
Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study
 , ,
, ,
Abstract
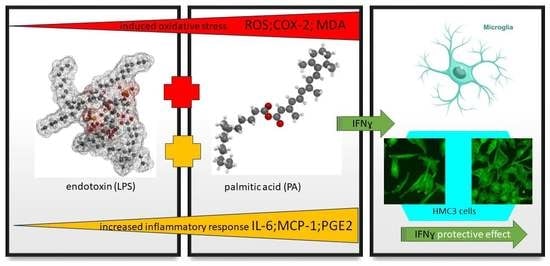
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals Used in the Study
2.2. Microglial Cell Culture
2.3. Palmitic Acid Preparation
2.4. Microglial Cell Viability and Morphology
2.5. Conditioned Media
2.6. Oxidative Stress Assessment
2.7. IL-6 and MCP-1 Assays
2.8. Statistical Analysis
3. Results
3.1. HMC3 Viability and Morphology
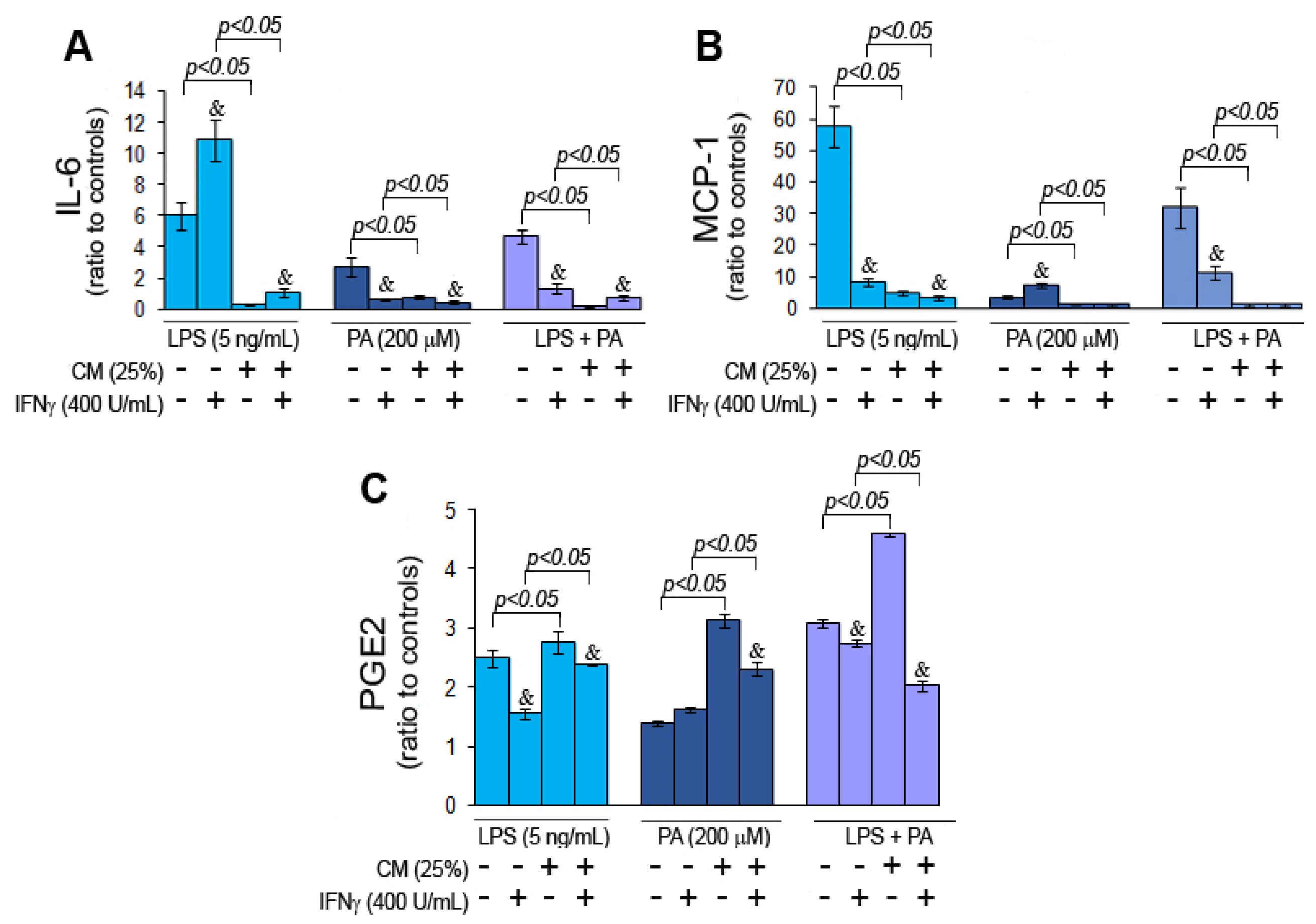
3.2. Palmitic Acid and Endotoxin Modulate Cytokine Secretion by Microglial Cells
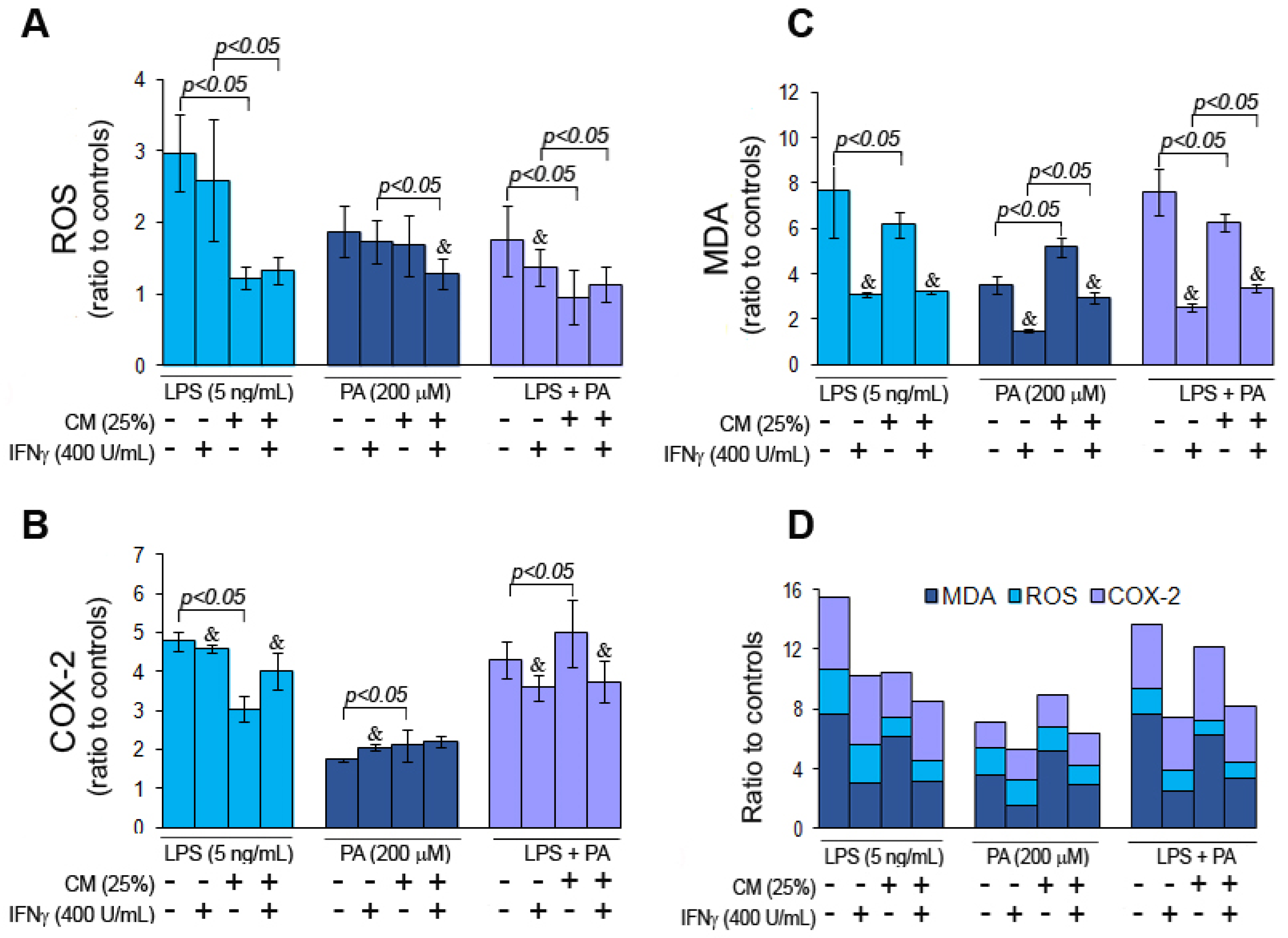
3.3. Palmitic Acid and Endotoxin Induce Oxidative Stress in Microglial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T. Altered Gut Microbiota: A Link Between Diet and the Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2018, 16, 321–328. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalby, M.J.; Aviello, G.; Ross, A.W.; Walker, A.W.; Barrett, P.; Morgan, P.J. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci. Rep. 2018, 8, 15648. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.-Y.; Jang, H.-M.; Kim, D.-H. High-fat diet causes psychiatric disorders in mice by increasing Proteobacteria population. Neurosci. Lett. 2019, 698, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, P.; Laugerette, F.; Féart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11, 1887. [Google Scholar] [CrossRef] [Green Version]
- Brown, B. Nutritional management of metabolic endotoxemia: A clinical review. Altern. Ther. Health Med. 2017, 23, 42–54. [Google Scholar] [PubMed]
- Lasselin, J.; Lekander, M.; Benson, S.; Schedlowski, M.; Engler, H. Sick for science: Experimental endotoxemia as a translational tool to develop and test new therapies for inflammation-associated depression. Mol. Psychiatry 2021, 26, 3672–3683. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugerette, F.; Vors, C.; Géloën, A.; Chauvin, M.-A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischof, G.N.; Park, D.C. Obesity and Aging: Consequences for cognition, brain structure and brain function. Psychosom. Med. 2015, 77, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Howe, A.-M.; Burke, S.; O’reilly, M.E.; McGillicuddy, F.C.; Costello, D.A. Palmitic Acid and Oleic Acid Differently Modulate TLR2-Mediated Inflammatory Responses in Microglia and Macrophages. Mol. Neurobiol. 2022, 59, 2348–2362. [Google Scholar] [CrossRef]
- Sabia, S.; Kivimaki, M.; Shipley, M.J.; Marmot, M.G.; Singh-Manoux, A. Body mass index over the adult life course and cognition in late midlife: The Whitehall II Cohort Study. Am. J. Clin. Nutr. 2009, 89, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Mannan, M.; Mamun, A.; Doi, S.; Clavarino, A. Prospective Associations between Depression and Obesity for Adolescent Males and Females- A Systematic Review and Meta-Analysis of Longitudinal Studies. PLoS ONE 2016, 11, e0157240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflammation 2019, 16, 180. [Google Scholar] [CrossRef] [Green Version]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Hernandes, M.S. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation 2021, 44, 2143–2150. [Google Scholar] [CrossRef]
- Pifferi, F.; Laurent, B.; Plourde, M. Lipid Transport and Metabolism at the Blood-Brain Interface: Implications in Health and Disease. Front. Physiol. 2021, 12, 645646. [Google Scholar] [CrossRef]
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-Brain Barriers in Obesity. AAPS J. 2017, 19, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Vesga-Jiménez, D.J.; Martin, C.; Barreto, G.E.; Aristizábal-Pachón, A.F.; Pinzón, A.; González, J. Fatty Acids: An Insight into the Pathogenesis of Neurodegenerative Diseases and Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 2577. [Google Scholar] [CrossRef] [PubMed]
- Schommer, J.; Marwarha, G.; Nagamoto-Combs, K.; Ghribi, O. Palmitic Acid-Enriched Diet Increases α-Synuclein and Tyrosine Hydroxylase Expression Levels in the Mouse Brain. Front. Neurosci. 2018, 12, 552. [Google Scholar] [CrossRef]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Custers, E.M.; Kiliaan, A.J. Dietary lipids from body to brain. Prog. Lipid Res. 2022, 85, 101144. [Google Scholar] [CrossRef]
- Desale, S.E.; Chinnathambi, S. Role of dietary fatty acids in microglial polarization in Alzheimer’s disease. J. Neuroinflammation 2020, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Melo, H.M.; Santos, L.E.; Ferreira, S.T. Diet-Derived Fatty Acids, Brain Inflammation, and Mental Health. Front. Neurosci. 2019, 13, 265. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Liu, S.; Lopes-Virella, M.F.; Wang, Z. LPS and palmitic acid Co-upregulate microglia activation and neuroinflammatory response. Compr. Psychoneuroendocrinology 2021, 6, 100048. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, A.L.; Stephens, J.W.; Harris, D.A. A review on gut microbiota: A central factor in the pathophysiology of obesity. Lipids Heal. Dis. 2021, 20, 65. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Winocur, G.; Greenwood, C.E. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol. Aging 2005, 26, 46–49. [Google Scholar] [CrossRef]
- Karmi, A.; Iozzo, P.; Viljanen, A.; Hirvonen, J.; Fielding, B.A.; Virtanen, K.; Oikonen, V.; Kemppainen, J.; Viljanen, T.; Guiducci, L.; et al. Increased Brain Fatty Acid Uptake in Metabolic Syndrome. Diabetes 2010, 59, 2171–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, N.M. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Arch. Intern. Med. 1989, 149, 1514–1520. [Google Scholar] [CrossRef]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Ruga, S.; Zito, M.C.; et al. From Metabolic Syndrome to Neurological Diseases: Role of Autophagy. Front. Cell Dev. Biol. 2021, 9, 651021. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol. Aging 2014, 35, S59–S64. [Google Scholar] [CrossRef] [Green Version]
- Dragano, N.R.; Monfort-Pires, M.; Velloso, L.A. Mechanisms Mediating the Actions of Fatty Acids in the Hypothalamus. Neuroscience 2020, 447, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Rothhaar, T.L.; Grösgen, S.; Burg, V.K.; Hundsdörfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Dahl, A.K.; Hassing, L.B. Obesity and Cognitive Aging. Epidemiologic Rev. 2013, 35, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Karlsson, C.; Skoog, I.; Rosengren, L.; Lissner, L.; Blennow, K. Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J. Intern. Med. 2007, 262, 643–650. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, X.; Deng, B.; Xiao, J.; Jin, J.; Huang, Z. Lipopolysaccharide and palmitic acid synergistically induced MCP-1 production via MAPK-meditated TLR4 signaling pathway in RAW264.7 cells. Lipids Health Dis. 2019, 18, 71. [Google Scholar] [CrossRef] [Green Version]
- Tucureanu, M.M.; Rebleanu, D.; Constantinescu, C.A.; Deleanu, M.; Voicu, G.; Butoi, E.; Calin, M.; Manduteanu, I. Lipopolysaccharide-induced inflammation in monocytes/macrophages is blocked by liposomal delivery of Gi-protein inhibitor. Int. J. Nanomed. 2017, 13, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Gutteridge, J.M.C. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.W.; Goodwin, M.E.; Porter, M.M.; Beggs, M.L. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 2007, 101, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Page, M.J.; Kell, D.B.; Pretorius, E. The Role of Lipopolysaccharide-Induced Cell Signalling in Chronic Inflammation. Chronic Stress 2022, 6, 24705470221076390. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Madhavpeddi, L.; Gonzales, R.J. Palmitate induces glycosylation of cyclooxygenase-2 in primary human vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2018, 314, C545–C553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Chang, S.L. Meta-analysis of the effects of palmitic acid on microglia activation and neurodegeneration. NeuroImmune Pharmacol. Ther. 2022. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Gruol, D.L. IL-6 regulation of synaptic function in the CNS. Neuropharmacology 2015, 96, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Kummer, K.K.; Zeidler, M.; Kalpachidou, T.; Kress, M. Role of IL-6 in the regulation of neuronal development, survival and function. Cytokine 2021, 144, 155582. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, J.; Yu, Y.; Li, Z. Gene silencing of MCP-1 prevents microglial activation and inflammatory injury after intracerebral hemorrhage. Int. Immunopharmacol. 2016, 33, 18–23. [Google Scholar] [CrossRef]
- Wojcieszak, J.; Kuczyńska, K.; Zawilska, J.B. Role of Chemokines in the Development and Progression of Alzheimer’s Disease. J. Mol. Neurosci. 2022, 72, 1929–1951. [Google Scholar] [CrossRef]
- Lee, W.-J.; Liao, Y.-C.; Wang, Y.-F.; Lin, I.-F.; Wang, S.-J.; Fuh, J.-L. Plasma MCP-1 and Cognitive Decline in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: A Two-year Follow-up Study. Sci. Rep. 2018, 8, 1280. [Google Scholar] [CrossRef] [Green Version]
- Niraula, A.; Fasnacht, R.D.; Ness, K.M.; Frey, J.M.; Cuschieri, S.A.; Dorfman, M.D.; Thaler, J.P. Prostaglandin PGE2 Receptor EP4 Regulates Microglial Phagocytosis and Increases Susceptibility to Diet-Induced Obesity. Diabetes 2023, 72, 233–244. [Google Scholar] [CrossRef]
- Jin, K.; Qian, C.; Lin, J.; Liu, B. Cyclooxygenase-2-Prostaglandin E2 pathway: A key player in tumor-associated immune cells. Front. Oncol. 2023, 13, 1099811. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Nicolini, A.; Minghetti, L. Prolonged exposure of microglia to lipopolysaccharide modifies the intracellular signaling pathways and selectively promotes prostaglandin E2 synthesis. J. Neurochem. 2003, 87, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boje, K.M.K.; Jaworowicz, D.; Raybon, J.J. Neuroinflammatory Role of Prostaglandins during Experimental Meningitis: Evidence Suggestive of an in Vivo Relationship between Nitric Oxide and Prostaglandins. J. Pharmacol. Exp. Ther. 2003, 304, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.-C.; Lee, K.-F.; Chen, S.-L.; Chiu, S.-C.; Lee, L.-Y.; Chen, W.-P.; Chen, C.-C.; Chu, C.-H. Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis. Biomedicines 2022, 10, 419. [Google Scholar] [CrossRef]
- Miller, N.N.; Wang, J.; Tan, Y.; Dittel, B.N. Anti-inflammatory mechanisms of IFN-γ studied in experimental autoimmune encephalomyelitis reveal neutrophils as a potential target in multiple sclerosis. Front. Neurosci. 2015, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Shaked, I.; Tchoresh, D.; Gersner, R.; Meiri, G.; Mordechai, S.; Xiao, X.; Hart, R.P.; Schwartz, M. Protective autoimmunity: Interferon-γ enables microglia to remove glutamate without evoking inflammatory mediators. J. Neurochem. 2005, 92, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Hu, S.; Deshpande, A.; Munir, S.; May, B.J.; Baker, C.A.; Peterson, P.K.; Kapur, V. Transcriptional response of human microglial cells to interferon-γ. Genes Immun. 2005, 6, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10, a028571. [Google Scholar] [CrossRef]
- Sosa, R.A.; Murphey, C.; Robinson, R.R.; Forsthuber, T.G. IFN-γ ameliorates autoimmune encephalomyelitis by limiting myelin lipid peroxidation. Proc. Natl. Acad. Sci. USA 2015, 112, 5038–5047. [Google Scholar] [CrossRef]
- Christen, S.; Peterhans, E.; Stocker, R. Antioxidant activities of some tryptophan metabolites: Possible implication for inflammatory diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 2506–2510. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmielarz, M.; Sobieszczańska, B.; Teisseyre, A.; Wawrzyńska, M.; Bożemska, E.; Środa-Pomianek, K. Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study. Nutrients 2023, 15, 3463. https://doi.org/10.3390/nu15153463
Chmielarz M, Sobieszczańska B, Teisseyre A, Wawrzyńska M, Bożemska E, Środa-Pomianek K. Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study. Nutrients. 2023; 15(15):3463. https://doi.org/10.3390/nu15153463
Chicago/Turabian StyleChmielarz, Mateusz, Beata Sobieszczańska, Andrzej Teisseyre, Magdalena Wawrzyńska, Edyta Bożemska, and Kamila Środa-Pomianek. 2023. "Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study" Nutrients 15, no. 15: 3463. https://doi.org/10.3390/nu15153463
APA StyleChmielarz, M., Sobieszczańska, B., Teisseyre, A., Wawrzyńska, M., Bożemska, E., & Środa-Pomianek, K. (2023). Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study. Nutrients, 15(15), 3463. https://doi.org/10.3390/nu15153463