA | 0.062 | 0.856 | 0.645 | 0.024 | 0.026 | 0.937 | 0.507 | 0.929 | 0.409 | 0.275 | −0.116 | 0.719 | −0.043 | 0.873 | 0.081 | 0.765 |
| OEA | 0.121 | 0.724 | 0.396 | 0.202 | 0.339 | 0.282 | 0.461 | 0.131 | 0.598 | 0.089 | −0.023 | 0.943 | 0.674 | 0.004 | 0.270 | 0.312 | |
| POEA | 0.045 | 0.895 | 0.530 | 0.076 | 0.068 | 0.834 | 0.461 | 0.132 | 0.188 | 0.655 | −0.167 | 0.605 | −0.043 | 0.873 | 0.081 | 0.765 | |
| 18:1n9 | AEA | −0.847 | 0.001 | −0.460 | 0.133 | 0.144 | 0.655 | 0.225 | 0.482 | 0.210 | 0.561 | −0.644 | 0.024 | 0.255 | 0.358 | −0.362 | 0.169 |
| 2-AG | −0.401 | 0.221 | −0.351 | 0.290 | −0.740 | 0.006 | −0.710 | 0.010 | −0.244 | 0.527 | 0.086 | 0.792 | −0.256 | 0.338 | −0.212 | 0.430 | |
| PEA | −0.768 | 0.006 | −0.431 | 0.162 | 0.222 | 0.489 | 0.086 | 0.790 | −0.472 | 0.200 | −0.491 | 0.105 | 0.058 | 0.831 | −0.125 | 0.646 | |
| OEA | −0.086 | 0.801 | −0.172 | 0.594 | 0.236 | 0.460 | 0.383 | 0.219 | −0.117 | 0.765 | −0.423 | 0.171 | 0.799 | 0.0002 | 0.507 | 0.045 | |
| POEA | 0.296 | 0.376 | −0.183 | 0.570 | −0.023 | 0.944 | 0.179 | 0.578 | 0.264 | 0.528 | −0.752 | 0.005 | 0.058 | 0.831 | −0.125 | 0.646 | |
| 18:2n6 | AEA | 0.650 | 0.031 | 0.090 | 0.793 | −0.457 | 0.135 | −0.479 | 0.115 | −0.289 | 0.418 | −0.419 | 0.176 | −0.198 | 0.480 | 0.250 | 0.350 |
| 2-AG | 0.169 | 0.619 | 0.672 | 0.024 | 0.624 | 0.030 | 0.898 | 0.0001 | −0.255 | 0.477 | 0.003 | 0.992 | 0.251 | 0.349 | 0.199 | 0.460 | |
| PEA | 0.159 | 0.640 | −0.312 | 0.324 | −0.741 | 0.006 | −0.352 | 0.263 | −0.043 | 0.913 | −0.271 | 0.394 | −0.011 | 0.968 | 0.097 | 0.721 | |
| OEA | −0.424 | 0.194 | −0.585 | 0.046 | −0.803 | 0.002 | −0.719 | 0.008 | −0.423 | 0.257 | −0.333 | 0.290 | −0.739 | 0.001 | −0.544 | 0.029 | |
| POEA | −0.211 | 0.533 | −0.603 | 0.038 | −0.243 | 0.447 | −0.339 | 0.281 | −0.617 | 0.103 | −0.512 | 0.089 | −0.011 | 0.968 | 0.097 | 0.721 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carta, G.; Murru, E.; Trinchese, G.; Cavaliere, G.; Manca, C.; Mollica, M.P.; Banni, S. Reducing Dietary Polyunsaturated to Saturated Fatty Acids Ratio Improves Lipid and Glucose Metabolism in Obese Zucker Rats. Nutrients 2023, 15, 4761. https://doi.org/10.3390/nu15224761
Carta G, Murru E, Trinchese G, Cavaliere G, Manca C, Mollica MP, Banni S. Reducing Dietary Polyunsaturated to Saturated Fatty Acids Ratio Improves Lipid and Glucose Metabolism in Obese Zucker Rats. Nutrients. 2023; 15(22):4761. https://doi.org/10.3390/nu15224761
Chicago/Turabian StyleCarta, Gianfranca, Elisabetta Murru, Giovanna Trinchese, Gina Cavaliere, Claudia Manca, Maria Pina Mollica, and Sebastiano Banni. 2023. "Reducing Dietary Polyunsaturated to Saturated Fatty Acids Ratio Improves Lipid and Glucose Metabolism in Obese Zucker Rats" Nutrients 15, no. 22: 4761. https://doi.org/10.3390/nu15224761
APA StyleCarta, G., Murru, E., Trinchese, G., Cavaliere, G., Manca, C., Mollica, M. P., & Banni, S. (2023). Reducing Dietary Polyunsaturated to Saturated Fatty Acids Ratio Improves Lipid and Glucose Metabolism in Obese Zucker Rats. Nutrients, 15(22), 4761. https://doi.org/10.3390/nu15224761