

Effectiveness of Anthocyanin-Rich Sour Cherry Extract on Gliadin-Induced Caco-2 Barrier Damage

 ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Pepsin-Trypsin-Resistant Gliadin (PT-Gliadin)
2.3. Cell Culturing for Permeability Studies
2.4. Real-Time Monitoring of Cell Index (RTCA)
2.5. UV Spectroscopy and Determination of AC Adsorption by PT-Gliadin
2.6. Transepithelial Electrical Resistance (TEER) Measurements
2.7. Immunofluorescence Staining of ZO-1 and Occludin
2.8. Permeability Study
2.9. NF-κB Pathway Activation Study
2.10. Measurement of TNF-α, IFN-γ, and IL-8 Production with the ELISA Method
2.11. Assessment of Mitochondrial ROS
2.12. Statistical Analysis
3. Results
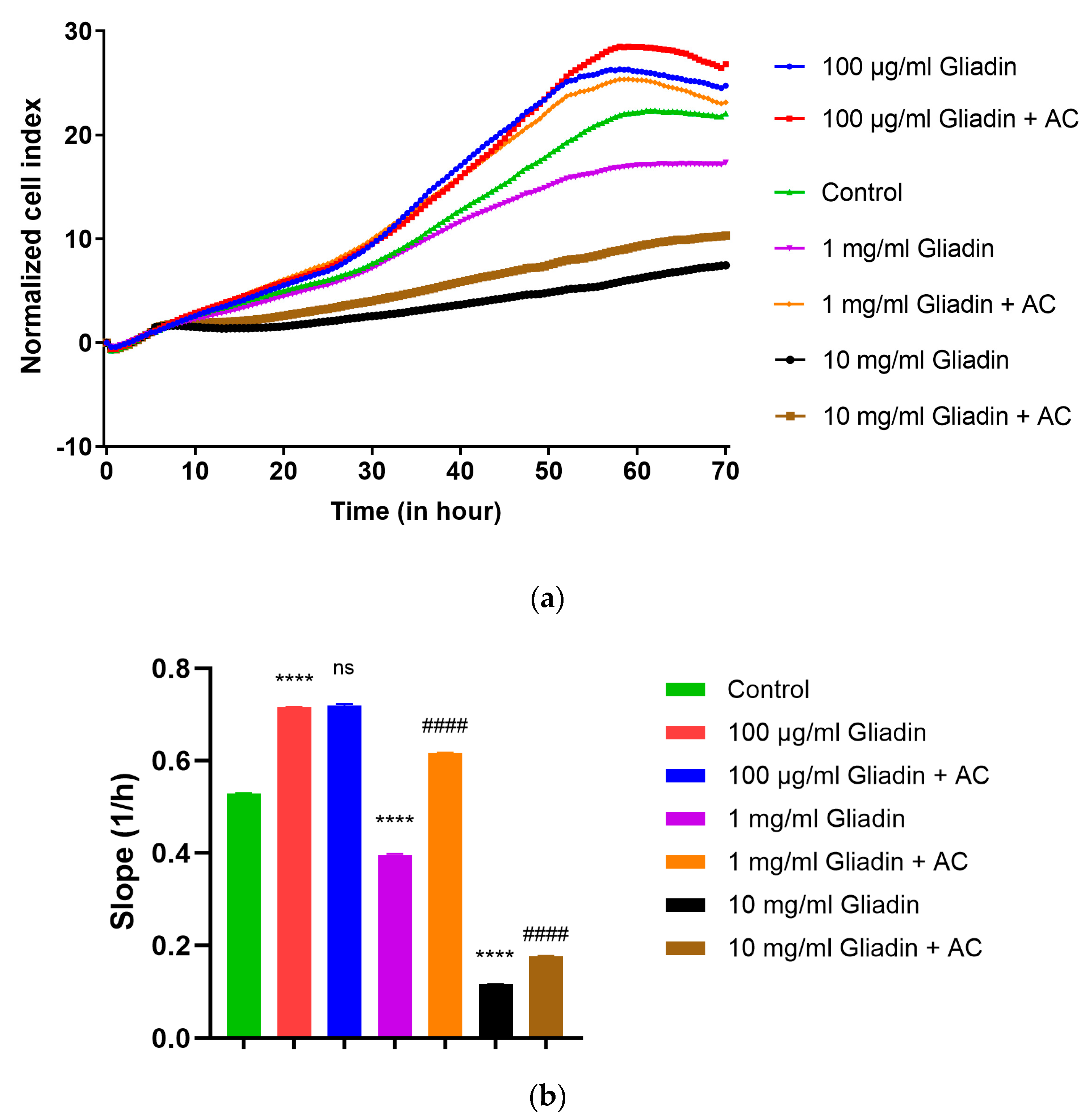
3.1. Real-Time Monitoring of Cell Index
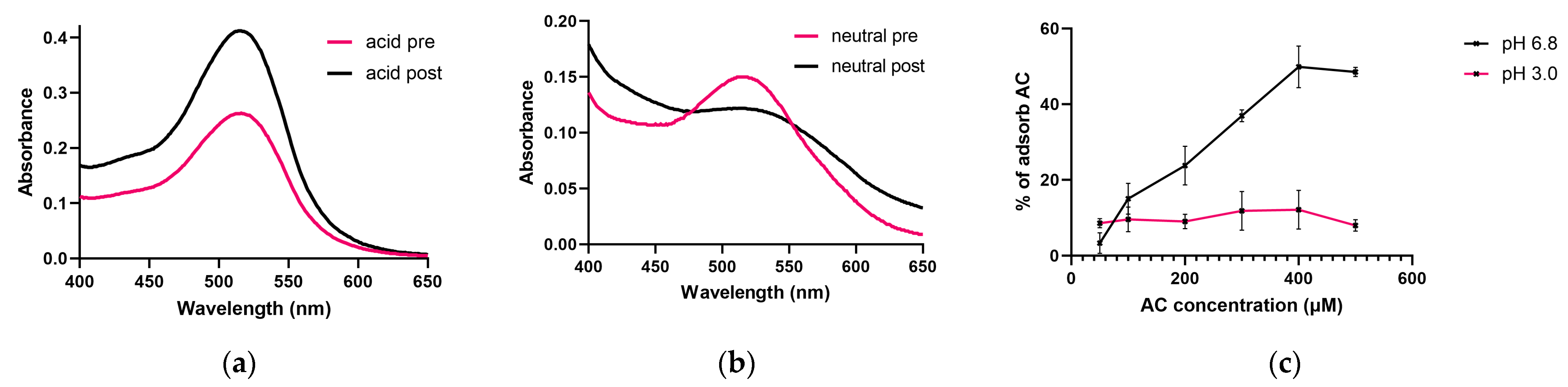
3.2. Investigation of the AC Adsorption of PT-Gliadin
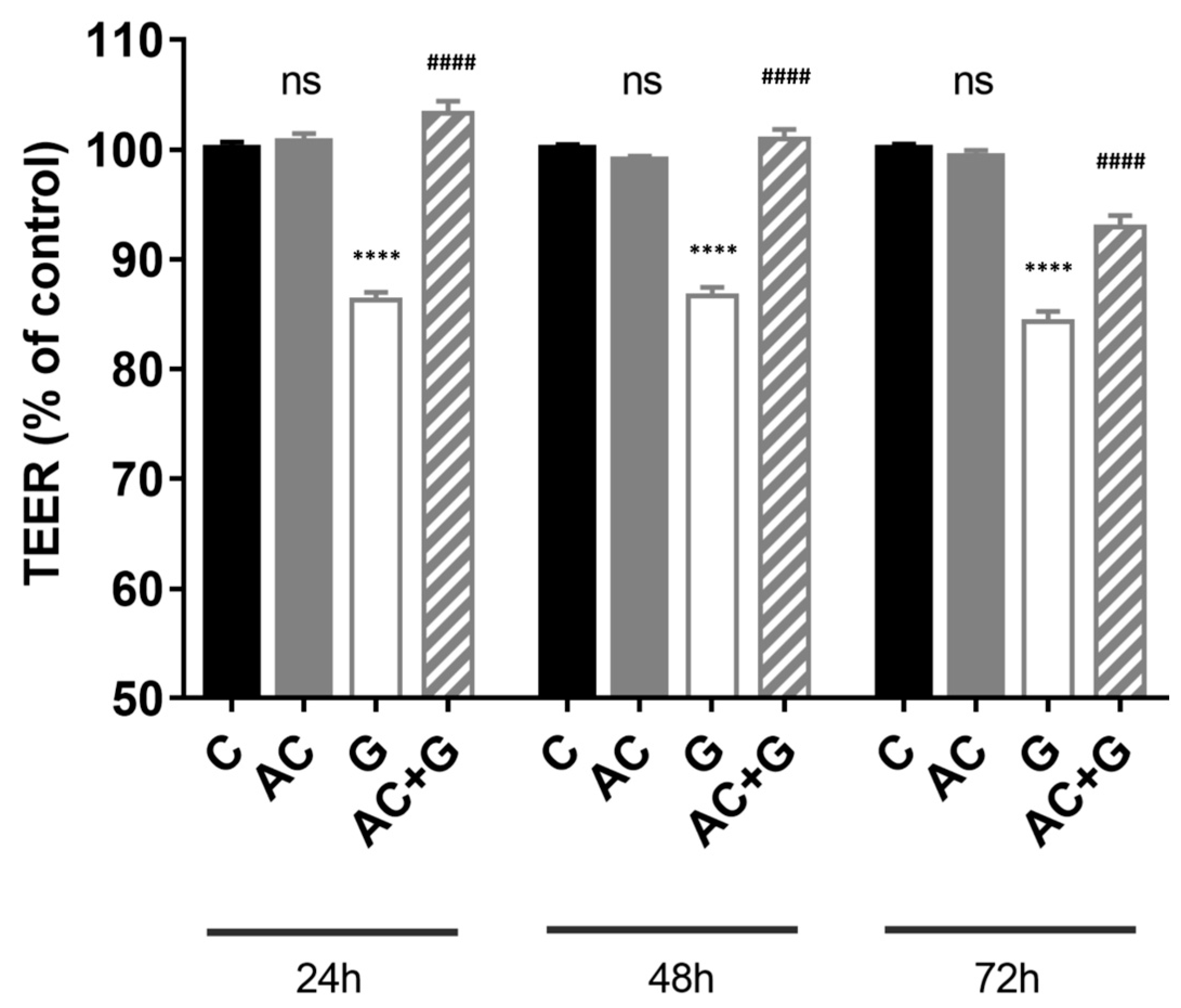
3.3. Measurement of Transepithelial Electric Resistance (TEER)
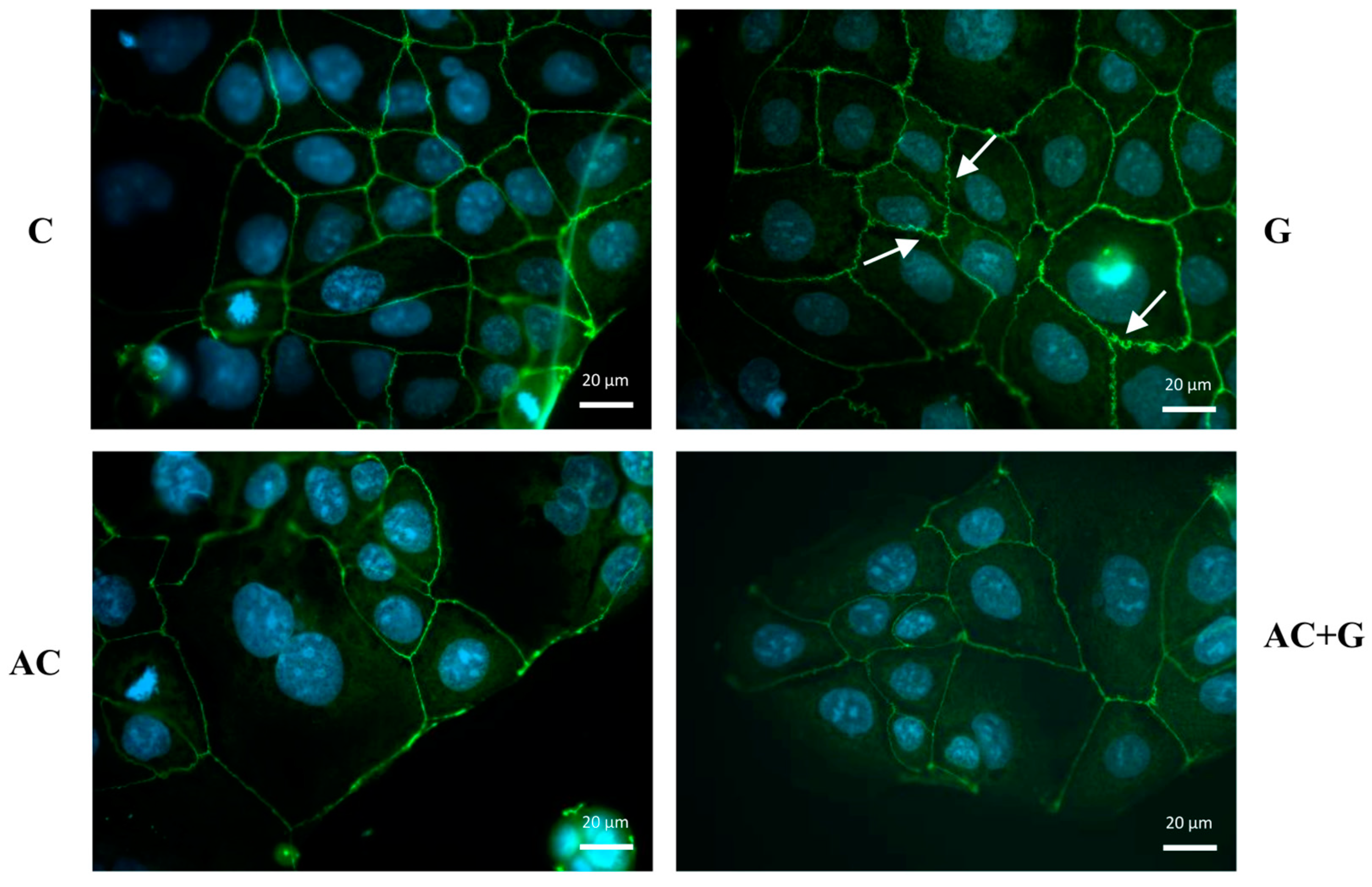
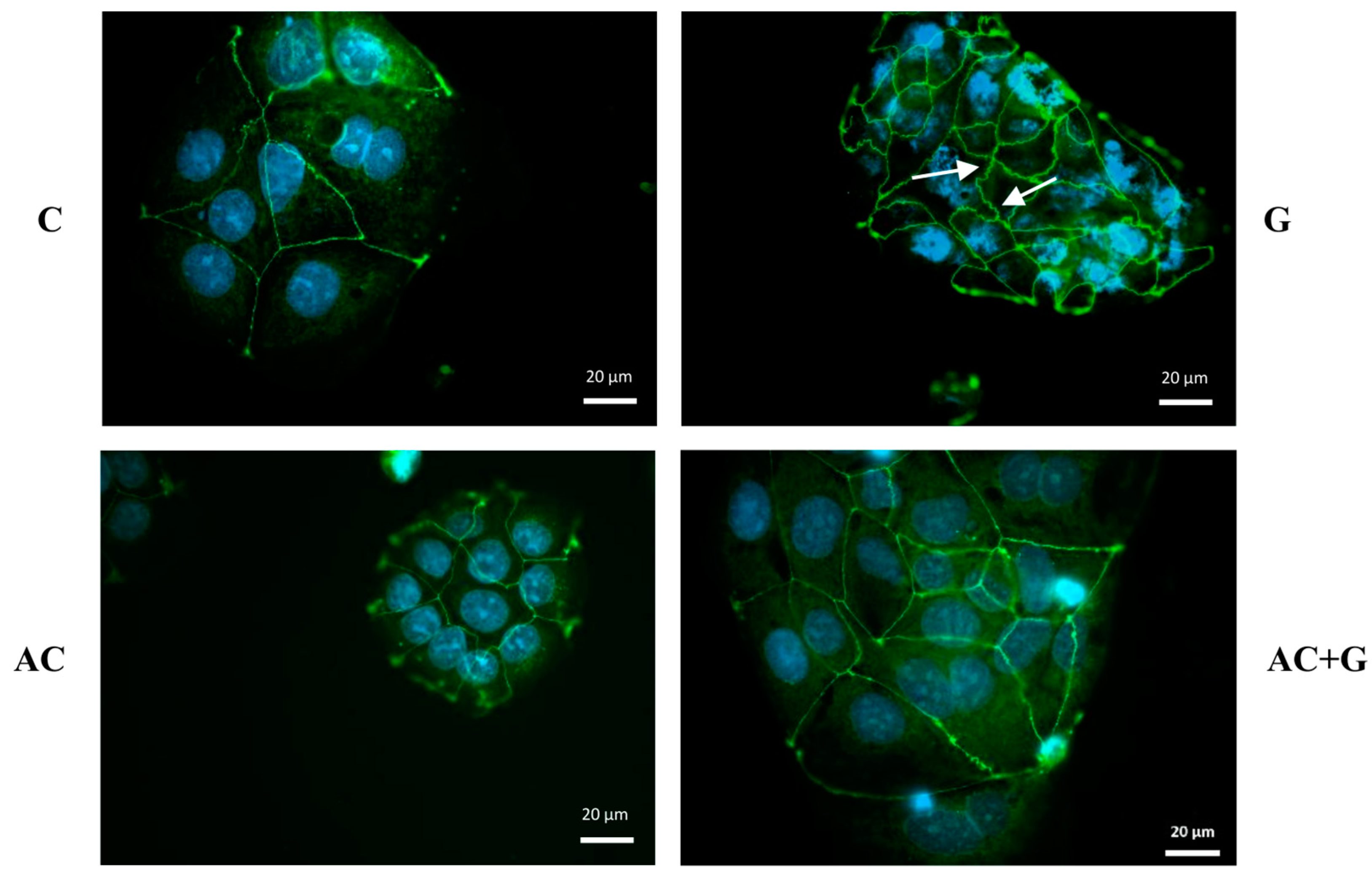
3.4. Immunohistochemical Staining of TJ Proteins
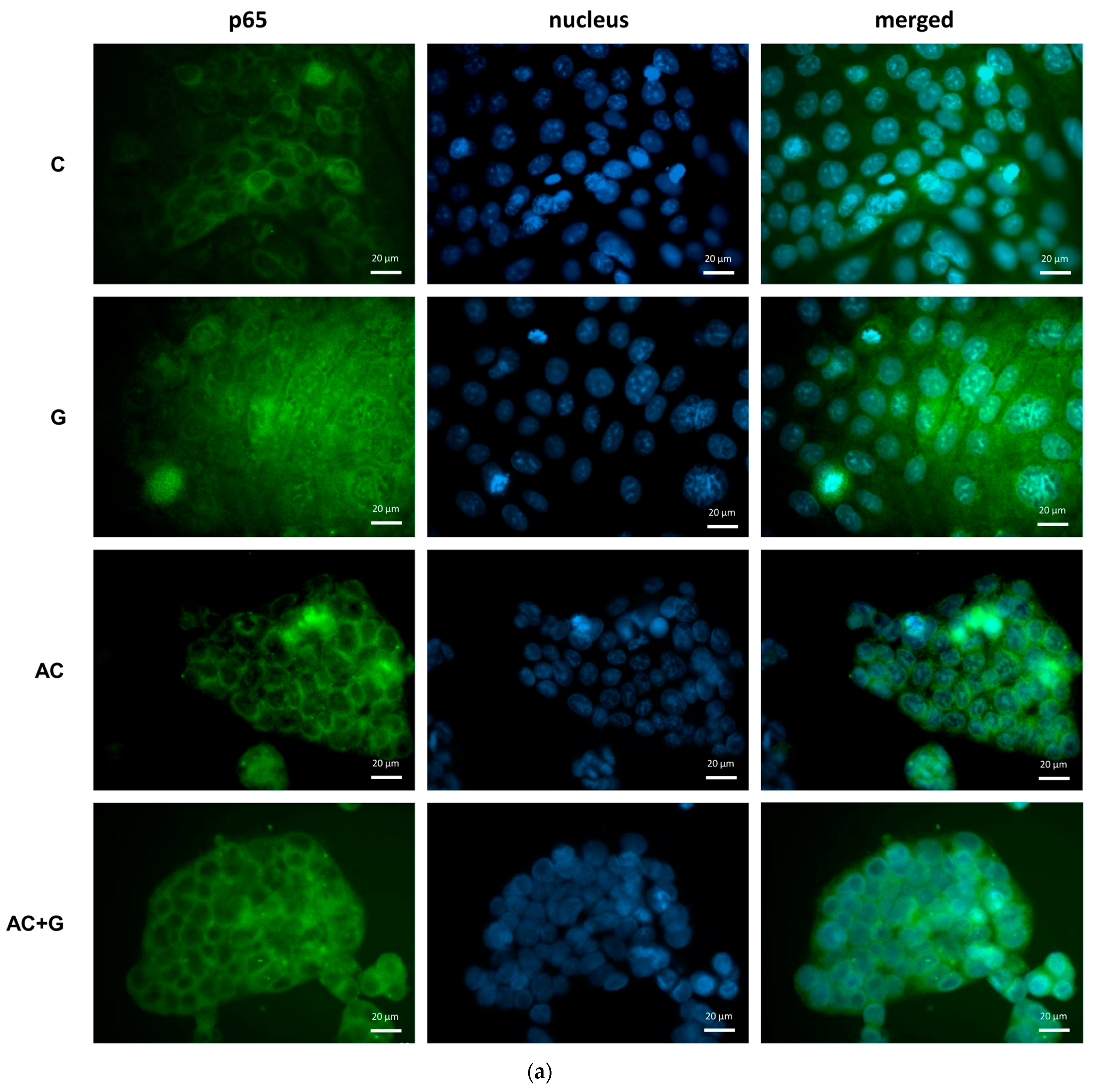
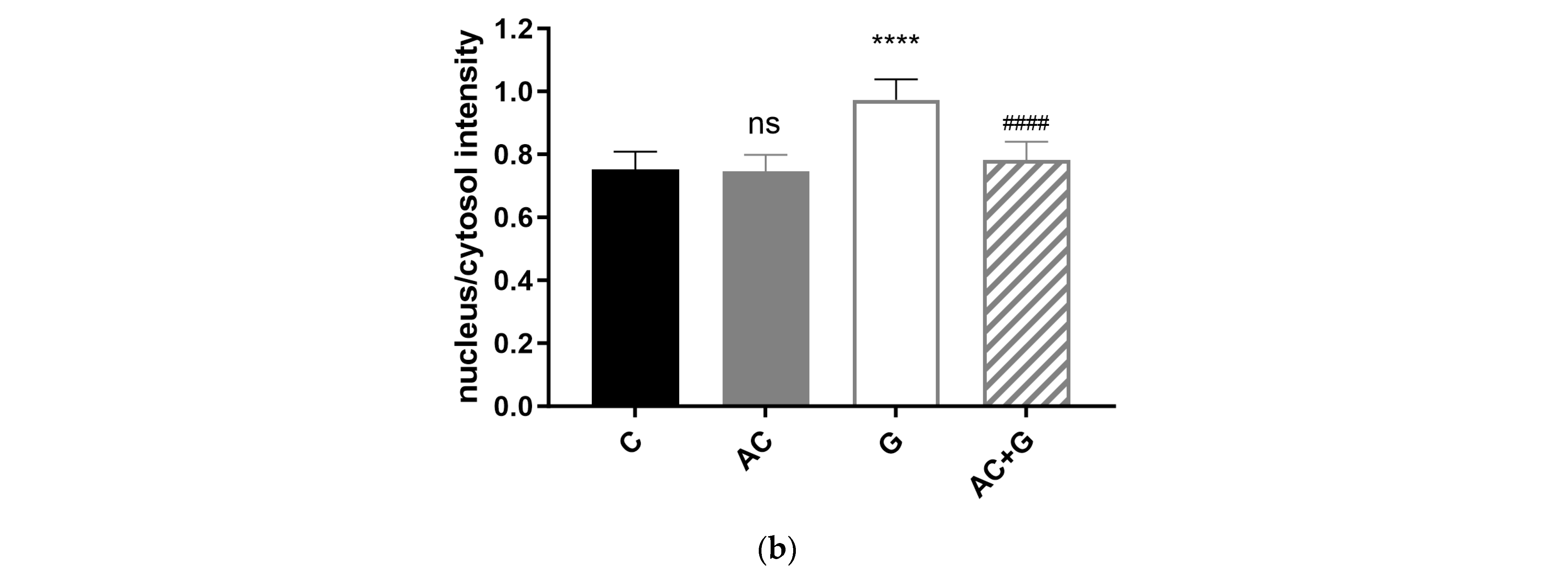
3.5. NF-κB Activation Pathway Study
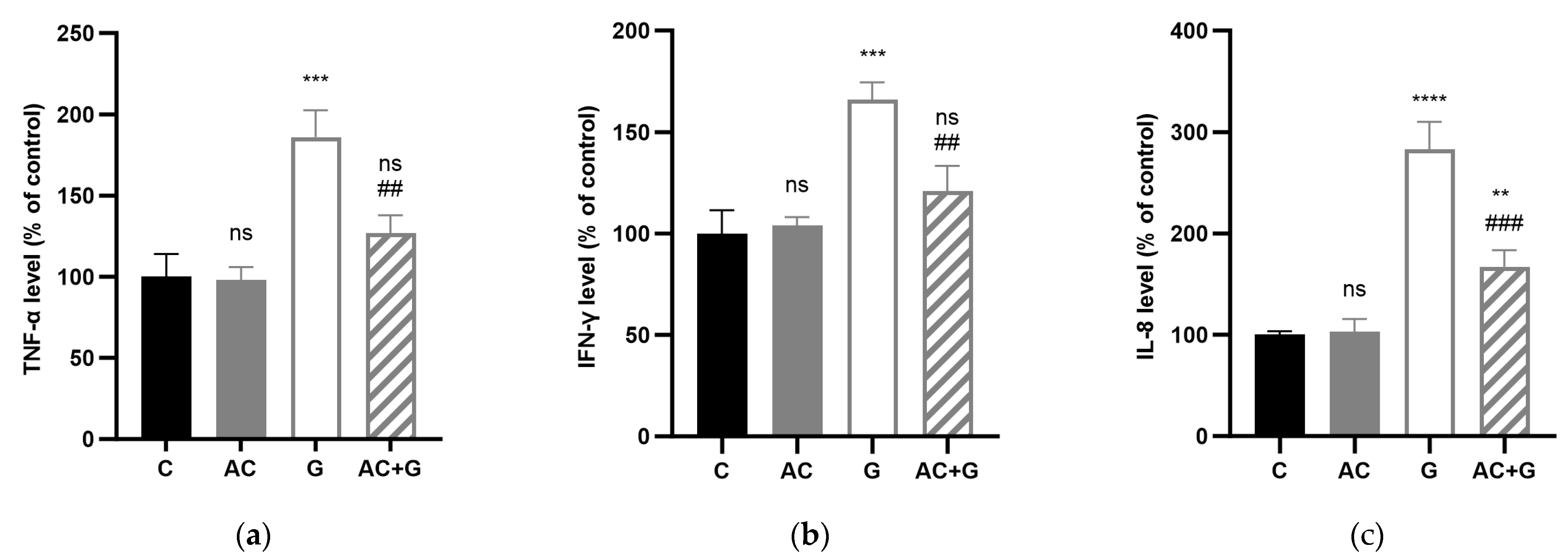
3.6. Cytokine Levels
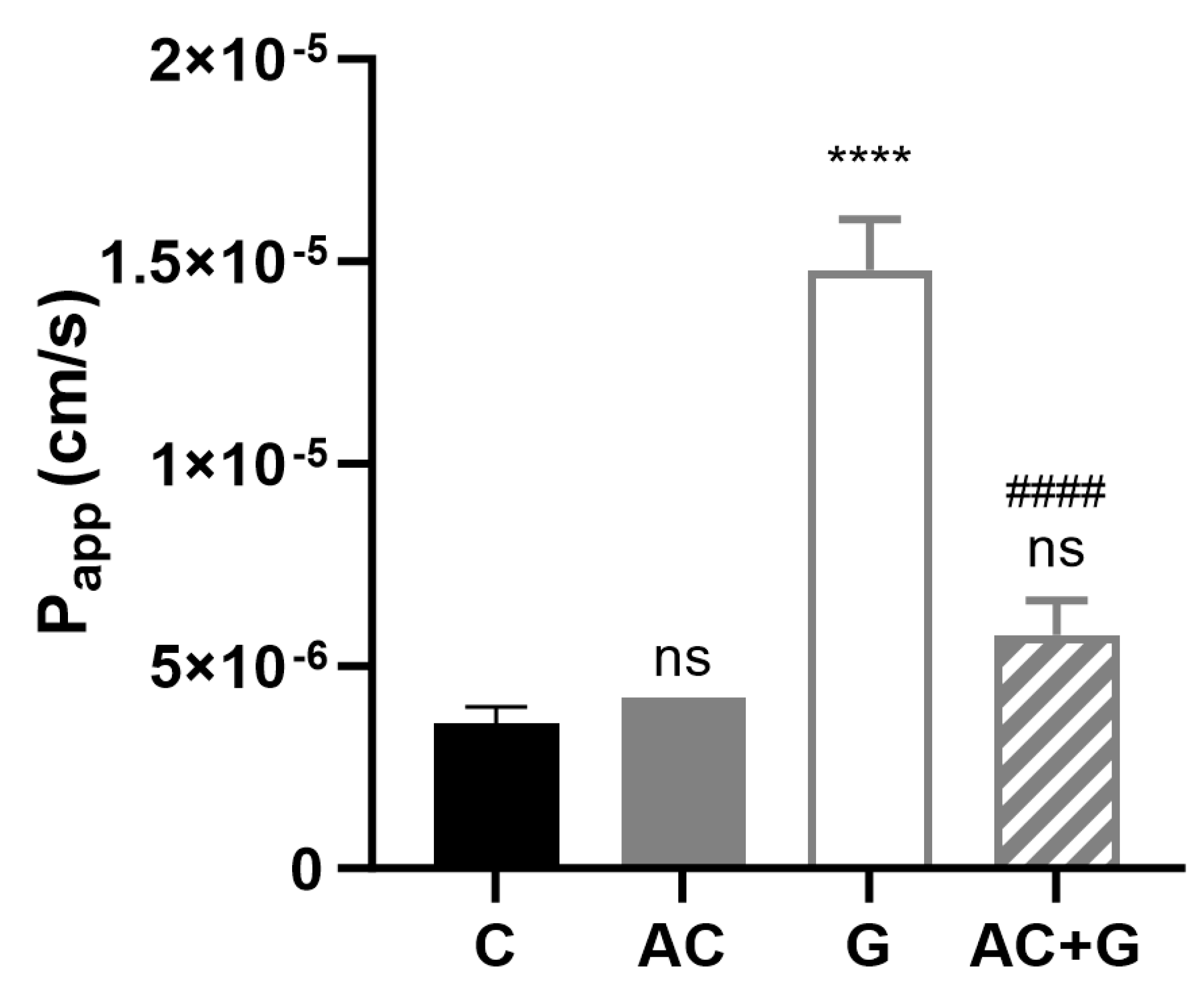
3.7. Permeability Study of Small Molecular Markers
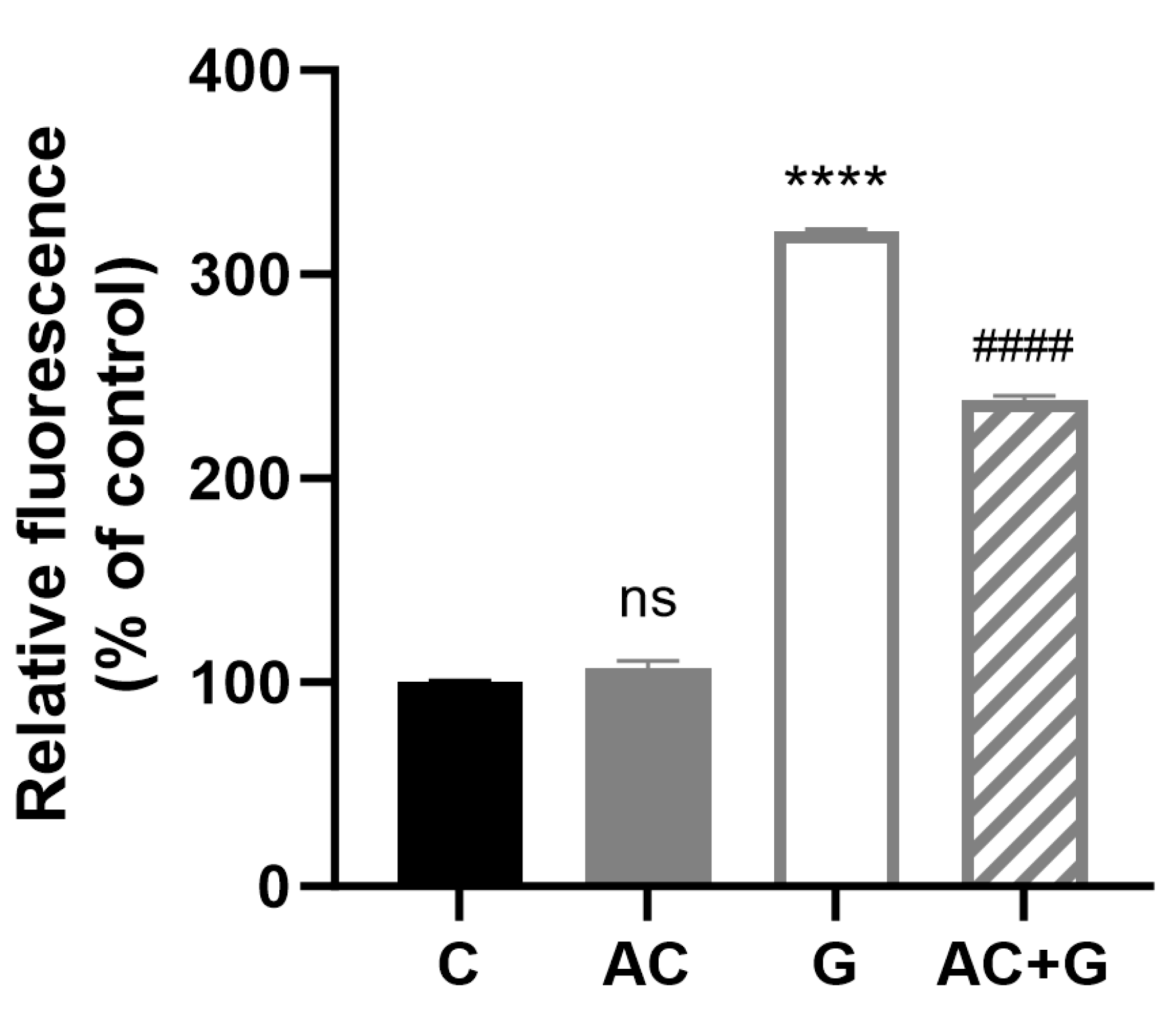
3.8. Investigation of Mitochondrial ROS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sollid, L.M.; Jabri, B. Triggers and drivers of autoimmunity: Lessons from coeliac disease. Nat. Rev. Immunol. 2013, 13, 294–302. [Google Scholar] [PubMed]
- Lindfors, K.; Ciacci, C.; Kurppa, K.; Lundin, K.E.A.; Makharia, G.K.; Mearin, M.L.; Murray, J.A.; Verdu, E.F.; Kaukinen, K. Coeliac disease. Nat. Rev. Dis. Prim. 2019, 5, 3. [Google Scholar] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [PubMed]
- Caminero, A.; Galipeau, H.J.; McCarville, J.L.; Johnston, C.W.; Bernier, S.P.; Russell, A.K.; Jury, J.; Herran, A.R.; Casqueiro, J.; Tye-Din, J.A.; et al. Duodenal Bacteria from Patients with Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity. Gastroenterology 2016, 151, 670–683. [Google Scholar] [CrossRef]
- Molberg, Ø.; Solheim Flaete, N.; Jensen, T.; Lundin, K.E.A.; Arentz-Hansen, H.; Anderson, O.D.; Kjersti Uhlen, A.; Sollid, L.M. Intestinal T-cell responses to high-molecular-weight glutenins in celiac disease. Gastroenterology 2003, 125, 337–344. [Google Scholar] [CrossRef]
- Thomas, K.E.; Sapone, A.; Fasano, A.; Vogel, S.N. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: Role of the innate immune response in Celiac disease. J. Immunol. 2006, 176, 2512–2521. [Google Scholar] [CrossRef]
- Boivin, M.A.; Roy, P.K.; Bradley, A.; Kennedy, J.C.; Rihani, T.; Ma, T.Y. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J. Interferon Cytokine Res. 2009, 29, 45–54. [Google Scholar] [CrossRef]
- Asri, N.; Rostami-Nejad, M.; Anderson, R.P.; Rostami, K. The Gluten Gene: Unlocking the Understanding of Gluten Sensitivity and Intolerance. Appl. Clin. Genet. 2021, 14, 37–50. [Google Scholar] [CrossRef]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef]
- Cardoso-Silva, D.; Delbue, D.; Itzlinger, A.; Moerkens, R.; Withoff, S.; Branchi, F.; Schumann, M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients 2019, 11, 2325. [Google Scholar] [CrossRef]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e3. [Google Scholar] [PubMed]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [PubMed]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar]
- Clemente, M.G.; De Virgiliis, S.; Kang, J.S.; Macatagney, R.; Musu, M.P.; Di Pierro, M.R.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223. [Google Scholar] [PubMed]
- Sander, G.R.; Cummins, A.G.; Henshall, T.; Powell, B.C. Rapid disruption of intestinal barrier function by gliadin involves altered expression of apical junctional proteins. FEBS Lett. 2005, 579, 4851–4855. [Google Scholar]
- Nemes, A.; Szőllősi, E.; Stündl, L.; Biró, A.; Homoki, J.R.; Szarvas, M.M.; Balogh, P.; Cziáky, Z.; Remenyik, J. Determination of Flavonoid and Proanthocyanidin Profile of Hungarian Sour Cherry. Molecules 2018, 23, 3278. [Google Scholar]
- Homoki, J.R.; Nemes, A.; Fazekas, E.; Gyémánt, G.; Balogh, P.; Gál, F.; Al-Asri, J.; Mortier, J.; Wolber, G.; Babinszky, L.; et al. Anthocyanin composition, antioxidant efficiency, and α-amylase inhibitor activity of different Hungarian sour cherry varieties (Prunus cerasus L.). Food Chem. 2016, 194, 222–229. [Google Scholar]
- Markovics, A.; Biró, A.; Kun-Nemes, A.; Fazekas, M.É.; Rácz, A.A.; Paholcsek, M.; Lukács, J.; Stündl, L.; Remenyik, J. Effect of Anthocyanin-Rich Extract of Sour Cherry for Hyperglycemia-Induced Inflammatory Response and Impaired Endothelium-Dependent Vasodilation. Nutrients 2020, 12, 3373. [Google Scholar]
- Varga, B.; Priksz, D.; Lampé, N.; Bombicz, M.; Kurucz, A.; Szabó, A.M.; Pósa, A.; Szabó, R.; Kemény-Beke, Á.; Remenyik, J.; et al. Protective Effect of Prunus Cerasus (Sour Cherry) Seed Extract on the Recovery of Ischemia/Reperfusion-Induced Retinal Damage in Zucker Diabetic Fatty Rat. Molecules 2017, 22, 1782. [Google Scholar]
- Remenyik, J.; Biró, A.; Klusóczki, Á.; Juhász, K.Z.; Szendi-Szatmári, T.; Kenesei, Á.; Szőllősi, E.; Vasvári, G.; Stündl, L.; Fenyvesi, F.; et al. Comparison of the Modulating Effect of Anthocyanin-Rich Sour Cherry Extract on Occludin and ZO-1 on Caco-2 and HUVEC Cultures. Int. J. Mol. Sci. 2022, 23, 9036. [Google Scholar]
- Le Phuong Nguyen, T.; Fenyvesi, F.; Remenyik, J.; Homoki, J.R.; Gogolák, P.; Bácskay, I.; Fehér, P.; Ujhelyi, Z.; Vasvári, G.; Vecsernyés, M.; et al. Protective Effect of Pure Sour Cherry Anthocyanin Extract on Cytokine-Induced Inflammatory Caco-2 Monolayers. Nutrients 2018, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Mazzaracchio, P.; Tozzi, S.; Boga, C.; Forlani, L.; Pifferi, P.G.; Barbiroli, G. Interaction between gliadins and anthocyan derivatives. Food Chem. 2011, 129, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, S.; Zanna, N.; Taddei, P. Study on the interaction between gliadins and a coumarin as molecular model system of the gliadins-anthocyanidins complexes. Food Chem. 2013, 141, 3586–3597. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Huang, Y.; Huang, J.; Li, S.; Zhu, Z.; Deng, Q.; Cheng, S. Formation of protein-anthocyanin complex induced by grape skin extracts interacting with wheat gliadins: Multi-spectroscopy and molecular docking analysis. Food Chem. 2022, 385, 132702. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J.E.; Murray, M.T. (Eds.) Textbook of Natural Medicine, 5th ed.; Elsevier: St. Louis, MO, USA, 2020; pp. I–1–I–51. ISBN 978-0-323-52342-4. [Google Scholar]
- Hundsberger, H.; Koppensteiner, A.; Hofmann, E.; Ripper, D.; Pflüger, M.; Stadlmann, V.; Klein, C.T.; Kreiseder, B.; Katzlinger, M.; Eger, A.; et al. A Screening Approach for Identifying Gliadin Neutralizing Antibodies on Epithelial Intestinal Caco-2 Cells. SLAS Discov. Adv. Life Sci. R D 2017, 22, 1035–1043. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orlando, A.; Chimienti, G.; Pesce, V.; Fracasso, F.; Lezza, A.M.S.; Russo, F. An In Vitro Study on Mitochondrial Compensatory Response Induced by Gliadin Peptides in Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 1862. [Google Scholar] [CrossRef]
- Van Buiten, C.B.; Lambert, J.D.; Elias, R.J. Green Tea Polyphenols Mitigate Gliadin-Mediated Inflammation and Permeability in Vitro. Mol. Nutr. Food Res. 2018, 62, e1700879. [Google Scholar] [CrossRef]
- Houghton, A.; Appelhagen, I.; Martin, C. Natural Blues: Structure Meets Function in Anthocyanins. Plants 2021, 10, 726. [Google Scholar] [CrossRef]
- Khaleghi, S.; Ju, J.M.; Lamba, A.; Murray, J.A. The potential utility of tight junction regulation in celiac disease: Focus on larazotide acetate. Therap. Adv. Gastroenterol. 2016, 9, 37–49. [Google Scholar] [CrossRef]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Mitic, L.L.; Van Itallie, C.M.; Anderson, J.M. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: Lessons from mutant animals and proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G250–G254. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Morales, P.; Gotteland, M. Polyphenols protect the epithelial barrier function of Caco-2 cells exposed to indomethacin through the modulation of occludin and zonula occludens-1 expression. J. Agric. Food Chem. 2013, 61, 5291–5297. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, J.; Park, S.-H.; Do, K.H.; Yang, H.; Moon, Y. Pro-inflammatory NF-κB and early growth response gene 1 regulate epithelial barrier disruption by food additive carrageenan in human intestinal epithelial cells. Toxicol. Lett. 2012, 211, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cui, C.; Xu, C.; Ren, H.; Wang, F.; Yu, Q.; Zhang, G. Effect and mechanism of peanut skin proanthocyanidins on gliadin-induced Caco-2 celiac disease model cells. Clin. Immunol. 2022, 245, 109100. [Google Scholar] [CrossRef]
- Maiuri, M.C.; De Stefano, D.; Mele, G.; Fecarotta, S.; Greco, L.; Troncone, R.; Carnuccio, R. Nuclear factor kappa B is activated in small intestinal mucosa of celiac patients. J. Mol. Med. 2003, 81, 373–379. [Google Scholar] [CrossRef]
- Li, J.; Jia, Q.; Liu, Y.; Chen, D.; Fang, Z.; Liu, Y.; Li, S.; Hu, B.; Wang, C.; Chen, H. Different Structures of Arabinoxylan Hydrolysates Alleviated Caco-2 Cell Barrier Damage by Regulating the TLRs/MyD88/NF-κB Pathway. Foods 2022, 11, 3535. [Google Scholar] [CrossRef]
- Ferrari, D.; Cimino, F.; Fratantonio, D.; Molonia, M.S.; Bashllari, R.; Busà, R.; Saija, A.; Speciale, A. Cyanidin-3-O-Glucoside Modulates the In Vitro Inflammatory Crosstalk between Intestinal Epithelial and Endothelial Cells. Mediat. Inflamm. 2017, 2017, 3454023. [Google Scholar] [CrossRef]
- Zimmer, K.-P.; Fischer, I.; Mothes, T.; Weissen-Plenz, G.; Schmitz, M.; Wieser, H.; Büning, J.; Lerch, M.M.; Ciclitira, P.C.; Weber, P.; et al. Endocytotic segregation of gliadin peptide 31–49 in enterocytes. Gut 2010, 59, 300–310. [Google Scholar] [CrossRef]
- Lim, S.D.; Min, H.; Youn, E.; Kawasaki, I.; Shim, Y.-H. Gliadin intake induces oxidative-stress responses in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2018, 503, 2139–2145. [Google Scholar] [CrossRef]
- Procházková, D.; Boušová, I.; Wilhelmová, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klusóczki, Á.; Oláh, B.; Hosszú, D.; Fenyvesi, F.; Remenyik, J.; Homoki, J.; Gyöngyösi, A.; Bácskay, I.; Váradi, J. Effectiveness of Anthocyanin-Rich Sour Cherry Extract on Gliadin-Induced Caco-2 Barrier Damage. Nutrients 2023, 15, 4022. https://doi.org/10.3390/nu15184022
Klusóczki Á, Oláh B, Hosszú D, Fenyvesi F, Remenyik J, Homoki J, Gyöngyösi A, Bácskay I, Váradi J. Effectiveness of Anthocyanin-Rich Sour Cherry Extract on Gliadin-Induced Caco-2 Barrier Damage. Nutrients. 2023; 15(18):4022. https://doi.org/10.3390/nu15184022
Chicago/Turabian StyleKlusóczki, Ágnes, Boglárka Oláh, Dominik Hosszú, Ferenc Fenyvesi, Judit Remenyik, Judit Homoki, Alexandra Gyöngyösi, Ildikó Bácskay, and Judit Váradi. 2023. "Effectiveness of Anthocyanin-Rich Sour Cherry Extract on Gliadin-Induced Caco-2 Barrier Damage" Nutrients 15, no. 18: 4022. https://doi.org/10.3390/nu15184022
APA StyleKlusóczki, Á., Oláh, B., Hosszú, D., Fenyvesi, F., Remenyik, J., Homoki, J., Gyöngyösi, A., Bácskay, I., & Váradi, J. (2023). Effectiveness of Anthocyanin-Rich Sour Cherry Extract on Gliadin-Induced Caco-2 Barrier Damage. Nutrients, 15(18), 4022. https://doi.org/10.3390/nu15184022