
How Does CBG Administration Affect Sphingolipid Deposition in the Liver of Insulin-Resistant Rats?

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
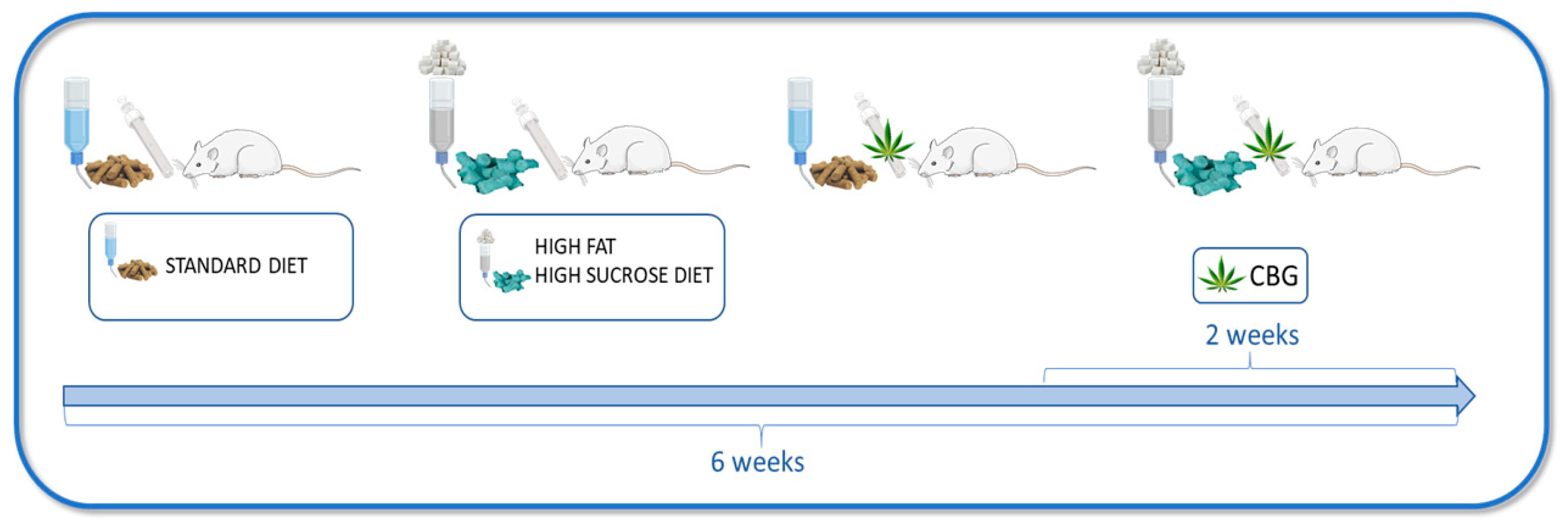
2.1. Animals and Experimental Protocol
2.2. Insulin Tolerance Test
2.3. Determination of the Plasma and Tissue Sphingolipid Concentration
2.4. Determination of the Plasma and Tissue Sphingomyelin Concentration
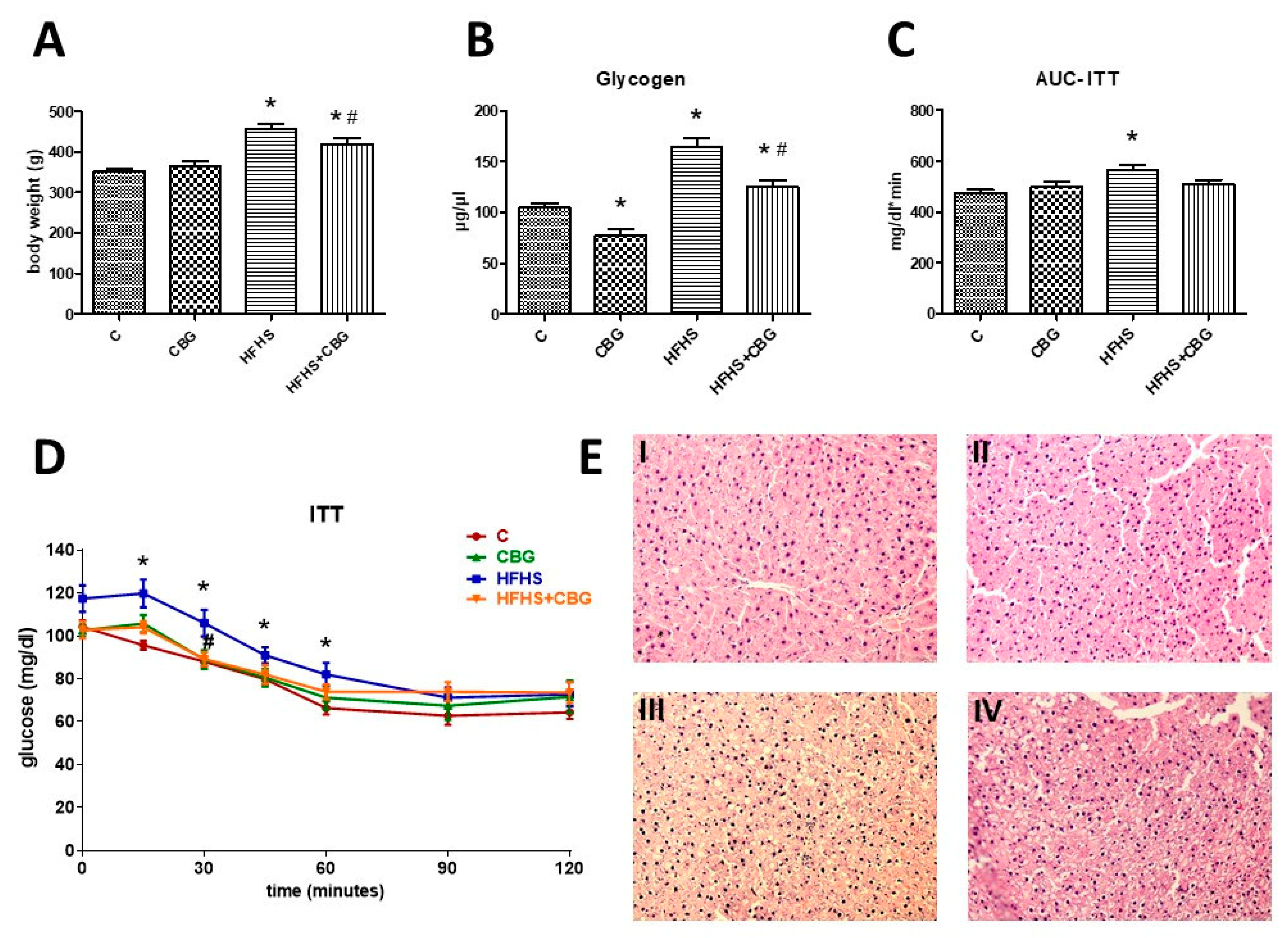
2.5. Determination of Glycogen Concentration
2.6. Western Blotting
2.7. Histology Analysis
2.8. Statistical Analysis
3. Results
3.1. The Effect of CBG Treatment on Sphingolipid Concentration in the Liver
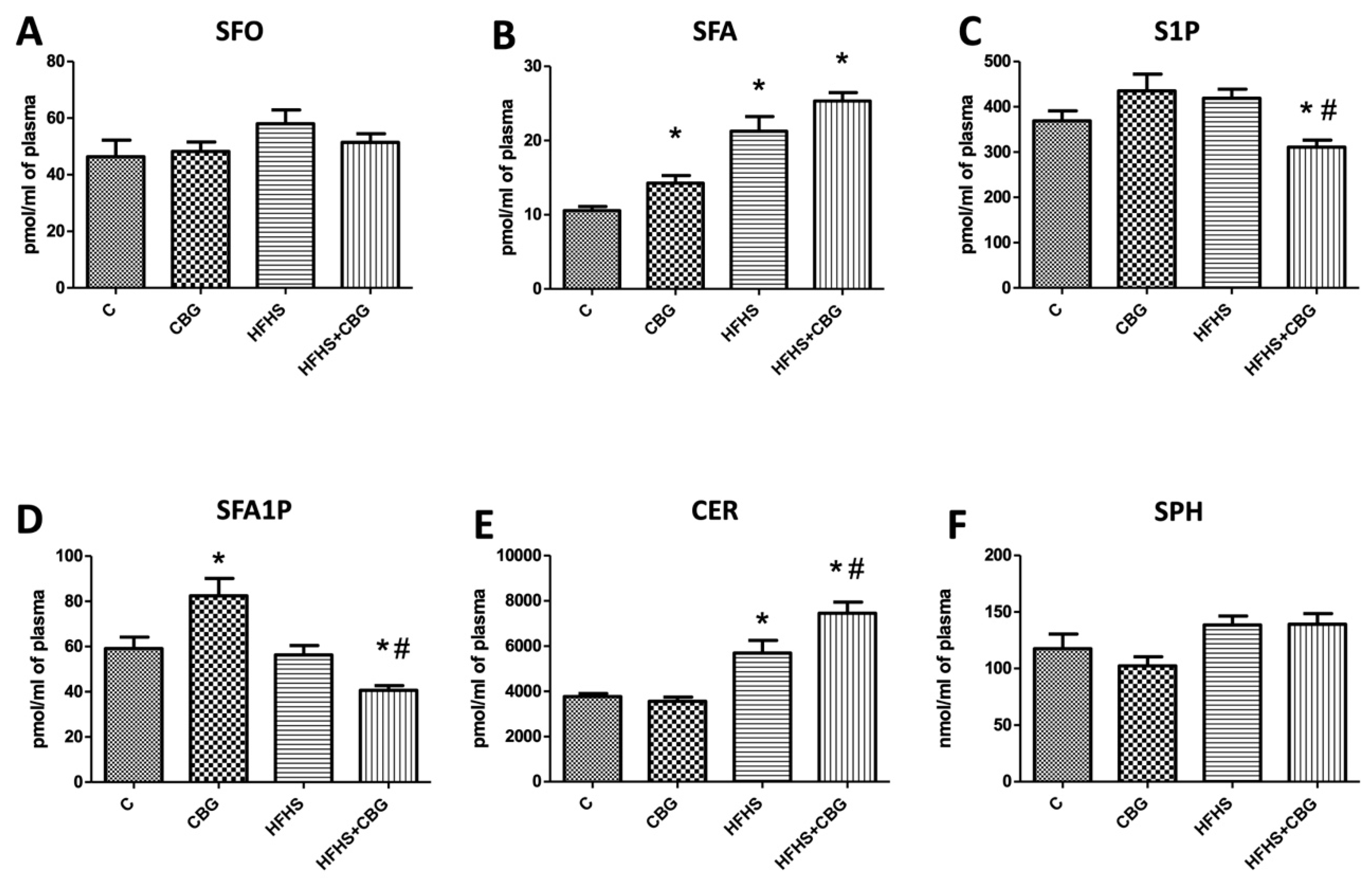
3.2. The Effect of CBG Treatment on Sphingolipid Concentration in the Plasma
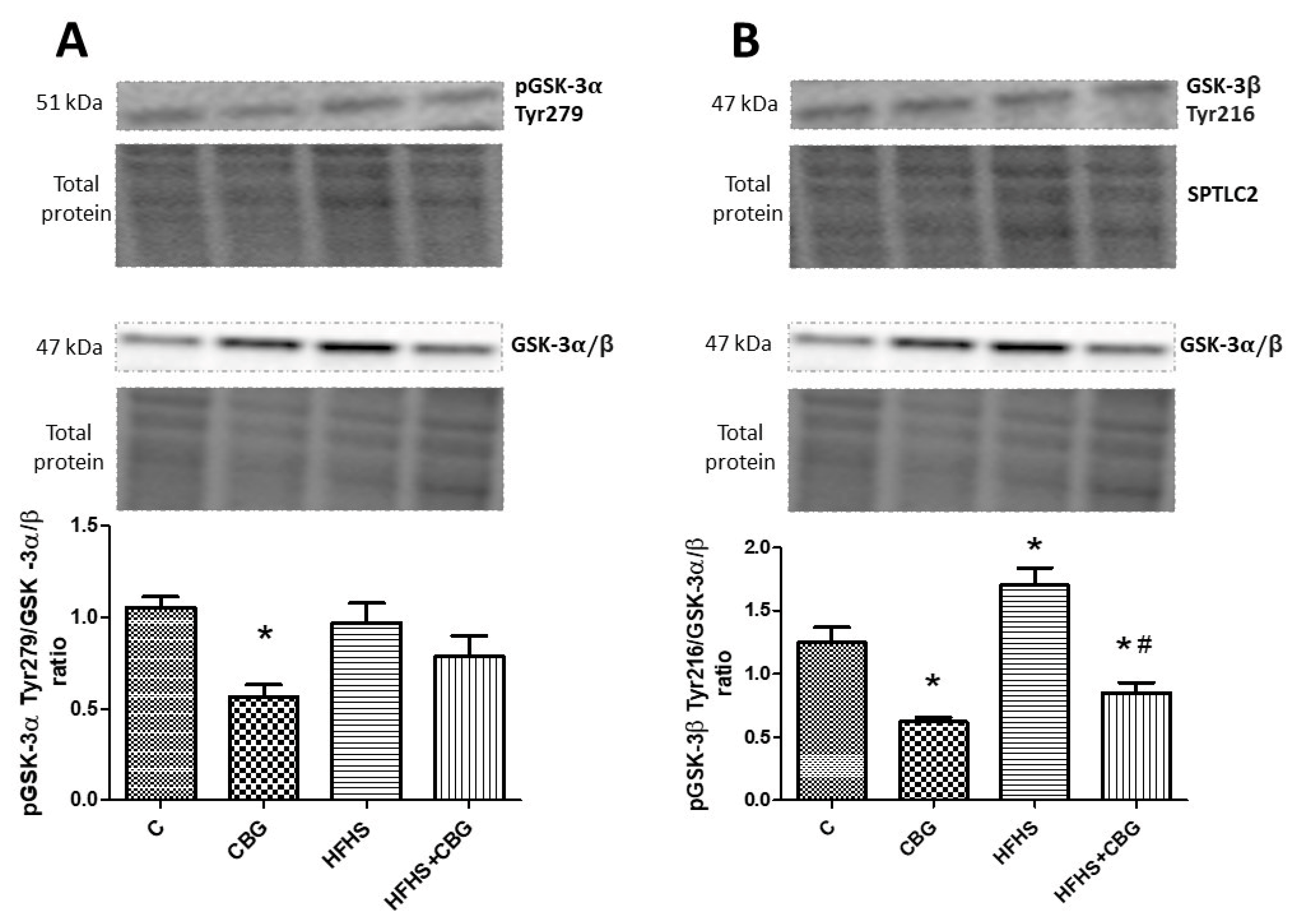
3.3. The Effect of CBG Treatment on the Expression of Enzymes Specific to the De Novo Ceramide Synthesis Pathway
3.4. The Effect of CBG Treatment on the Expression of Enzymes That Share Salvage and De Novo Ceramide Synthesis Pathways
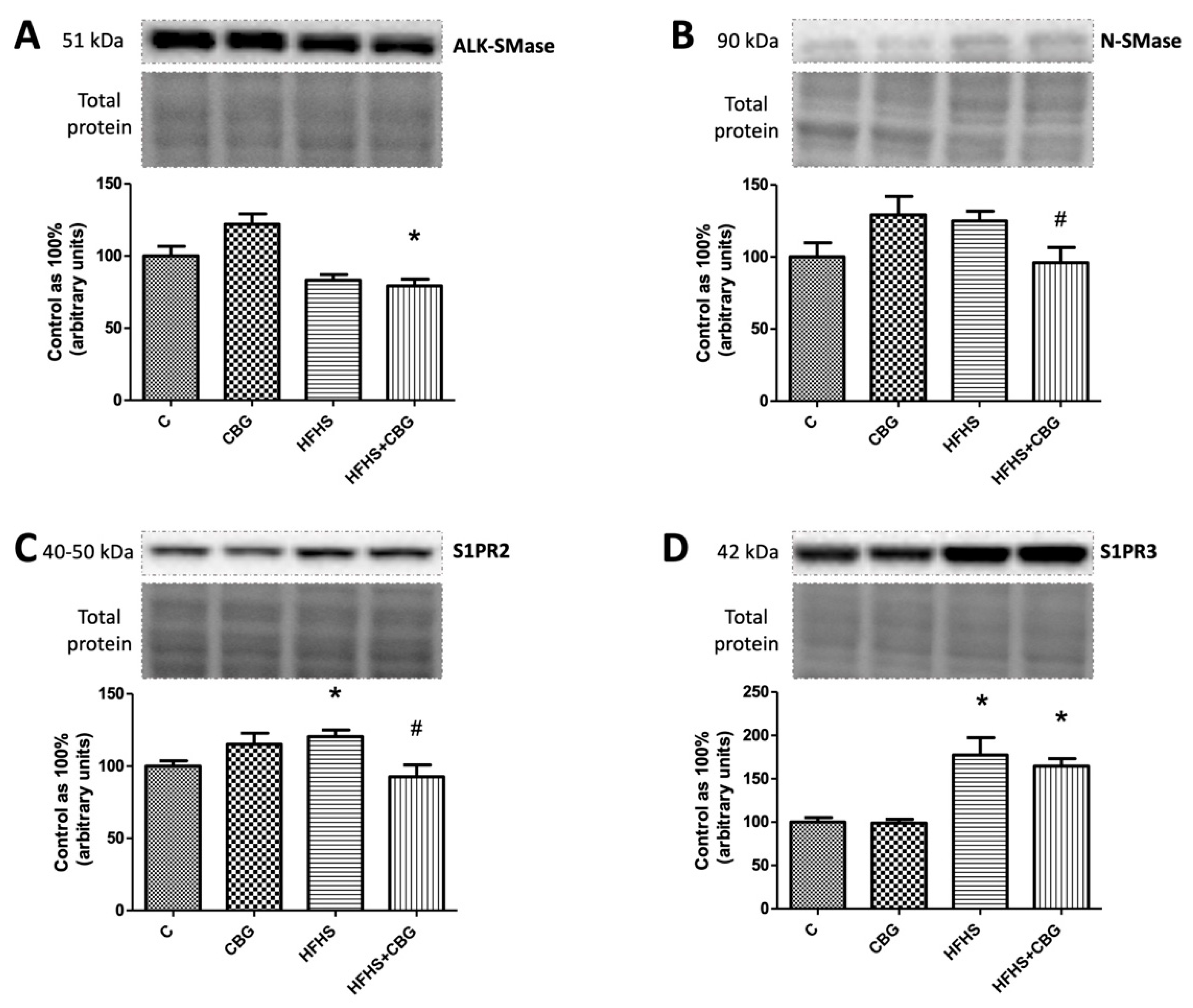
3.5. The Effect of CBG Treatment on the Expression of Enzymes from the Ceramide Catabolic Pathway
3.6. The Effect of CBG Treatment on the Expression of Enzymes from Hydrolysis of the Sphingomyelin Pathway and Receptors for S1P
3.7. The Effect of CBG Treatment on the Expression of S1P-Degrading Enzyme and Sphingolipid Transporting Proteins
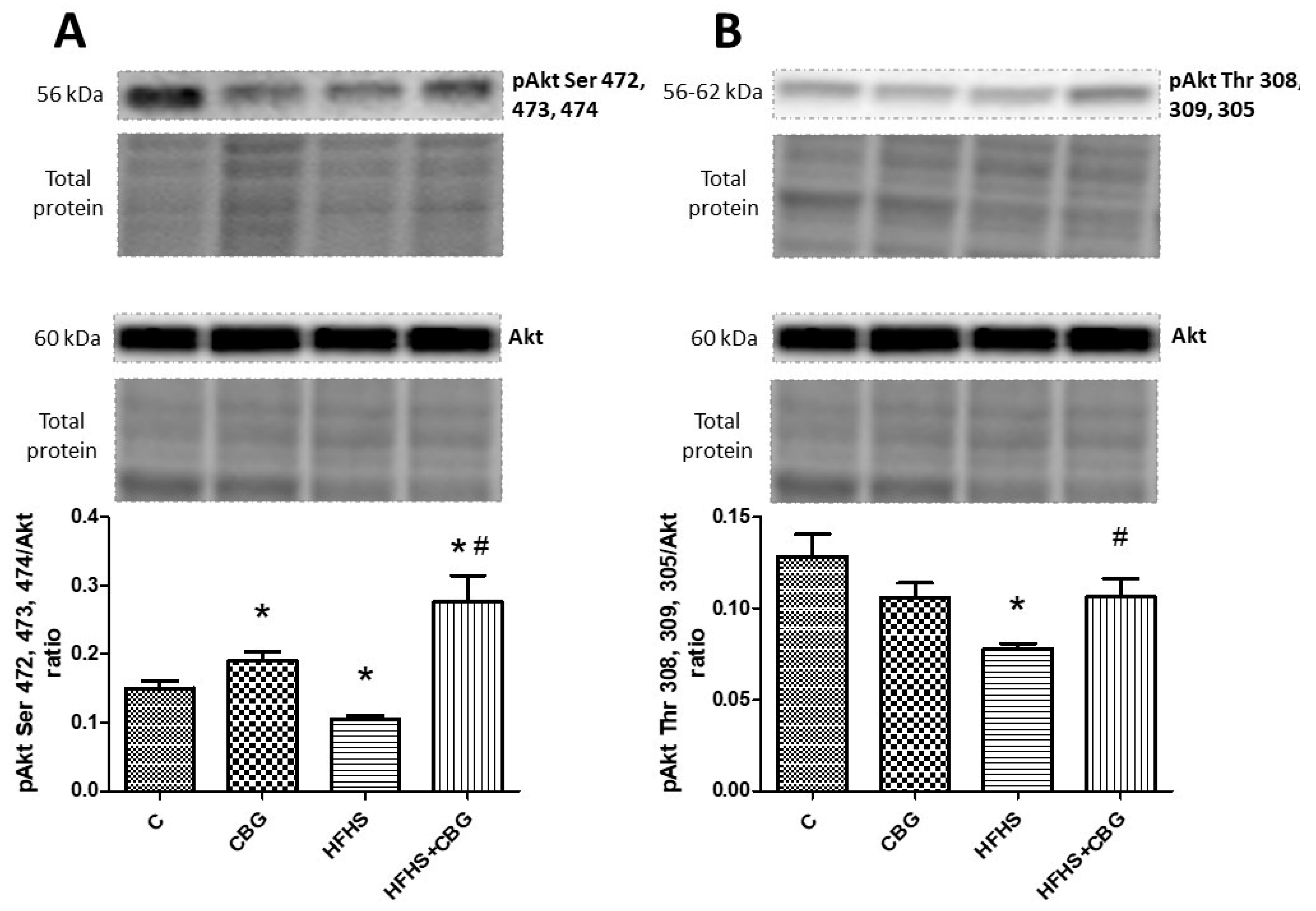
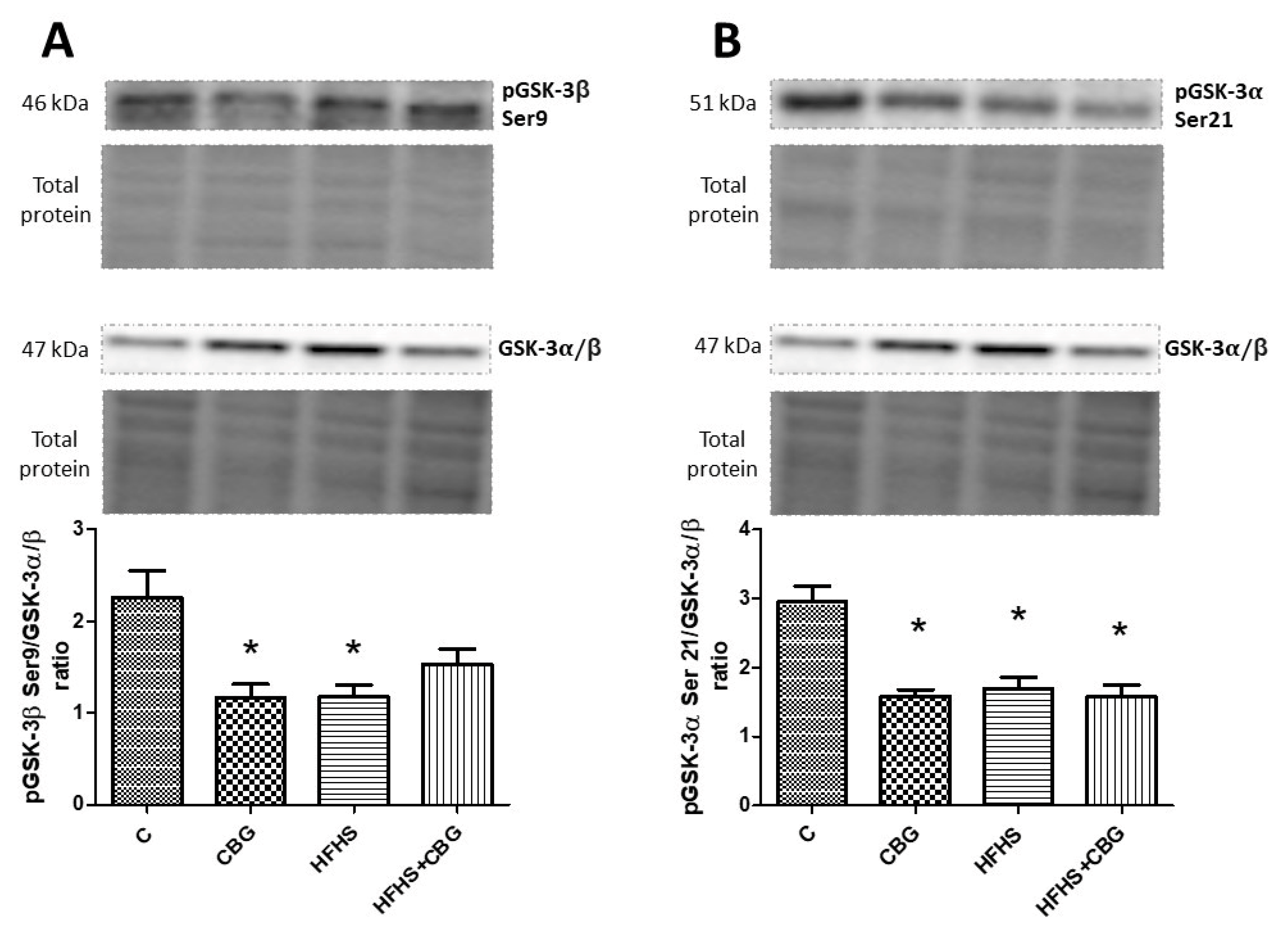
3.8. The Effect of CBG Treatment on the Expression of Proteins from the Insulin Signaling Pathway
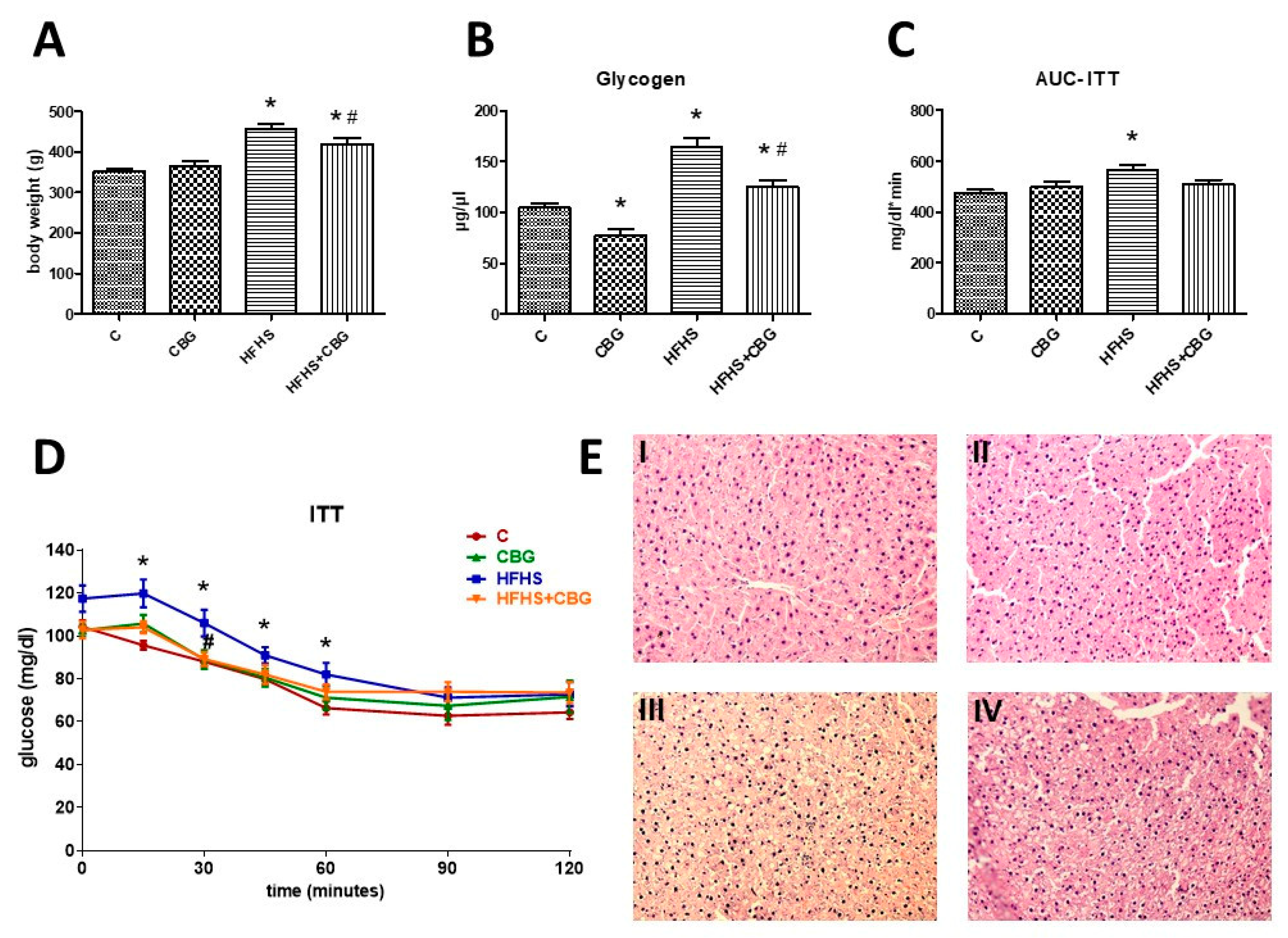
3.9. The Effect of CBG Treatment on Insulin Sensitivity and Body Weight
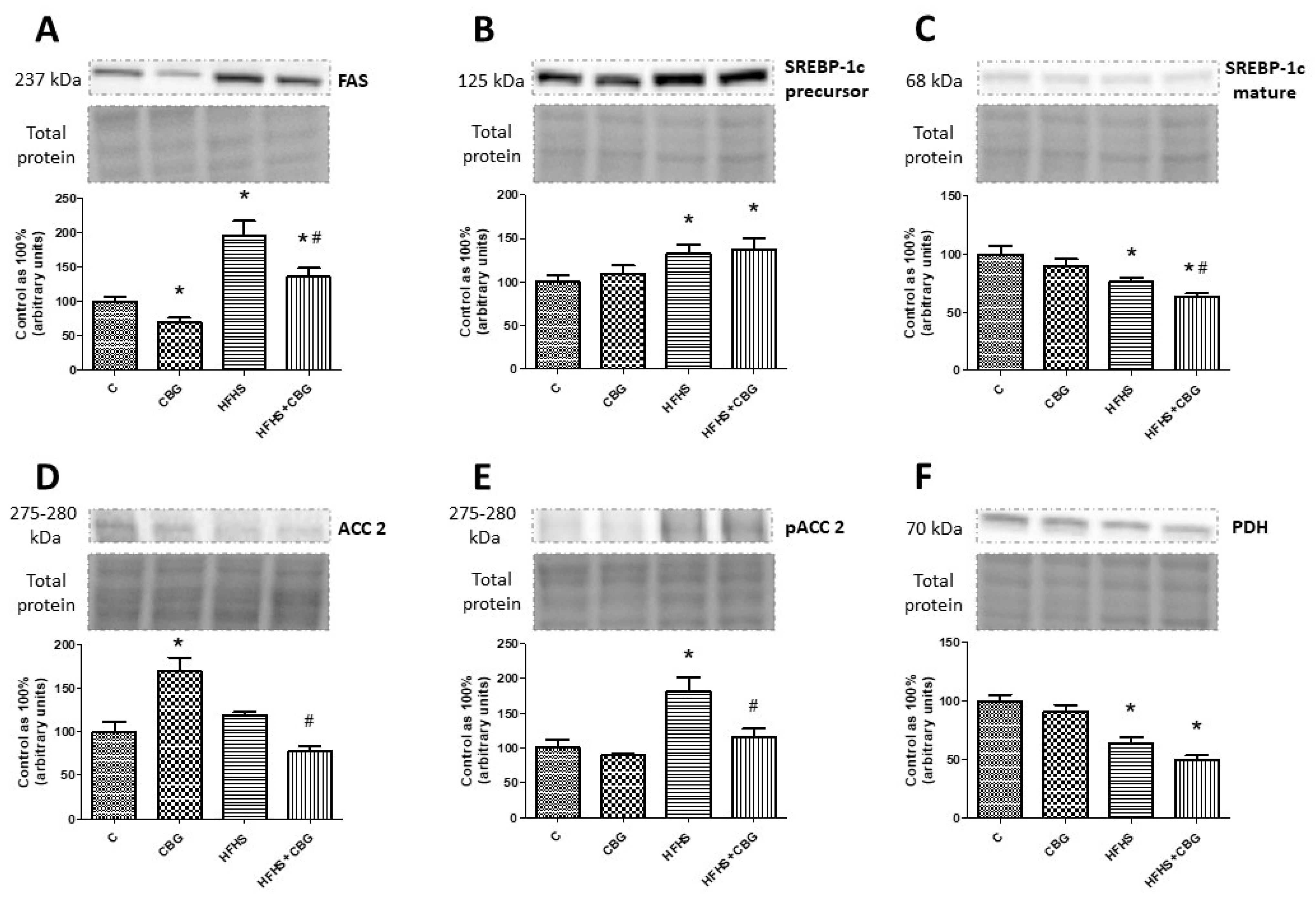
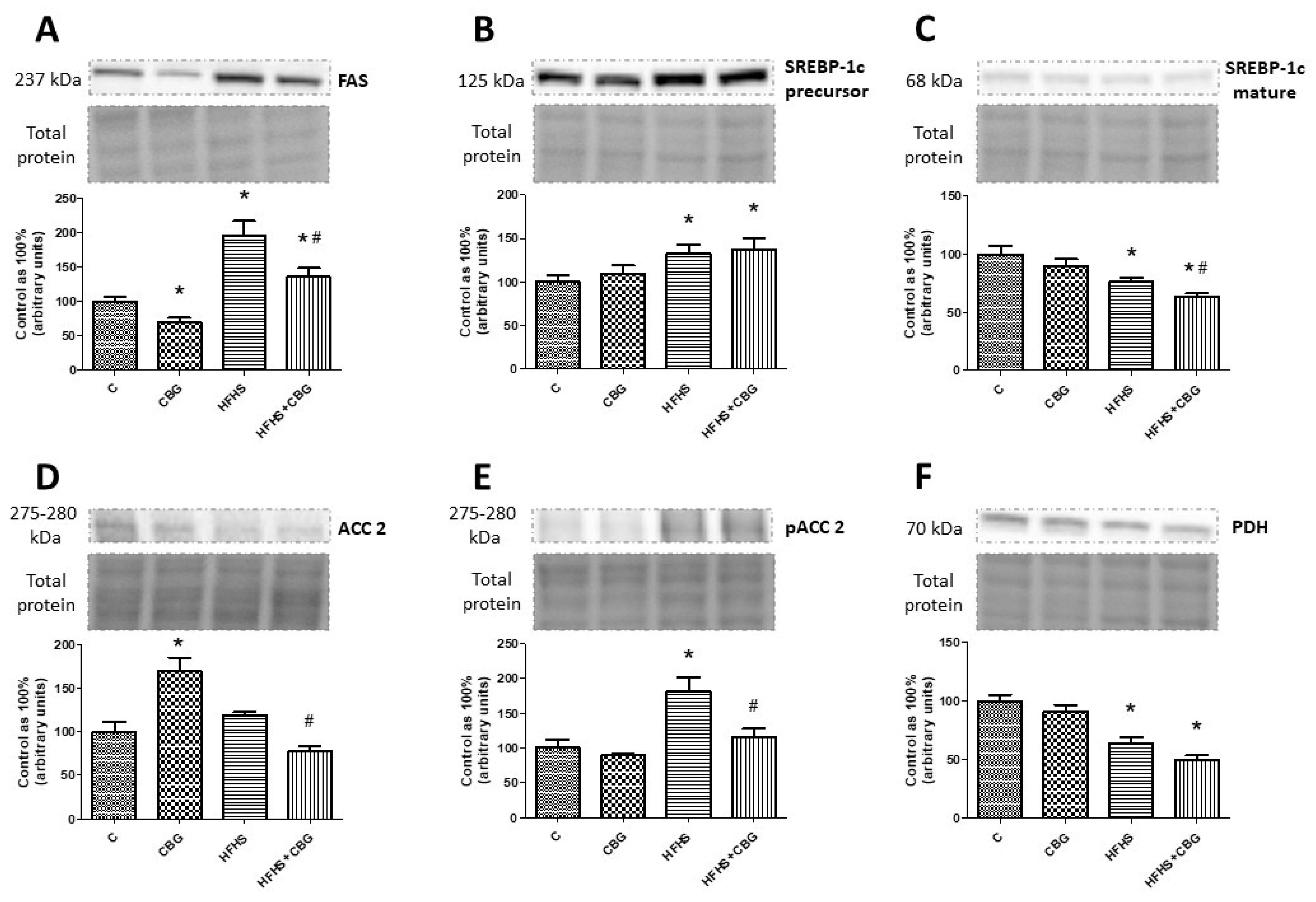
3.10. The Effect of CBG Treatment on the Expression of Proteins Associated with Fatty Acids and Glucose Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CBG | cannabigerol |
| IR | insulin resistance |
| HFHS | high-fat, high-sucrose diet |
| TRP | transient receptor potential channel |
| COX-1, COX-2 | cyclooxygenase 1,2 |
| 5-HT1A | the serotonin 1A receptor |
| PPARγ | peroxisome proliferator-activated nuclear receptor γ |
| NF-kb | nuclear factor kb |
| IκB-α | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha |
| SOD-1 | superoxide dismutase 1 |
| iNOS] | inducible nitric oxide synthase |
| MAFLD | metabolic-associated fatty liver disease |
| TLC | Thin-Layer Chromatography |
| GLC | gas–liquid chromatography |
| RIPA | radioimmunoprecipitation assay |
| BCA | bicinchoninic acid |
| PVDF | polyvinylidene fluoride |
| SFO | sphingosine |
| SFA | sphinganine |
| SPH | sphingomyelin |
| S1P | sphingosine-1-phosphate |
| SFA1P | sphinganine-1-phosphate |
| CER | ceramide |
| SPTLC1, SPTLC2 | serine palmitoyltransferase 1,2 |
| SPHK1, SPHK2 | sphingosine kinase 1,2 |
| CerS2, CerS4, CerS5, CerS6 | dihydroceramide synthase 2,4,5,6 |
| ASAH1 | acid ceramidase |
| ASAH2 | neutral ceramidase |
| ASAH3 | alkaline ceramidase |
| N-SMase | neutral sphingomyelinase |
| Alk-SMase | alkaline sphingomyelinase |
| S1PR2, S1PR3 | sphingosine-1-phosphate receptor 2,3 |
| SGPL1 | sphingosine-1-phosphate lyase 1 |
| CERT | ceramide transport protein |
| SPSN2 | sphingolipid transporter 2/sphingosine phosphate-specific phosphatase 2 |
| ABCA1 | ATP-binding cassette transporter |
| MCD | methionine/choline-deficient diet |
| LCcer | long-chain ceramides |
| VLCcer | very long-chain ceramides |
| ER | endoplasmic reticulum |
| ApoM | apolipoprotein M |
| HDL | high-density lipoprotein |
| LPS | Lipopolysaccharides |
References
- ElSohly, M.A.; Slade, D. Chemical Constituents of Marijuana: The Complex Mixture of Natural Cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Varani, K.; Reyes-Resina, I.; de Medina, V.S.; Rivas-Santisteban, R.; Callado, C.S.C.; Vincenzi, F.; Casano, S.; Ferreiro-Vera, C.; Canela, E.I.; et al. Cannabigerol Action at Cannabinoid CB1 and CB2 Receptors and at CB1–CB2 Heteroreceptor Complexes. Front. Pharmacol. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.E.; Williams, C.M.; Iversen, L.; Whalley, B.J. Molecular Pharmacology of Phytocannabinoids. Prog. Chem. Org. Nat. Prod. 2017, 103, 61–101. [Google Scholar] [CrossRef]
- Calapai, F.; Cardia, L.; Esposito, E.; Ammendolia, I.; Mondello, C.; lo Giudice, R.; Gangemi, S.; Calapai, G.; Mannucci, C. Pharmacological Aspects and Biological Effects of Cannabigerol and Its Synthetic Derivatives. Evid. Based Complement. Alternat Med. 2022, 2022, 3336516. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Gugliandolo, A.; Trubiani, O.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Cannabinoid CB2 Receptors Are Involved in the Protection of RAW264.7 Macrophages against the Oxidative Stress: An In Vitro Study. Eur. J. Histochem. 2017, 61, 2749. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Fasolino, I.; Romano, B.; Capasso, R.; Maiello, F.; Coppola, D.; Orlando, P.; Battista, G.; Pagano, E.; di Marzo, V.; et al. Beneficial Effect of the Non-Psychotropic Plant Cannabinoid Cannabigerol on Experimental Inflammatory Bowel Disease. Biochem. Pharmacol. 2013, 85, 1306–1316. [Google Scholar] [CrossRef]
- Nachnani, R.; Raup-Konsavage, W.M.; Vrana, K.E. The Pharmacological Case for Cannabigerol. J. Pharmacol. Exp. Ther. 2021, 376, 204–212. [Google Scholar] [CrossRef]
- Granja, A.G.; Carrillo-Salinas, F.; Pagani, A.; Gómez-Cañas, M.; Negri, R.; Navarrete, C.; Mecha, M.; Mestre, L.; Fiebich, B.L.; Cantarero, I.; et al. A Cannabigerol Quinone Alleviates Neuroinflammation in a Chronic Model of Multiple Sclerosis. J. Neuroimmune Pharmacol. 2012, 7, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castro, C.; Garvey, W.T. Intracellular Lipid Accumulation in Liver and Muscle and the Insulin Resistance Syndrome. Endocrinol. Metab. Clin. North Am. 2008, 37, 841. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in Insulin Resistance: Insights into Mechanisms and Therapeutic Strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Qu, H.Q.; Li, Q.; Rentfro, A.R.; Fisher-Hoch, S.P.; McCormick, J.B. The Definition of Insulin Resistance Using HOMA-IR for Americans of Mexican Descent Using Machine Learning. PLoS ONE 2011, 6, e21041. [Google Scholar] [CrossRef]
- Fahed, M.; Abou Jaoudeh, M.G.; Merhi, S.; Mosleh, J.M.B.; Ghadieh, R.; Ghadieh, R.; al Hayek, S.; al Hayek, S.; el Hayek Fares, J.E. Evaluation of Risk Factors for Insulin Resistance: A Cross Sectional Study among Employees at a Private University in Lebanon. BMC Endocr. Disord. 2020, 20, 85. [Google Scholar] [CrossRef]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Fontanesi, F.; Merscher, S.; Fornoni, A. The Vicious Cycle of Renal Lipotoxicity and Mitochondrial Dysfunction. Front. Physiol. 2020, 11, 732. [Google Scholar] [CrossRef] [PubMed]
- Zywno, H.; Bzdega, W.; Kolakowski, A.; Kurzyna, P.; Harasim-Symbor, E.; Sztolsztener, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The Influence of Coumestrol on Sphingolipid Signaling Pathway and Insulin Resistance Development in Primary Rat Hepatocytes. Biomolecules 2021, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Konstantynowicz-Nowicka, K.; Harasim, E.; Baranowski, M.; Chabowski, A. New Evidence for the Role of Ceramide in the Development of Hepatic Insulin Resistance. PLoS ONE 2015, 10, e0116858. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Marco, E.M.; Romero-Zerbo, S.Y.; Viveros, M.P.; Bermudez-Silva, F.J. The Role of the Endocannabinoid System in Eating Disorders: Pharmacological Implications. Behav. Pharmacol. 2012, 23, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Wargent, E.T.; Zaibi, M.S.; Silvestri, C.; Hislop, D.C.; Stocker, C.J.; Stott, C.G.; Guy, G.W.; Duncan, M.; Di Marzo, V.; Cawthorne, M.A. The Cannabinoid Δ(9)-Tetrahydrocannabivarin (THCV) Ameliorates Insulin Sensitivity in Two Mouse Models of Obesity. Nutr. Diabetes 2013, 3, e68. [Google Scholar] [CrossRef] [PubMed]
- Baranowski, M.; Zabielski, P.; Blachnio, A.; Gorski, J. Effect of Exercise Duration on Ceramide Metabolism in the Rat Heart. Acta Physiol. 2008, 192, 519–529. [Google Scholar] [CrossRef]
- FOLCH, J.; LEES, M.; SLOANE STANLEY, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Stain-Free Total Protein Staining Is a Superior Loading Control to β-Actin for Western Blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis Sativa: A Comprehensive Ethnopharmacological Review of a Medicinal Plant with a Long History. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef]
- Dörnyei, G.; Vass, Z.; Juhász, C.B.; Nádasy, G.L.; Hunyady, L.; Szekeres, M. Role of the Endocannabinoid System in Metabolic Control Processes and in the Pathogenesis of Metabolic Syndrome: An Update. Biomedicines 2023, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown; Sringer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Choi, S.; Snider, A.J. Sphingolipids in High Fat Diet and Obesity-Related Diseases. Mediat. Inflamm. 2015, 2015, 520618. [Google Scholar] [CrossRef] [PubMed]
- Torretta, E.; Barbacini, P.; Al-Daghri, N.M.; Gelfi, C. Sphingolipids in Obesity and Correlated Comorbidities: The Contribution of Gender, Age and Environment. Int. J. Mol. Sci. 2019, 20, 5901. [Google Scholar] [CrossRef] [PubMed]
- Berk, K.; Konstantynowicz-Nowicka, K.; Charytoniuk, T.; Harasim-Symbor, E.; Chabowski, A. Distinct Effects of Cannabidiol on Sphingolipid Metabolism in Subcutaneous and Visceral Adipose Tissues Derived from High-Fat-Diet-Fed Male Wistar Rats. Int. J. Mol. Sci. 2022, 23, 5382. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Godlewski, G.; Liu, J.; Tam, J.; Jourdan, T.; Mukhopadhyay, B.; Harvey-White, J.; Kunos, G. Hepatic Cannabinoid-1 Receptors Mediate Diet-Induced Insulin Resistance by Increasing de Novo Synthesis of Long-Chain Ceramides. Hepatology 2014, 59, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A. Mast Cell Infiltration in Liver of Cannabigerol Treated Methionine/Choline Deficient Diet Induced Mice Non-Alcoholic Steatohepatitis Model. Bachelor’s Thesis, University of Northern Colorado, Greeley, CO, USA, 2021. [Google Scholar]
- Rinella, M.E.; Green, R.M. The Methionine-Choline Deficient Dietary Model of Steatohepatitis Does Not Exhibit Insulin Resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Bielawiec, P.; Harasim-Symbor, E.; Sztolsztener, K.; Konstantynowicz-Nowicka, K.; Chabowski, A. Attenuation of Oxidative Stress and Inflammatory Response by Chronic Cannabidiol Administration Is Associated with Improved N-6/n-3 Pufa Ratio in the White and Red Skeletal Muscle in a Rat Model of High-Fat Diet-Induced Obesity. Nutrients 2021, 13, 1603. [Google Scholar] [CrossRef] [PubMed]
- Charytoniuk, T.; Sztolsztener, K.; Harasim-Symbor, E.; Berk, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. Cannabidiol—A Phytocannabinoid That Widely Affects Sphingolipid Metabolism under Conditions of Brain Insulin Resistance. Biomed. Pharmacother. 2021, 142, 112057. [Google Scholar] [CrossRef] [PubMed]
- Charytoniuk, T.; Sztolsztener, K.; Bielawiec, P.; Chabowski, A.; Konstantynowicz-Nowicka, K.; Harasim-Symbor, E. Cannabidiol Downregulates Myocardial De Novo Ceramide Synthesis Pathway in a Rat Model of High-Fat Diet-Induced Obesity. Int. J. Mol. Sci. 2022, 23, 2232. [Google Scholar] [CrossRef]
- Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of Muscle Insulin Resistance and the Cross-talk with Liver and Adipose Tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef]
- Wiciński, M.; Fajkiel-Madajczyk, A.; Kurant, Z.; Gryczka, K.; Kurant, D.; Szambelan, M.; Malinowski, B.; Falkowski, M.; Zabrzyński, J.; Słupski, M. The Use of Cannabidiol in Metabolic Syndrome-An Opportunity to Improve the Patient’s Health or Much Ado about Nothing? J. Clin. Med. 2023, 12, 4620. [Google Scholar] [CrossRef]
- Tidhar, R.; Futerman, A.H. The Complexity of Sphingolipid Biosynthesis in the Endoplasmic Reticulum. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 2511–2518. [Google Scholar] [CrossRef]
- Bielawiec, P.; Harasim-Symbor, E.; Konstantynowicz-Nowicka, K.; Sztolsztener, K.; Chabowski, A. Chronic Cannabidiol Administration Attenuates Skeletal Muscle De Novo Ceramide Synthesis Pathway and Related Metabolic Effects in a Rat Model of High-Fat Diet-Induced Obesity. Biomolecules 2020, 10, 1241. [Google Scholar] [CrossRef]
- Taltavull, N.; Ras, R.; Mariné, S.; Romeu, M.; Giralt, M.; Méndez, L.; Medina, I.; Ramos-Romero, S.; Torres, J.L.; Nogués, M.R. Protective Effects of Fish Oil on Pre-Diabetes: A Lipidomic Analysis of Liver Ceramides in Rats. Food Funct. 2016, 7, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Angrish, M.M.; McQueen, C.A.; Cohen-Hubal, E.; Bruno, M.; Ge, Y.; Chorley, B.N. Editor’s Highlight: Mechanistic Toxicity Tests Based on an Adverse Outcome Pathway Network for Hepatic Steatosis. Toxicol. Sci. 2017, 159, 159–169. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-Phosphate: Lipid Signaling in Pathology and Therapy. Science 2019, 366, 323. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.; Zhang, C.; Ding, P.; Tian, S.; Chen, J.; Ji, G.; Wu, T. Sphingosine 1-Phosphate Receptor, a New Therapeutic Direction in Different Diseases. Biomed. Pharmacother. 2022, 153, 113341. [Google Scholar] [CrossRef]
- Kleuser, B. Divergent Role of Sphingosine 1-Phosphate in Liver Health and Disease. Int. J. Mol. Sci. 2018, 19, 722. [Google Scholar] [CrossRef]
- Kurano, M.; Tsuneyama, K.; Morimoto, Y.; Shimizu, T.; Jona, M.; Kassai, H.; Nakao, K.; Aiba, A.; Yatomi, Y. Apolipoprotein M Protects Lipopolysaccharide-Treated Mice from Death and Organ Injury. Thromb. Haemost. 2018, 118, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Freeman, C.M.; Schuster, R.M.; Japtok, L.; Kleuser, B.; Edwards, M.J.; Gulbins, E.; Lentsch, A.B. Hepatocyte Exosomes Mediate Liver Repair and Regeneration via Sphingosine-1-Phosphate. J. Hepatol. 2016, 64, 60–68. [Google Scholar] [CrossRef]
- Zheng, D.M.; Kitamura, T.; Ikejima, K.; Enomoto, N.; Yamashina, S.; Suzuki, S.; Takei, Y.; Sato, N. Sphingosine 1-Phosphate Protects Rat Liver Sinusoidal Endothelial Cells from Ethanol-Induced Apoptosis: Role of Intracellular Calcium and Nitric Oxide. Hepatology 2006, 44, 1278–1287. [Google Scholar] [CrossRef]
- Osawa, Y.; Uchinami, H.; Bielawski, J.; Schwabe, R.F.; Hannun, Y.A.; Brenner, D.A. Roles for C16-Ceramide and Sphingosine 1-Phosphate in Regulating Hepatocyte Apoptosis in Response to Tumor Necrosis Factor-α. J. Biol. Chem. 2005, 280, 27879–27887. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ikeda, H.; Uranbileg, B.; Kurano, M.; Saigusa, D.; Aoki, J.; Maki, H.; Kudo, H.; Hasegawa, K.; Kokudo, N.; et al. Sphingosine Kinase-1, S1P Transporter Spinster Homolog 2 and S1P2 MRNA Expressions Are Increased in Liver with Advanced Fibrosis in Human. Sci. Rep. 2016, 6, 32119. [Google Scholar] [CrossRef] [PubMed]
- Karimian, G.; Buist-Homan, M.; Schmidt, M.; Tietge, U.J.F.; de Boer, J.F.; Klappe, K.; Kok, J.W.; Combettes, L.; Tordjmann, T.; Faber, K.N.; et al. Sphingosine Kinase-1 Inhibition Protects Primary Rat Hepatocytes against Bile Salt-Induced Apoptosis. Biochim. Biophys. Acta 2013, 1832, 1922–1929. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bzdęga, W.; Kurzyna, P.F.; Harasim-Symbor, E.; Hołownia, A.; Chabowski, A.; Konstantynowicz-Nowicka, K. How Does CBG Administration Affect Sphingolipid Deposition in the Liver of Insulin-Resistant Rats? Nutrients 2023, 15, 4350. https://doi.org/10.3390/nu15204350
Bzdęga W, Kurzyna PF, Harasim-Symbor E, Hołownia A, Chabowski A, Konstantynowicz-Nowicka K. How Does CBG Administration Affect Sphingolipid Deposition in the Liver of Insulin-Resistant Rats? Nutrients. 2023; 15(20):4350. https://doi.org/10.3390/nu15204350
Chicago/Turabian StyleBzdęga, Wiktor, Piotr Franciszek Kurzyna, Ewa Harasim-Symbor, Adam Hołownia, Adrian Chabowski, and Karolina Konstantynowicz-Nowicka. 2023. "How Does CBG Administration Affect Sphingolipid Deposition in the Liver of Insulin-Resistant Rats?" Nutrients 15, no. 20: 4350. https://doi.org/10.3390/nu15204350
APA StyleBzdęga, W., Kurzyna, P. F., Harasim-Symbor, E., Hołownia, A., Chabowski, A., & Konstantynowicz-Nowicka, K. (2023). How Does CBG Administration Affect Sphingolipid Deposition in the Liver of Insulin-Resistant Rats? Nutrients, 15(20), 4350. https://doi.org/10.3390/nu15204350