
The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg?

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Insulin Resistance
2.1. Pathophysiology of Insulin Resistance
2.1.1. Skeletal Muscle
2.1.2. Liver
2.1.3. Adipose Tissue
2.2. Metabolic Inflexibility
3. Direct Effects of Weight Loss on Insulin Sensitivity
- (1)
- Worsen lipid ectopic accumulation (see previous paragraph);
- (2)
- Interfere negatively with the release of adipokines;
- (3)
- Cause inflammation and adipose tissue macrophage accumulation.
4. Ketogenic Diet
- -
- VLCKD (very-low-calorie ketogenic diet) with less than 700 to 800 kcal/day, carbohydrate intake < 30–50 g/day, lipid intake of up to 30 to 40 g/day and protein intake of 0.8 to 1.2 g/kg of body weight per day;
- -
- LCKD (low-calorie ketogenic diet) with at least 700 to 800 kcal/day, but less than the daily caloric requirement total energy expenditure(TEE), carbohydrate intake < 30–50 g/day, lipid intake > 30 to 40 g/day;
- -
- ICKD (isocaloric ketogenic diet) with caloric intake in line with the daily TEE requirement, carbohydrate intake < 30–50 g/day, lipid intake > 70–80% of daily calorie intake.
4.1. Biochemical and Physiological Aspects of Ketogenic Diets
4.2. Effects of Ketogenic Diets on Weight Loss
4.3. Effects of KDs on Insulin Sensitivity Mediated by Fat Changes
4.3.1. KDs and Visceral vs. Subcutaneous Fat
4.3.2. KD and Skeletal Muscle Insulin Sensitivity
4.3.3. KDs and Liver Fat Depots and Insulin Sensitivity
4.4. Direct Effects of KDs on Insulin Sensitivity
4.4.1. Oxidative Stress
4.4.2. G-Protein-Coupled Receptors (GPCRs)
4.4.3. Inflammation
4.4.4. Sirtuin Mediated Signals
4.4.5. Mitochondrial Efficiency
4.4.6. The Microbiome Connection
5. Safety of KDs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 31 May 2022).
- James, D.E.; Stockli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef]
- Hostalek, U. Global epidemiology of prediabetes-present and future perspectives. Clin. Diabetes Endocrinol. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Guthold, R.; Stevens, G.A.; Riley, L.M.; Bull, F.C. Worldwide trends in insufficient physical activity from 2001 to 2016: A pooled analysis of 358 population-based surveys with 1.9 million participants. Lancet Glob. Health 2018, 6, e1077–e1086. [Google Scholar] [CrossRef]
- Katzmarzyk, P.T.; Friedenreich, C.; Shiroma, E.J.; Lee, I.-M. Physical inactivity and non-communicable disease burden in low-income, middle-income and high-income countries. Br. J. Sport. Med. 2022, 56, 101–106. [Google Scholar] [CrossRef]
- Ushula, T.W.; Mamun, A.; Darssan, D.; Wang, W.Y.S.; Williams, G.M.; Whiting, S.J.; Najman, J.M. Dietary patterns and the risks of metabolic syndrome and insulin resistance among young adults: Evidence from a longitudinal study. Clin. Nutr. 2022, 41, 1523–1531. [Google Scholar] [CrossRef]
- Parcha, V.; Heindl, B.; Kalra, R.; Li, P.; Gower, B.; Arora, G.; Arora, P. Insulin Resistance and Cardiometabolic Risk Profile Among Nondiabetic American Young Adults: Insights From NHANES. J. Clin. Endocrinol. Metab. 2022, 107, e25–e37. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.; Salazar, J.; Martinez, M.S.; Chavez-Castillo, M.; Olivar, L.C.; Calvo, M.J.; Palmar, J.; Bautista, J.; Ramos, E.; Cabrera, M.; et al. Prevalence and Associated Factors of Insulin Resistance in Adults from Maracaibo City, Venezuela. Adv. Prev. Med. 2016, 2016, 9405105. [Google Scholar] [CrossRef] [PubMed]
- Adany, R.; Piko, P.; Fiatal, S.; Kosa, Z.; Sandor, J.; Biro, E.; Kosa, K.; Paragh, G.; Bacsne Baba, E.; Veres-Balajti, I.; et al. Prevalence of Insulin Resistance in the Hungarian General and Roma Populations as Defined by Using Data Generated in a Complex Health (Interview and Examination) Survey. Int. J. Environ. Res. Public Health 2020, 17, 4833. [Google Scholar] [CrossRef]
- Friedrich, N.; Thuesen, B.; Jorgensen, T.; Juul, A.; Spielhagen, C.; Wallaschofksi, H.; Linneberg, A. The association between IGF-I and insulin resistance: A general population study in Danish adults. Diabetes Care 2012, 35, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.; Turan, E.; Uyar, M.; Bayram, F.; Turan, Y. The Prevalence of Insulin Resistance in the Turkish Population: A Study Conducted with 3331 Participants. Eurasian J. Med. Oncol. 2017, 1, 202–206. [Google Scholar] [CrossRef]
- Dowis, K.; Banga, S. The potential health benefits of the ketogenic diet: A narrative review. Nutrients 2021, 13, 1654. [Google Scholar] [CrossRef] [PubMed]
- Moreno, B.; Crujeiras, A.B.; Bellido, D.; Sajoux, I.; Casanueva, F.F. Obesity treatment by very low-calorie-ketogenic diet at two years: Reduction in visceral fat and on the burden of disease. Endocrine 2016, 54, 681–690. [Google Scholar] [CrossRef]
- Colica, C.; Merra, G.; Gasbarrini, A.; De Lorenzo, A.; Cioccoloni, G.; Gualtieri, P.; Perrone, M.A.; Bernardini, S.; Bernardo, V.; Di Renzo, L.; et al. Efficacy and safety of very-low-calorie ketogenic diet: A double blind randomized crossover study. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2274–2289. [Google Scholar]
- Paoli, A.; Mancin, L.; Giacona, M.C.; Bianco, A.; Caprio, M. Effects of a ketogenic diet in overweight women with polycystic ovary syndrome. J. Transl. Med. 2020, 18, 104. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath, C.A., III; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M., Jr.; Gower, B.A. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.M.; Hakkarainen, A.; Lehtimaki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Jarvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef]
- Badman, M.K.; Kennedy, A.R.; Adams, A.C.; Pissios, P.; Maratos-Flier, E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1197–E1204. [Google Scholar] [CrossRef]
- Gannon, M.C.; Nuttall, F.Q. Control of blood glucose in type 2 diabetes without weight loss by modification of diet composition. Nutr. Metab. 2006, 3, 16. [Google Scholar] [CrossRef]
- Hite, A.H.; Berkowitz, V.G.; Berkowitz, K. Low-carbohydrate diet review: Shifting the paradigm. Nutr. Clin. Pract. 2011, 26, 300–308. [Google Scholar] [CrossRef]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef]
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef]
- Sesti, G. Pathophysiology of insulin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Flannery, C.; Dufour, S.; Rabol, R.; Shulman, G.I.; Petersen, K.F. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012, 61, 2711–2717. [Google Scholar] [CrossRef]
- Cross, E.; Dearlove, D.J.; Hodson, L. Nutritional regulation of hepatic de novo lipogenesis in humans. Curr. Opin. Clin. Nutr. Metab. Care 2023, 26, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Chen, N.; Shi, X.; Tsang, B.; Yu, Y.H. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Investig. 2007, 117, 1679–1689. [Google Scholar] [CrossRef]
- Koves, T.R.; Li, P.; An, J.; Akimoto, T.; Slentz, D.; Ilkayeva, O.; Dohm, G.L.; Yan, Z.; Newgard, C.B.; Muoio, D.M. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005, 280, 33588–33598. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Obesity-related derangements in metabolic regulation. Annu. Rev. Biochem. 2006, 75, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Pender, C.; Trentadue, A.R.; Pories, W.J.; Dohm, G.L.; Houmard, J.A.; Youngren, J.F. Expression of genes regulating malonyl-CoA in human skeletal muscle. J. Cell. Biochem. 2006, 99, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef]
- Reitman, M.L.; Gavrilova, O. A-ZIP/F-1 mice lacking white fat: A model for understanding lipoatrophic diabetes. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. S4), S11–S14. [Google Scholar] [CrossRef]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef]
- Saponaro, C.; Sabatini, S.; Gaggini, M.; Carli, F.; Rosso, C.; Positano, V.; Armandi, A.; Caviglia, G.P.; Faletti, R.; Bugianesi, E.; et al. Adipose tissue dysfunction and visceral fat are associated to hepatic insulin resistance and severity of NASH even in lean individuals. Liver Int. 2022, 42, 2418–2427. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Monzon, J.R.; Basile, R.; Heneghan, S.; Udupi, V.; Green, A. Lipolysis in adipocytes isolated from deep and superficial subcutaneous adipose tissue. Obes. Res. 2002, 10, 266–269. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Amador, M.; Meza, C.A.; McAinch, A.J.; King, G.A.; Covington, J.D.; Bajpeyi, S. Exercise-Induced Improvements in Insulin Sensitivity Are Not Attenuated by a Family History of Type 2 Diabetes. Front. Endocrinol. 2020, 11, 120. [Google Scholar] [CrossRef]
- Rudwill, F.; O’Gorman, D.; Lefai, E.; Chery, I.; Zahariev, A.; Normand, S.; Pagano, A.F.; Chopard, A.; Damiot, A.; Laurens, C.; et al. Metabolic Inflexibility Is an Early Marker of Bed-Rest-Induced Glucose Intolerance Even When Fat Mass Is Stable. J. Clin. Endocrinol. Metab. 2018, 103, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Waldman, H.S.; Bryant, A.R.; Knight, S.N.; Killen, L.G.; Davis, B.A.; Robinson, M.A.; O’Neal, E.K. Assessment of Metabolic Flexibility by Substrate Oxidation Responses and Blood Lactate in Women Expressing Varying Levels of Aerobic Fitness and Body Fat. J. Strength Cond. Res. 2022, 37, 581–588. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef]
- Faerch, K.; Vaag, A. Metabolic inflexibility is a common feature of impaired fasting glycaemia and impaired glucose tolerance. Acta Diabetol. 2011, 48, 349–353. [Google Scholar] [CrossRef]
- Eriksson-Hogling, D.; Andersson, D.P.; Backdahl, J.; Hoffstedt, J.; Rossner, S.; Thorell, A.; Arner, E.; Arner, P.; Ryden, M. Adipose tissue morphology predicts improved insulin sensitivity following moderate or pronounced weight loss. Int. J. Obes. 2015, 39, 893–898. [Google Scholar] [CrossRef]
- McLaughlin, T.; Abbasi, F.; Lamendola, C.; Yee, G.; Carter, S.; Cushman, S.W. Dietary weight loss in insulin-resistant non-obese humans: Metabolic benefits and relationship to adipose cell size. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 62–68. [Google Scholar] [CrossRef]
- Mileti, E.; Kwok, K.H.M.; Andersson, D.P.; Mathelier, A.; Raman, A.; Backdahl, J.; Jalkanen, J.; Massier, L.; Thorell, A.; Gao, H.; et al. Human White Adipose Tissue Displays Selective Insulin Resistance in the Obese State. Diabetes 2021, 70, 1486–1497. [Google Scholar] [CrossRef]
- Riis, S.; Christensen, B.; Nellemann, B.; Moller, A.B.; Husted, A.S.; Pedersen, S.B.; Schwartz, T.W.; Jorgensen, J.O.L.; Jessen, N. Molecular adaptations in human subcutaneous adipose tissue after ten weeks of endurance exercise training in healthy males. J. Appl. Physiol. 2019, 126, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Ding, J.; Wang, K.; Ni, Z.; Yu, J. Mediterranean Diet Combined with a Low-Carbohydrate Dietary Pattern in the Treatment of Overweight Polycystic Ovary Syndrome Patients. Front. Nutr. 2022, 9, 876620. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Gortan Cappellari, G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight Disord. 2018, 23, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Cheng, K.K.; Lam, K.S.; Wang, B.; Xu, A. Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Adiponectin: Action, regulation and association to insulin sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Monda, V.; Polito, R.; Lovino, A.; Finaldi, A.; Valenzano, A.; Nigro, E.; Corso, G.; Sessa, F.; Asmundo, A.; Nunno, N.D.; et al. Short-Term Physiological Effects of a Very Low-Calorie Ketogenic Diet: Effects on Adiponectin Levels and Inflammatory States. Int. J. Mol. Sci. 2020, 21, 3228. [Google Scholar] [CrossRef]
- Reinehr, T.; Roth, C.; Menke, T.; Andler, W. Adiponectin before and after weight loss in obese children. J. Clin. Endocrinol. Metab. 2004, 89, 3790–3794. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, W.J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.L.; Chen, C.L.; Tai, T.Y.; Chuang, L.M. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J. Clin. Endocrinol. Metab. 2001, 86, 3815–3819. [Google Scholar] [CrossRef]
- Hajri, T.; Tao, H.; Wattacheril, J.; Marks-Shulman, P.; Abumrad, N.N. Regulation of adiponectin production by insulin: Interactions with tumor necrosis factor-alpha and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E350–E360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Spagnoli, A.; Torquati, A. Omental gene expression of adiponectin correlates with degree of insulin sensitivity before and after gastric bypass surgery. Obes. Surg. 2012, 22, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.P.; Eriksson Hogling, D.; Thorell, A.; Toft, E.; Qvisth, V.; Naslund, E.; Thorne, A.; Wiren, M.; Lofgren, P.; Hoffstedt, J.; et al. Changes in subcutaneous fat cell volume and insulin sensitivity after weight loss. Diabetes Care 2014, 37, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Moullec, G.; Santosa, S. Factors associated with adipocyte size reduction after weight loss interventions for overweight and obesity: A systematic review and meta-regression. Metabolism 2017, 67, 31–40. [Google Scholar] [CrossRef]
- Merlotti, C.; Ceriani, V.; Morabito, A.; Pontiroli, A.E. Subcutaneous fat loss is greater than visceral fat loss with diet and exercise, weight-loss promoting drugs and bariatric surgery: A critical review and meta-analysis. Int. J. Obes. 2017, 41, 672–682. [Google Scholar] [CrossRef]
- Taylor, R.; Al-Mrabeh, A.; Sattar, N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 726–736. [Google Scholar] [CrossRef]
- Petersen, M.C.; Gallop, M.R.; Flores Ramos, S.; Zarrinpar, A.; Broussard, J.L.; Chondronikola, M.; Chaix, A.; Klein, S. Complex physiology and clinical implications of time-restricted eating. Physiol. Rev. 2022, 102, 1991–2034. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V. Obesity and dyslipidemia. Metabolism 2019, 92, 71–81. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; O’Rourke, R.W.; Lumeng, C.N.; Sandoval, D.A.; Seeley, R.J. Does bariatric surgery improve adipose tissue function? Obes. Rev. 2016, 17, 795–809. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, W.; Kwon, H.S.; Baek, K.H.; Kim, E.K.; Song, K.H. Effects of bariatric surgery on metabolic and nutritional parameters in severely obese Korean patients with type 2 diabetes: A prospective 2-year follow up. J. Diabetes Investig. 2014, 5, 221–227. [Google Scholar] [CrossRef]
- Reynisdottir, S.; Langin, D.; Carlstrom, K.; Holm, C.; Rossner, S.; Arner, P. Effects of weight reduction on the regulation of lipolysis in adipocytes of women with upper-body obesity. Clin. Sci. 1995, 89, 421–429. [Google Scholar] [CrossRef]
- Perez-Martinez, P.; Mikhailidis, D.P.; Athyros, V.G.; Bullo, M.; Couture, P.; Covas, M.I.; de Koning, L.; Delgado-Lista, J.; Diaz-Lopez, A.; Drevon, C.A.; et al. Lifestyle recommendations for the prevention and management of metabolic syndrome: An international panel recommendation. Nutr. Rev. 2017, 75, 307–326. [Google Scholar] [CrossRef]
- Gardner, C.D.; Vadiveloo, M.K.; Petersen, K.S.; Anderson, C.A.M.; Springfield, S.; Van Horn, L.; Khera, A.; Lamendola, C.; Mayo, S.M.; Joseph, J.J.; et al. Popular Dietary Patterns: Alignment with American Heart Association 2021 Dietary Guidance: A Scientific Statement from the American Heart Association. Circulation 2023, 147, 1715–1730. [Google Scholar] [CrossRef] [PubMed]
- Wilder, R.M. The effects of ketonemia on the course of epilepsy. Mayo Clin. Proc. 1921, 2, 307–308. [Google Scholar]
- Peterman, M. The ketogenic diet in the treatment of epilepsy: A preliminary report. J. Nerv. Ment. Dis. 1926, 64, 96–97. [Google Scholar] [CrossRef]
- Trimboli, P.; Castellana, M.; Bellido, D.; Casanueva, F.F. Confusion in the nomenclature of ketogenic diets blurs evidence. Rev. Endocr. Metab. Disord. 2020, 21, 1–3. [Google Scholar] [CrossRef]
- Paoli, A. Ketogenic diet for obesity: Friend or foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef]
- Paoli, A.; Bosco, G.; Camporesi, E.M.; Mangar, D. Ketosis, ketogenic diet and food intake control: A complex relationship. Front. Psychol. 2015, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; Migliaccio, S.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Barrea, L.; Laudisio, D.; Pugliese, G.; Salzano, C.; Savastano, S.; Colao, A. The management of very low-calorie ketogenic diet in obesity outpatient clinic: A practical guide. J. Transl. Med. 2019, 17, 356. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Barrea, L.; Caprio, M.; Watanabe, M.; Cammarata, G.; Feraco, A.; Muscogiuri, G.; Verde, L.; Colao, A.; Savastano, S.; and on behalf of Obesity Programs of Nutrition, Education, Research and Assessment (OPERA) Group. Could very low-calorie ketogenic diets turn off low grade inflammation in obesity? Emerging evidence. Crit. Rev. Food Sci. Nutr. 2022, 1–17. [Google Scholar] [CrossRef]
- Ferraris, C.; Guglielmetti, M.; Neri, L.C.L.; Allehdan, S.; Mohsin Albasara, J.M.; Fareed Alawadhi, H.H.; Trentani, C.; Perna, S.; Tagliabue, A. A Review of Ketogenic Dietary Therapies for Epilepsy and Neurological Diseases: A Proposal to Implement an Adapted Model to Include Healthy Mediterranean Products. Foods 2023, 12, 1743. [Google Scholar] [CrossRef]
- Paoli, A.; Moro, T.; Bosco, G.; Bianco, A.; Grimaldi, K.A.; Camporesi, E.; Mangar, D. Effects of n-3 polyunsaturated fatty acids (omega-3) supplementation on some cardiovascular risk factors with a ketogenic Mediterranean diet. Mar. Drugs 2015, 13, 996–1009. [Google Scholar] [CrossRef]
- Paoli, A.; Bianco, A.; Grimaldi, K.A.; Lodi, A.; Bosco, G. Long term successful weight loss with a combination biphasic ketogenic mediterranean diet and mediterranean diet maintenance protocol. Nutrients 2013, 5, 5205–5217. [Google Scholar] [CrossRef]
- Paoli, A.; Grimaldi, K.; Bianco, A.; Lodi, A.; Cenci, L.; Parmagnani, A. Medium term effects of a ketogenic diet and a Mediterranean diet on resting energy expenditure and respiratory ratio. BMC Proc. 2012, 6, P37. [Google Scholar] [CrossRef]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49 (Suppl. S8), 3–5. [Google Scholar] [CrossRef]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Patikorn, C.; Saidoung, P.; Pham, T.; Phisalprapa, P.; Lee, Y.Y.; Varady, K.A.; Veettil, S.K.; Chaiyakunapruk, N. Effects of ketogenic diet on health outcomes: An umbrella review of meta-analyses of randomized clinical trials. BMC Med. 2023, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. The regulation of the release of ketone bodies by the liver. Adv. Enzym. Regul. 1966, 4, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, R.S.; Hendler, R.G.; Felig, P. Effect of diabetes mellitus and insulin on the turnover and metabolic response to ketones in man. Diabetes 1976, 25, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Thaele-Franz, M.A.; Steckhan, N.; Michalsen, A.; Stange, R. Ketosis in patients undergoing medically supervised therapeutic fasting-results from an observational trial. Eur. J. Clin. Nutr. 2020, 74, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Antonio Paoli, A.; Mancin, L.; Caprio, M.; Monti, E.; Narici, M.V.; Cenci, L.; Piccini, F.; Pincella, M.; Grigoletto, D.; Marcolin, G. Effects of 30 days of ketogenic diet on body composition, muscle strength, muscle area, metabolism, and performance in semi-professional soccer players. J. Int. Soc. Sport. Nutr. 2021, 18, 62. [Google Scholar] [CrossRef]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic Diet in Neuromuscular and Neurodegenerative Diseases. BioMed Res. Int. 2014, 2014, 474296. [Google Scholar] [CrossRef]
- Hirobata, T.; Inaba, H.; Kaido, Y.; Kosugi, D.; Itoh, S.; Matsuoka, T.; Inoue, G. Serum ketone body measurement in patients with diabetic ketoacidosis. Diabetol. Int. 2022, 13, 624–630. [Google Scholar] [CrossRef]
- Lodi, A.; Zarantonello, L.; Bisiacchi, P.S.; Cenci, L.; Paoli, A. Ketonemia and Glycemia Affect Appetite Levels and Executive Functions in Overweight Females During Two Ketogenic Diets. Obesity 2020, 28, 1868–1877. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr.; Veech, R.L. Ketoacids? Good medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–161; discussion 162–143. [Google Scholar]
- Paoli, A.; Bianco, A.; Grimaldi, K.A. The Ketogenic Diet and Sport: A Possible Marriage? Exerc. Sport. Sci. Rev. 2015, 43, 153–162. [Google Scholar] [CrossRef]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Puchowicz, M.A.; Zechel, J.L.; Valerio, J.; Emancipator, D.S.; Xu, K.; Pundik, S.; LaManna, J.C.; Lust, W.D. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Fukao, T.; Mitchell, G.; Sass, J.O.; Hori, T.; Orii, K.; Aoyama, Y. Ketone body metabolism and its defects. J. Inherit. Metab. Dis. 2014, 37, 541–551. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Hormonal control of ketogenesis. Biochemical considerations. Arch. Intern. Med. 1977, 137, 495–501. [Google Scholar] [CrossRef]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F., Jr. Liver and kidney metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 574–583. [Google Scholar] [CrossRef]
- Barrea, L.; Verde, L.; Camajani, E.; Sojat, A.S.; Marina, L.; Savastano, S.; Colao, A.; Caprio, M.; Muscogiuri, G. Effects of very low-calorie ketogenic diet on hypothalamic-pituitary-adrenal axis and renin-angiotensin-aldosterone system. J. Endocrinol. Investig. 2023. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Park, H.; Kaushik, V.K.; Dean, D.; Constant, S.; Prentki, M.; Saha, A.K. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol. Scand. 2003, 178, 435–442. [Google Scholar] [CrossRef]
- Ness, G.C.; Zhao, Z.; Wiggins, L. Insulin and glucagon modulate hepatic 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity by affecting immunoreactive protein levels. J. Biol. Chem. 1994, 269, 29168–29172. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Ebbeling, C.B. The Carbohydrate-Insulin Model of Obesity: Beyond “Calories In, Calories Out”. JAMA Intern. Med. 2018, 178, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Schoeller, D.A. The energy balance equation: Looking back and looking forward are two very different views. Nutr. Rev. 2009, 67, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Mutch, D.M.; Clement, K. Unraveling the genetics of human obesity. PLoS Genet. 2006, 2, e188. [Google Scholar] [CrossRef] [PubMed]
- Bueno, N.B.; de Melo, I.S.; de Oliveira, S.L.; da Rocha Ataide, T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef]
- Muscogiuri, G.; El Ghoch, M.; Colao, A.; Hassapidou, M.; Yumuk, V.; Busetto, L.; Obesity Management Task Force of the European Association for the Study of Obesity. European Guidelines for Obesity Management in Adults with a Very Low-Calorie Ketogenic Diet: A Systematic Review and Meta-Analysis. Obes. Facts 2021, 14, 222–245. [Google Scholar] [CrossRef]
- Castellana, M.; Conte, E.; Cignarelli, A.; Perrini, S.; Giustina, A.; Giovanella, L.; Giorgino, F.; Trimboli, P. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: A systematic review and meta-analysis. Rev. Endocr. Metab. Disord. 2020, 21, 5–16. [Google Scholar] [CrossRef]
- Hall, K.D. A review of the carbohydrate-insulin model of obesity. Eur. J. Clin. Nutr. 2017, 71, 323–326. [Google Scholar] [CrossRef]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy expenditure and body composition changes after an isocaloric ketogenic diet in overweight and obese men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef]
- Veldhorst, M.; Smeets, A.; Soenen, S.; Hochstenbach-Waelen, A.; Hursel, R.; Diepvens, K.; Lejeune, M.; Luscombe-Marsh, N.; Westerterp-Plantenga, M. Protein-induced satiety: Effects and mechanisms of different proteins. Physiol. Behav. 2008, 94, 300–307. [Google Scholar] [CrossRef]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Ketosis and appetite-mediating nutrients and hormones after weight loss. Eur. J. Clin. Nutr. 2013, 67, 759–764. [Google Scholar] [CrossRef]
- Gibson, A.A.; Seimon, R.V.; Lee, C.M.; Ayre, J.; Franklin, J.; Markovic, T.P.; Caterson, I.D.; Sainsbury, A. Do ketogenic diets really suppress appetite? A systematic review and meta-analysis. Obes. Rev. 2015, 16, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Klein, G.L.; Luoto, P.K.; Wong, J.M.W.; Bielak, L.; Eddy, R.G.; Steltz, S.K.; Devlin, C.; Sandman, M.; Hron, B.; et al. A randomized study of dietary composition during weight-loss maintenance: Rationale, study design, intervention, and assessment. Contemp. Clin. Trials 2018, 65, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Rubini, A.; Bosco, G.; Lodi, A.; Cenci, L.; Parmagnani, A.; Grimaldi, K.; Zhongjin, Y.; Paoli, A. Erratum to: Effects of Twenty Days of the Ketogenic Diet on Metabolic and Respiratory Parameters in Healthy Subjects. Lung 2017, 195, 155. [Google Scholar] [CrossRef] [PubMed]
- Barkeling, B.; Rossner, S.; Bjorvell, H. Effects of a high-protein meal (meat) and a high-carbohydrate meal (vegetarian) on satiety measured by automated computerized monitoring of subsequent food intake, motivation to eat and food preferences. Int. J. Obes. 1990, 14, 743–751. [Google Scholar] [PubMed]
- Stubbs, R.J.; van Wyk, M.C.; Johnstone, A.M.; Harbron, C.G. Breakfasts high in protein, fat or carbohydrate: Effect on within-day appetite and energy balance. Eur. J. Clin. Nutr. 1996, 50, 409–417. [Google Scholar]
- Astrup, A. The satiating power of protein—A key to obesity prevention? Am. J. Clin. Nutr. 2005, 82, 1–2. [Google Scholar] [CrossRef]
- Weigle, D.S.; Breen, P.A.; Matthys, C.C.; Callahan, H.S.; Meeuws, K.E.; Burden, V.R.; Purnell, J.Q. A high-protein diet induces sustained reductions in appetite, ad libitum caloric intake, and body weight despite compensatory changes in diurnal plasma leptin and ghrelin concentrations. Am. J. Clin. Nutr. 2005, 82, 41–48. [Google Scholar] [CrossRef]
- Johnstone, A.M.; Horgan, G.W.; Murison, S.D.; Bremner, D.M.; Lobley, G.E. Effects of a high-protein ketogenic diet on hunger, appetite, and weight loss in obese men feeding ad libitum. Am. J. Clin. Nutr. 2008, 87, 44–55. [Google Scholar] [CrossRef]
- Hall, K.D.; Guo, J.; Courville, A.B.; Boring, J.; Brychta, R.; Chen, K.Y.; Darcey, V.; Forde, C.G.; Gharib, A.M.; Gallagher, I.; et al. Effect of a plant-based, low-fat diet versus an animal-based, ketogenic diet on ad libitum energy intake. Nat. Med. 2021, 27, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Valassi, E.; Scacchi, M.; Cavagnini, F. Neuroendocrine control of food intake. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Long-term persistence of hormonal adaptations to weight loss. N. Engl. J. Med. 2011, 365, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, J.; Mutungi, G.; Puglisi, M.J.; Volek, J.S.; Fernandez, M.L. Carbohydrate restriction (with or without additional dietary cholesterol provided by eggs) reduces insulin resistance and plasma leptin without modifying appetite hormones in adult men. Nutr. Res. 2009, 29, 262–268. [Google Scholar] [CrossRef]
- Cipryan, L.; Dostal, T.; Plews, D.J.; Hofmann, P.; Laursen, P.B. Adiponectin/leptin ratio increases after a 12-week very low-carbohydrate, high-fat diet, and exercise training in healthy individuals: A non-randomized, parallel design study. Nutr. Res. 2021, 87, 22–30. [Google Scholar] [CrossRef]
- Suyama, S.; Maekawa, F.; Maejima, Y.; Kubota, N.; Kadowaki, T.; Yada, T. Glucose level determines excitatory or inhibitory effects of adiponectin on arcuate POMC neuron activity and feeding. Sci. Rep. 2016, 6, 30796. [Google Scholar] [CrossRef]
- Deemer, S.E.; Plaisance, E.P.; Martins, C. Impact of ketosis on appetite regulation—A review. Nutr. Res. 2020, 77, 1–11. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Cyranka, M.; Clarke, K.; de Wet, H. A Ketone Ester Drink Lowers Human Ghrelin and Appetite. Obesity 2017, 26, 269–273. [Google Scholar] [CrossRef]
- Paoli, A. Booster Ketones: Battling Hunger. Obesity 2018, 26, 252–253. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Dickinson, S.L.; Henschel, B.; Ebbeling, C.B.; Allison, D.B. Do Lower-Carbohydrate Diets Increase Total Energy Expenditure? An Updated and Reanalyzed Meta-Analysis of 29 Controlled-Feeding Studies. J. Nutr. 2021, 151, 482–490. [Google Scholar] [CrossRef]
- Ebbeling, C.B.; Feldman, H.A.; Klein, G.L.; Wong, J.M.W.; Bielak, L.; Steltz, S.K.; Luoto, P.K.; Wolfe, R.R.; Wong, W.W.; Ludwig, D.S. Effects of a low carbohydrate diet on energy expenditure during weight loss maintenance: Randomized trial. BMJ 2018, 363, k4583. [Google Scholar] [CrossRef]
- Hall, K.D.; Bemis, T.; Brychta, R.; Chen, K.Y.; Courville, A.; Crayner, E.J.; Goodwin, S.; Guo, J.; Howard, L.; Knuth, N.D.; et al. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. Cell Metab. 2015, 22, 427–436. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Ebbeling, C.B. Raising the bar on the low-carbohydrate diet. Am. J. Clin. Nutr. 2016, 104, 1487–1488. [Google Scholar] [CrossRef]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; Ravussin, E. Reply to DS Ludwig and CB Ebbeling. Am. J. Clin. Nutr. 2016, 104, 1488–1490. [Google Scholar] [CrossRef]
- Gomez-Arbelaez, D.; Crujeiras, A.B.; Castro, A.I.; Martinez-Olmos, M.A.; Canton, A.; Ordoñez-Mayan, L.; Sajoux, I.; Galban, C.; Bellido, D.; Casanueva, F.F. Resting metabolic rate of obese patients under very low calorie ketogenic diet. Nutr. Metab. 2018, 15, 18. [Google Scholar] [CrossRef]
- Gomez-Arbelaez, D.; Bellido, D.; Castro, A.I.; Ordonez-Mayan, L.; Carreira, J.; Galban, C.; Martinez-Olmos, M.A.; Crujeiras, A.B.; Sajoux, I.; Casanueva, F.F. Body Composition Changes After Very-Low-Calorie Ketogenic Diet in Obesity Evaluated by 3 Standardized Methods. J. Clin. Endocrinol. Metab. 2017, 102, 488–498. [Google Scholar] [CrossRef]
- Paoli, A.; Cenci, L.; Fancelli, M.; Parmagnani, A.; Fratter, A.; Cucchi, A.; Bianco, A. Ketogenic diet and phytoextracts Comparison of the efficacy of Mediterranean, zone and tisanoreica diet on some health risk factors. Agro Food Ind. Hi-Tech 2010, 21, 24–29. [Google Scholar]
- Paoli, A.; Cenci, L.; Grimaldi, K.A. Effect of Ketogenic Mediterranean diet with phytoextracts and low carbohydrates/high-protein meals on weight, cardiovascular risk factors, body composition and diet compliance in Italian council employees. Nutr. J. 2011, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Milic, J.; Sebastiani, G.; Raggi, P. Sarcopenic obesity at the crossroad of pathogenesis of cardiometabolic diseases. Atherosclerosis 2021, 335, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Bortz, W.M.; Paul, P.; Haff, A.C.; Holmes, W.L. Glycerol turnover and oxidation in man. J. Clin. Investig. 1972, 51, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Bertoli, S.; Trentani, C.; Borrelli, P.; Veggiotti, P. Effects of the ketogenic diet on nutritional status, resting energy expenditure, and substrate oxidation in patients with medically refractory epilepsy: A 6-month prospective observational study. Clin. Nutr. 2012, 31, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Kolanowski, J.; Bodson, A.; Desmecht, P.; Bemelmans, S.; Stein, F.; Crabbe, J. On the relationship between ketonuria and natriuresis during fasting and upon refeeding in obese patients. Eur. J. Clin. Investig. 1978, 8, 277–282. [Google Scholar] [CrossRef]
- Romano, L.; Marchetti, M.; Gualtieri, P.; Di Renzo, L.; Belcastro, M.; De Santis, G.L.; Perrone, M.A.; De Lorenzo, A. Effects of a Personalized VLCKD on Body Composition and Resting Energy Expenditure in the Reversal of Diabetes to Prevent Complications. Nutrients 2019, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Zaki, H.A.; Iftikhar, H.; Bashir, K.; Gad, H.; Samir Fahmy, A.; Elmoheen, A. A Comparative Study Evaluating the Effectiveness Between Ketogenic and Low-Carbohydrate Diets on Glycemic and Weight Control in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Cureus 2022, 14, e25528. [Google Scholar] [CrossRef]
- Ashtary-Larky, D.; Bagheri, R.; Asbaghi, O.; Tinsley, G.M.; Kooti, W.; Abbasnezhad, A.; Afrisham, R.; Wong, A. Effects of resistance training combined with a ketogenic diet on body composition: A systematic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 2022, 62, 5717–5732. [Google Scholar] [CrossRef]
- Parry-Strong, A.; Wright-McNaughton, M.; Weatherall, M.; Hall, R.M.; Coppell, K.J.; Barthow, C.; Krebs, J.D. Very low carbohydrate (ketogenic) diets in type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2022, 24, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, M.; Liang, J.; He, G.; Chen, N. Ketogenic Diet Benefits to Weight Loss, Glycemic Control, and Lipid Profiles in Overweight Patients with Type 2 Diabetes Mellitus: A Meta-Analysis of Randomized Controlled Trails. Int. J. Environ. Res. Public Health 2022, 19, 10429. [Google Scholar] [CrossRef]
- Stefan, N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol. 2020, 8, 616–627. [Google Scholar] [CrossRef]
- Pi-Sunyer, X. Changes in body composition and metabolic disease risk. Eur. J. Clin. Nutr. 2019, 73, 231–235. [Google Scholar] [CrossRef]
- Al Aamri, K.S.; Alrawahi, A.H.; Al Busaidi, N.; Al Githi, M.S.; Al Jabri, K.; Al Balushi, F.; Ronquillo-Talara, R.; Al Balushi, S.; Waly, M. The effect of low-carbohydrate ketogenic diet in the management of obesity compared with low caloric, low-fat diet. Clin. Nutr. ESPEN 2022, 49, 522–528. [Google Scholar] [CrossRef]
- Paoli, A.; Cancellara, P.; Pompei, P.; Moro, T. Ketogenic Diet and Skeletal Muscle Hypertrophy: A Frenemy Relationship? J. Hum. Kinet. 2019, 68, 233–247. [Google Scholar] [CrossRef]
- Miller, V.J.; LaFountain, R.A.; Barnhart, E.; Sapper, T.S.; Short, J.; Arnold, W.D.; Hyde, P.N.; Crabtree, C.D.; Kackley, M.L.; Kraemer, W.J. A ketogenic diet combined with exercise alters mitochondrial function in human skeletal muscle while improving metabolic health. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E995–E1007. [Google Scholar] [CrossRef]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diabetes Rep. 2021, 21, 15. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging role of hepatic ketogenesis in fatty liver disease. Front. Physiol. 2022, 13, 1300. [Google Scholar] [CrossRef]
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739. [Google Scholar] [CrossRef]
- Cunha, G.M.; Guzman, G.; Correa De Mello, L.L.; Trein, B.; Spina, L.; Bussade, I.; Marques Prata, J.; Sajoux, I.; Countinho, W. Efficacy of a 2-Month Very Low-Calorie Ketogenic Diet (VLCKD) Compared to a Standard Low-Calorie Diet in Reducing Visceral and Liver Fat Accumulation in Patients with Obesity. Front. Endocrinol. 2020, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Tam, S.; Murray-Segal, L.; Huynh, K.; Meikle, P.J.; Scott, J.W.; van Denderen, B.; Chen, Z.; Steel, R.; LeBlond, N.D.; et al. Inhibition of Adenosine Monophosphate-Activated Protein Kinase-3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Signaling Leads to Hypercholesterolemia and Promotes Hepatic Steatosis and Insulin Resistance. Hepatol. Commun. 2019, 3, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Forsythe, C.E. Modification of lipoproteins by very low-carbohydrate diets. J. Nutr. 2005, 135, 1339–1342. [Google Scholar] [CrossRef]
- Kirk, E.; Reeds, D.N.; Finck, B.N.; Mayurranjan, S.M.; Patterson, B.W.; Klein, S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 2009, 136, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Jani, S.; Da Eira, D.; Stefanovic, M.; Ceddia, R.B. The ketogenic diet prevents steatosis and insulin resistance by reducing lipogenesis, diacylglycerol accumulation and protein kinase C activity in male rat liver. J. Physiol. 2022, 600, 4137–4151. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Cerullo, G. Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review. Antioxidants 2023, 12, 1065. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, J.O.; Jensen, M.D. Fat depots, free fatty acids, and dyslipidemia. Nutrients 2013, 5, 498–508. [Google Scholar] [CrossRef]
- Zhu, H.; Bi, D.; Zhang, Y.; Kong, C.; Du, J.; Wu, X.; Wei, Q.; Qin, H. Ketogenic diet for human diseases: The underlying mechanisms and potential for clinical implementations. Signal Transduct. Target. Ther. 2022, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, J.; Yang, S.; Gao, M.; Cao, L.; Li, X.; Hong, D.; Tian, S.; Sun, C. Effect of the ketogenic diet on glycemic control, insulin resistance, and lipid metabolism in patients with T2DM: A systematic review and meta-analysis. Nutr. Diabetes 2020, 10, 38. [Google Scholar] [CrossRef]
- Battezzati, A.; Foppiani, A.; Leone, A.; De Amicis, R.; Spadafranca, A.; Mari, A.; Bertoli, S. Acute Insulin Secretory Effects of a Classic Ketogenic Meal in Healthy Subjects: A Randomized Cross-Over Study. Nutrients 2023, 15, 1119. [Google Scholar] [CrossRef]
- Rafiullah, M.; Musambil, M.; David, S.K. Effect of a very low-carbohydrate ketogenic diet vs recommended diets in patients with type 2 diabetes: A meta-analysis. Nutr. Rev. 2022, 80, 488–502. [Google Scholar] [CrossRef]
- Hanners, A.; Melnyk, B.M.; Volek, J.; Kelley, M.M. Ketogenic diet, African American women, and cardiovascular health: A systematic review. Worldviews Evid. Based Nurs. 2022, 19, 35–41. [Google Scholar] [CrossRef]
- Brinkworth, G.D.; Noakes, M.; Buckley, J.D.; Keogh, J.B.; Clifton, P.M. Long-term effects of a very-low-carbohydrate weight loss diet compared with an isocaloric low-fat diet after 12 mo. Am. J. Clin. Nutr. 2009, 90, 23–32. [Google Scholar] [CrossRef]
- Nordmann, A.J.; Nordmann, A.; Briel, M.; Keller, U.; Yancy, W.S., Jr.; Brehm, B.J.; Bucher, H.C. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2006, 166, 285–293. [Google Scholar] [CrossRef]
- Tay, J.; Brinkworth, G.D.; Noakes, M.; Keogh, J.; Clifton, P.M. Metabolic effects of weight loss on a very-low-carbohydrate diet compared with an isocaloric high-carbohydrate diet in abdominally obese subjects. J. Am. Coll. Cardiol. 2008, 51, 59–67. [Google Scholar] [CrossRef]
- Mancin, L.; Piccini, F.; Paoli, A. Ketogenic diet and nafld: A great therapeutic opportunity? Acta Med. Mediterr. 2019, 35, 1909–1913. [Google Scholar]
- Sherrier, M.; Li, H. The impact of keto-adaptation on exercise performance and the role of metabolic-regulating cytokines. Am. J. Clin. Nutr. 2019, 110, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Yamaguchi, K.; Seko, Y.; Okishio, S.; Ishiba, H.; Tochiki, N.; Takahashi, A.; Kataoka, S.; Okuda, K.; Liu, Y.; et al. Hepatocyte-specific fibroblast growth factor 21 overexpression ameliorates high-fat diet-induced obesity and liver steatosis in mice. Lab. Investig. 2022, 102, 281–289. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Tiganis, T. Reactive oxygen species and insulin resistance: The good, the bad and the ugly. Trends Pharmacol. Sci. 2011, 32, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Veech, R.L. The determination of the redox states and phosphorylation potential in living tissues and their relationship to metabolic control of disease phenotypes. Biochem. Mol. Biol. Educ. 2006, 34, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Ipek, O.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD(+)/NADH Ratio in Healthy Human Brain: An in Vivo Study by (31)P-MRS. Front. Nutr. 2018, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The mechanisms by which the ketone body D-β-hydroxybutyrate may improve the multiple cellular pathologies of Parkinson’s disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, M.H.; Ha, S.; Bang, E.J.; Lee, Y.; Lee, A.K.; Lee, J.; Yu, B.P.; Chung, H.Y. Anti-inflammatory action of beta-hydroxybutyrate via modulation of PGC-1alpha and FoxO1, mimicking calorie restriction. Aging 2019, 11, 1283–1304. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed]
- Kenez, A.; Locher, L.; Rehage, J.; Danicke, S.; Huber, K. Agonists of the G protein-coupled receptor 109A-mediated pathway promote antilipolysis by reducing serine residue 563 phosphorylation of hormone-sensitive lipase in bovine adipose tissue explants. J. Dairy Sci. 2014, 97, 3626–3634. [Google Scholar] [CrossRef]
- Dobbins, R.; Byerly, R.; Gaddy, R.; Gao, F.; Mahar, K.; Napolitano, A.; Ambery, P.; Le Monnier de Gouville, A.C. GSK256073 acutely regulates NEFA levels via HCA2 agonism but does not achieve durable glycaemic control in type 2 diabetes. A randomised trial. Eur. J. Pharmacol. 2015, 755, 95–101. [Google Scholar] [CrossRef]
- Dobbins, R.L.; Shearn, S.P.; Byerly, R.L.; Gao, F.F.; Mahar, K.M.; Napolitano, A.; Nachbaur, G.J.; Le Monnier de Gouville, A.C. GSK256073, a selective agonist of G-protein coupled receptor 109A (GPR109A) reduces serum glucose in subjects with type 2 diabetes mellitus. Diabetes Obes. Metab. 2013, 15, 1013–1021. [Google Scholar] [CrossRef]
- Shaw, J.H.; Wolfe, R.R. Influence of beta-hydroxybutyrate infusion on glucose and free fatty acid metabolism in dogs. Am. J. Physiol. 1984, 247, E756–E764. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Lukasova, M.; Offermanns, S.; Zhang, Y.; Cao, G.; Judd, R.L. Niacin stimulates adiponectin secretion through the GPR109A receptor. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E549–E558. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Bajaj, M.; Suraamornkul, S.; Kashyap, S.; Cusi, K.; Mandarino, L.; DeFronzo, R.A. Sustained reduction in plasma free fatty acid concentration improves insulin action without altering plasma adipocytokine levels in subjects with strong family history of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol. Sci. 2006, 27, 384–390. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. beta-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Dilmore, A.H.; Martino, C.; Neth, B.J.; West, K.A.; Zemlin, J.; Rahman, G.; Panitchpakdi, M.; Meehan, M.J.; Weldon, K.C.; Blach, C.; et al. Effects of a ketogenic and low-fat diet on the human metabolome, microbiome, and foodome in adults at risk for Alzheimer’s disease. Alzheimer’s Dement. 2023. [Google Scholar] [CrossRef]
- Palmnas-Bedard, M.S.A.; Costabile, G.; Vetrani, C.; Aberg, S.; Hjalmarsson, Y.; Dicksved, J.; Riccardi, G.; Landberg, R. The human gut microbiota and glucose metabolism: A scoping review of key bacteria and the potential role of SCFAs. Am. J. Clin. Nutr. 2022, 116, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef]
- Gudan, A.; Skonieczna-Zydecka, K.; Palma, J.; Drozd, A.; Stachowska, E. Effects of dietary components on intestinal short-chain fatty acids (SCFAs) synthesis in healthy adult persons following a ketogenic diet. Rocz. Panstw. Zakl. Hig. 2022, 73, 51–69. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef]
- Prasun, P. Role of mitochondria in pathogenesis of type 2 diabetes mellitus. J. Diabetes Metab. Disord. 2020, 19, 2017–2022. [Google Scholar] [CrossRef] [PubMed]


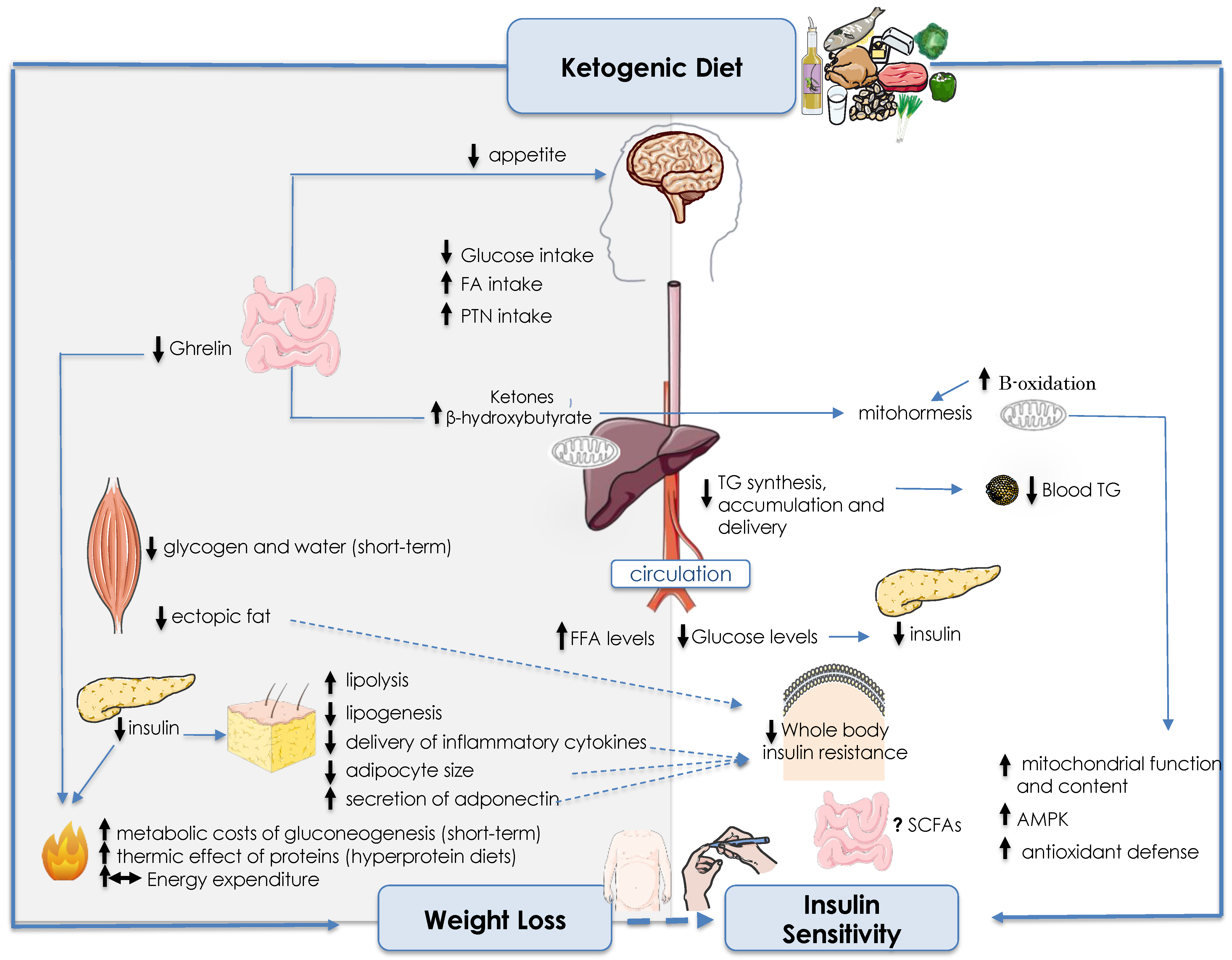{kind=link}
| Population Characteristics (N, Age, Sex) | Study DESIGN | Intervention | Control Diet | Duration | Body Composition Outcomes | Glycemic and Lipid Profile | Reference |
|---|---|---|---|---|---|---|---|
| N = 1415; >18 years; M and F; BMI > 27.5 kg/m2 | Meta-analysis of RCT | VLCKD (<50 g CHO/day or 10% of DE) | LFD (<30% energy from fat | ≥12 months | ↓ Greater weight loss in VLCKD groups WMD −0·91 (95% CI −1.65 to −0.17) kg | ↔ No differences between dietary groups for GLU, insulin, HbA1c, and CRP levels ↓ Greater TG reduction in VLCKD groups −0.18 mmol/L (WMD; 95% CI −0.27 to −0.08) ↑ Greater increase in HDL-C in VLCKD groups 0.09 mmol/L (WMD; 95 % CI 0.06 to 0.12) ↑ Greater increase in LDL-C in VLKD groups 0.12 mmol/L (WMD; 95 % CI 0.04 to 0.2) ↓ Greater reduction in DBP in VLKD groups −1.43 mmHg (WMD; 95 % CI −2.49 to −0.37) | [118] |
| N = 835 ≥18 years; M and F; overweight or obesity. | Meta-analysis of noncontrolled, controlled, and RCT | VLCK (30 to 50 g CHO/day and DE ≤ 800 kcal; protein, 0.8–1.2 g/day for an ideal body weight) | Any other weight loss diet | 3 weeks to 24 months | ↓ Greater mean weight loss and FM loss in KD groups respectively −7.06 kg (95% CI −11.16 to −2.97) and −9.35 kg (95% CI −13.29 to −5.41) ↔ No differences between dietary groups for FFM | ↔ No differences between dietary groups for GLU, HbA1c and LDL-c ↓ Greater reduction in HOMA-IR index in VLKD groups –1.36 (WMD; 95% CI –2.14 to –0.57) ↓ Greater reduction in cholesterol in VLKD groups –7.13 mg/dL (WMD; 95% CI –9.71 to –4.55) ↓ Greater reduction in TG in VLKD groups –29.90 mg/dL (WMD; 95% CI –42.47 to –17.32) | [119] |
| N = 801 M and F ≥18 years; overweight or obesity. | Meta-analysis of sentinel studies, including observational studies and RCT | VLCKD (CHO < 50 g/day and <800 kcal/day) | Other low or very low-calorie diets | 3 weeks to 24 months | ↔ No differences between VLCKD and very low-calorie diets for weight loss; −10.0 kg (95% CI −13.2 to −6.8) ↓ Greater mean weight loss in VLCKD groups compared to low calorie diet groups | [120] | |
| N = 1282 M and F patients with type 2 diabetes. | Meta-analysis of observational studies and RCT | KD or low carbohydrate diets (≤50 g CHO/day or ≤20% of DE from CHO) | Other diets | ↓ Greater weight loss in KD −2.67 kg (SMD; 95% CI −4.05 to −1.28 kg) ↔ No differences between KD and other diets for BMI | ↓ Greater reduction in HbA1c in KD diet compared to control diets −1.45% (SMD; 95% CI −2.73 to −0.17%) | [156] | |
| N = 567 ≥18 years; M and F patients with type 2 diabetes. | Meta-analysis of single-arm trials (pre-post studies) | VLCKD, LCF and VLCK (≤50 g CHO/day or ≤14% of DE from CHO) | No comparison groups | 1 to 56 weeks | ↓ body weight 8.66 kg (MC; 95% CI −11.40 to −5.92) after the intervention of KDs ↓ WC 9.17 cm (MC; 95% CI −10.67 to −7.66) after the intervention of KDs ↓ BMI −3.13 kg/m2 (MC; 95% CI −3.31 to 2.95 kg/m2) | ↓ GLU 1.29 mmol/L (MC; 95% CI −1.78 to −0.79) after the intervention of KDs ↓ HbA1c −1.07% (MC; 95% CI −1.37 to −0.78) after the intervention of KDs ↓ TG −0.72 mmol/L (MC; 95% CI −1.01 to −0.43) after the intervention of KDs ↓ TC −0.33 mmol/L (MC; 95% CI −0.66 to −0.01) after the intervention of KDs ↓ LDL-C −0.05 mmol/L (MC; 95% CI −0.25 to 0.15) after the intervention of KDs ↑ HDL-C 0.14 mmol/L (MC; 95% CI 0.03 to 0.25) after the intervention of KDs | [177] |
| N = 648 ≥18 years; M and F (65% and 100% of study participants were female) patients with type 2 diabetes, overweight or obesity. | Meta-analysis of RCT | VLCK (<50 g CHO/day or <10% of DE from CHO) | Any recommended diet for type 2 diabetes | 4 to 12 months | ↓ Greater weight loss in VLCK diets after 3 and 6 months respectively −2.91 kg (WMD; 95% CI −4.88 to −0.95) and −2.84 kg (WMD; 95% CI −5.29 to −0.39) ↔ No differences between dietary groups for weight loss after 12 months | ↓ Greater reduction in HbA1c in VLCK diets after 3 and 6 months respectively −6.7 mmol/mol (WMD; 95% CI −9.0 to −4.4) and −6.3 mmol/mol (WMD; 95% CI −9.3 to −3.5) ↔ No differences between dietary groups for HbA1c after 12 months ↓ Greater reduction in TG in VLCK diets after 6 and 12 months respectively −18.36 mg/dL (WMD; 95% CI −24.24 to −12.49) and −24.10 mg/dL (WMD; 95% CI −33.93 to −14.27) ↔ No differences between dietary groups for LDL-C after 3 and 6 months ↑ Greater increase in LDL-C in VLCKD groups after 12 months 6.35 mg/dL (WMD; 95% CI 2.02 to 10.69) ↑ Greater increase in HDL-C in VLCKD groups after 3, 6 and 12 months respectively 1.27 mg/dL (WMD; 95% CI 0.32 to 2.22); 3.01 mg/dL (WMD; 95% CI 0.41 to 5.61) and 1.88 mg/dL (WMD; 95% CI 0.37 to 3.39) | [179] |
| N = 322 ≥18 years; M and F; overweight or obesity. | Meta-analysis of experimental and quasi-experimental studies | KD (20–70 g CHO/day or ≤10% of a 2000 kcal/day diet) | No comparison groups or other types of diets or usual care | 6 weeks to 1 year | All studies showed a decrease in weight, BMI, and BFP in participants on a KD | GLU, HbA1c, and fasting insulin, decreased in all studies All studies showed a decrease in triglycerides Conflicting results were observed between studies for HDL-C and LDL-C. | [180] |
| N = 447 ≥16 years M and F overweight or obesity. | Meta-analysis of RCT | ILCD (≤60 g CHO/day) | Low-fat diet | 6 to 12 months | ↓ Greater weight loss in isocaloric low-carbohydrate diet compared to low fat diets after 6 months −3.3 kg (WMD; 95% CI −5.3 to −1.4) ↔ No differences between dietary groups for weight loss after 12 months | Conflicting results were observed between the dietary interventions for GLU and insulin. ↑ Greater increase in HDL-C in isocaloric low-carbohydrate diets after 6 months 4.6 mg/dL (WMD; 95% CI 1.5 to 8.1) ↓ Greater reduction in TG in isocaloric low-carbohydrate diet after 6 and 12 months respectively −22.1 mg/dL (WMD; 95% CI −38.1 to −5.3) and −31 mg/dL (WMD; 95% CI −59.3 to −2.7) ↓ Greater reduction in TC and LDL-C in low fat diets after 6 and 12 months. | [182] |
| N = 606 ≥16 years M and F people with pre-diabetes or type 2 diabetes | Meta-analysis of RCT | VLCK (≤50 g CHO/day) | Diet containing a carbohydrate content above 50 g/day (all comparison diets were low fat) | 3 to 24 months | ↔ No differences between dietary groups for weight loss, BMI, WC, FM, FFM, after 12 months. | ↔ No differences between dietary groups for HbA1c, GLU, fasting insulin, HOMA-IR, TC and LDL, after 12 months. ↓ Greater reduction in TG in VLCK after 12 months −0.28 mmol/L (MC; 95% CI −0.44 to −0.11) ↑ Greater increase in HDL-C in VLCK after 12 months 0.04 mmol/L (MC; 95% CI 0.01 to 0.08) | [158] |
| N = 611 ≥18 years M and F people with type 2 diabetes and overweight or obesity. | Meta-analysis of RCT | KD (<50 g CHO/day) | Other diets than KDs | 3 months to 2 years | ↓ Greater weight loss in KD groups −5.637 kg (SMD; 95% CI −9.76 to −1.49); ↓ Greater reduction in WC in KD groups −2.32 cm (SMD; 95% CI −4.58 to −0.06); ↔ No differences between dietary groups for BMI. | ↔ No differences between dietary groups for GLU, insulin, HOMA-IR, TC and LDL-C ↓ Greater reduction in HbA1c in KD groups −0.38% (SMD; 95% CI −0.61 to −0.16%) ↓ Greater reduction in TG in KD groups −0.36 mmol/L (SMD; 95% CI −0.55 to −0.18) ↑ Greater increase in HDL-C in KD groups 0.28 mmol/L (SMD; 95 % CI 0.09 to 0.46) | [159] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoli, A.; Bianco, A.; Moro, T.; Mota, J.F.; Coelho-Ravagnani, C.F. The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg? Nutrients 2023, 15, 3120. https://doi.org/10.3390/nu15143120
Paoli A, Bianco A, Moro T, Mota JF, Coelho-Ravagnani CF. The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg? Nutrients. 2023; 15(14):3120. https://doi.org/10.3390/nu15143120
Chicago/Turabian StylePaoli, Antonio, Antonino Bianco, Tatiana Moro, Joao Felipe Mota, and Christianne F. Coelho-Ravagnani. 2023. "The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg?" Nutrients 15, no. 14: 3120. https://doi.org/10.3390/nu15143120
APA StylePaoli, A., Bianco, A., Moro, T., Mota, J. F., & Coelho-Ravagnani, C. F. (2023). The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg? Nutrients, 15(14), 3120. https://doi.org/10.3390/nu15143120