

Advanced Glycation End Products (AGEs) in Diet and Skin in Relation to Stool Microbiota: The Rotterdam Study


 , and
, and
Abstract
1. Introduction
2. Subjects and Methods
2.1. Study Population
2.2. Estimation of Dietary AGEs and Other Dietary Characteristics
2.3. Measurement of Skin AGEs as SAF
2.4. Stool Microbiota Profiling and Processing
2.5. Assessment of Covariates and Population Characteristics
2.6. Statistical Analysis
2.6.1. Characteristics of Microbial Composition
2.6.2. Microbial Pathway Prediction
2.6.3. dAGEs and Microbiota Analyses
- (1)
- dAGEs and alpha diversity
- (2)
- dAGEs and beta dissimilarity
- (3)
- dAGEs and abundance of individual genera
- (4)
- dAGEs and predicted microbial pathways
2.6.4. Stool Microbiota and SAF
2.6.5. Sensitivity Analysis
2.6.6. Imputation of Missing Values
3. Results
3.1. dAGEs and Stool Microbiota
3.1.1. Descriptive statistics
3.1.2. dAGEs and Overall Diversity and Dissimilarity of the Stool Microbiota
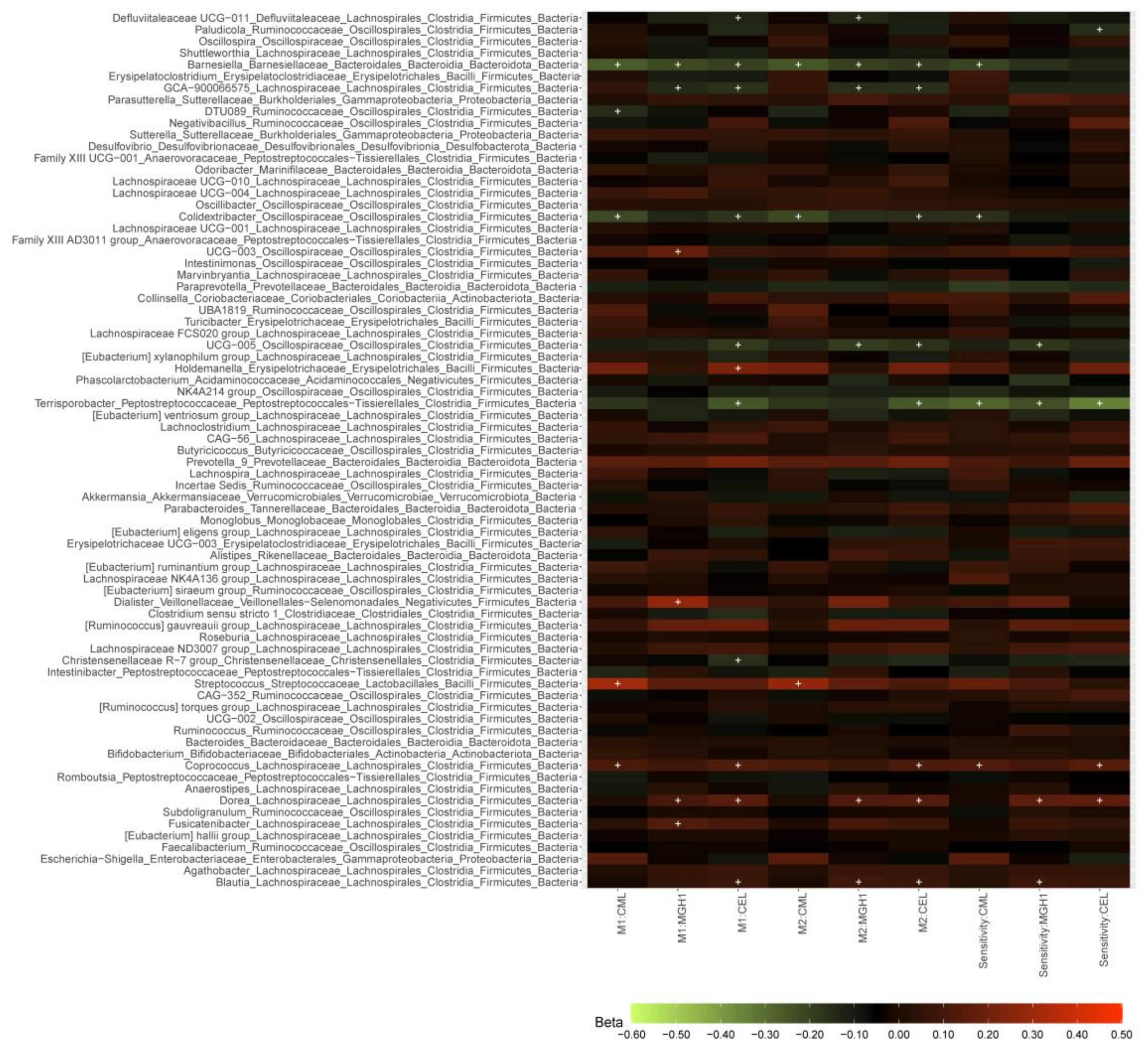
3.1.3. dAGEs and Microbial Abundance
3.1.4. dAGEs and Microbial Pathways
3.2. Stool Microbiota and SAF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henle, T. Protein-bound advanced glycation endproducts (AGEs) as bioactive amino acid derivatives in foods. Amino Acids. Dec. 2005, 29, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, N.; DeGroot, J.; Thorpe, S.R.; Bank, R.A.; Shaw, J.N.; Lyons, T.J.; Bijlsma, J.W.; Lafeber, F.P.; Baynes, J.W.; TeKoppele, J.M. Effect of collagen turnover on the accumulation of advanced glycation end products. J. Biol. Chem. 2000, 275, 39027–39031. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, Y.; Shen, Y.; Zhang, Y.; Liu, L.; Yang, X. Dietary polyphenols: Regulate the advanced glycation end products-RAGE axis and the microbiota-gut-brain axis to prevent neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2022, 1–27. [Google Scholar] [CrossRef]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does Accumulation of Advanced Glycation End Products Contribute to the Aging Phenotype? J. Gerontol. A Biol. 2010, 65, 963–975. [Google Scholar] [CrossRef]
- Twarda-Clapa, A.; Olczak, A.; Bialkowska, A.M.; Koziolkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef]
- Luevano-Contreras, C.; Gomez-Ojeda, A.; Macias-Cervantes, M.H.; Garay-Sevilla, M.E. Dietary Advanced Glycation End Products and Cardiometabolic Risk. Curr. Diabetes Rep. 2017, 17, 63. [Google Scholar] [CrossRef]
- Jiao, L.; Stolzenberg-Solomon, R.; Zimmerman, T.P.; Duan, Z.; Chen, L.; Kahle, L.; Risch, A.; Subar, A.F.; Cross, A.J.; Hollenbeck, A.; et al. Dietary consumption of advanced glycation end products and pancreatic cancer in the prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2015, 101, 126–134. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Seiquer, I.; Pilar Navarro, M. Study of the urinary and faecal excretion of Nepsilon-carboxymethyllysine in young human volunteers. Amino Acids 2012, 43, 595–602. [Google Scholar] [CrossRef]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef]
- Zhao, D.; Le, T.T.; Larsen, L.B.; Li, L.; Qin, D.; Su, G.; Li, B. Effect of glycation derived from alpha-dicarbonyl compounds on the in vitro digestibility of beta-casein and beta-lactoglobulin: A model study with glyoxal, methylglyoxal and butanedione. Food Res. Int. 2017, 102, 313–322. [Google Scholar] [CrossRef]
- Tadie, J.M.; Bae, H.B.; Banerjee, S.; Zmijewski, J.W.; Abraham, E. Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am. J. Physiol. -Cell. Physiol. 2012, 302, C249–C256. [Google Scholar] [CrossRef]
- Wang, J.; Cai, W.; Yu, J.; Liu, H.; He, S.; Zhu, L.; Xu, J. Dietary Advanced Glycation End Products Shift the Gut Microbiota Composition and Induce Insulin Resistance in Mice. Diabetes Metab. Syndr. Obes. 2022, 15, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.P.N.; Ritari, J.; Boeren, S.; de Waard, P.; Plugge, C.M.; de Vos, W.M. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat. Commun. 2015, 6, 10062. [Google Scholar] [CrossRef]
- Hellwig, M.; Auerbach, C.; Müller, N.; Samuel, P.; Kammann, S.; Beer, F.; Gunzer, F.; Henle, T. Metabolization of the Advanced Glycation End Product N-epsilon-Carboxymethyllysine (CML) by Different Probiotic E. coli Strains. J. Agric. Food Chem. 2019, 67, 1963–1972. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef]
- Nesreen, A.L.; Carbonero, F. Impact of Maillard reaction products on nutrition and health: Current knowledge and need to understand their fate in the human digestive system. Crit. Rev. Food Sci. Nutr. 2019, 59, 474–487. [Google Scholar]
- Phuong-Nguyen, K.; McNeill, B.A.; Aston-Mourney, K.; Rivera, L.R. Advanced Glycation End-Products and Their Effects on Gut Health. Nutrients 2023, 15, 405. [Google Scholar] [CrossRef]
- Cohen-Or, I.; Katz, C.; Ron, E.Z. AGEs Secreted by Bacteria Are Involved in the Inflammatory Response. PLoS ONE 2011, 6, e17974. [Google Scholar] [CrossRef]
- Rajaobelina, K.; Cougnard-Gregoire, A.; Delcourt, C.; Gin, H.; Barberger-Gateau, P.; Rigalleau, V. Autofluorescence of Skin Advanced Glycation End Products: Marker of Metabolic Memory in Elderly Population. J. Gerontol. A Biol. 2015, 70, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.A.; Brusselle, G.; Ghanbari, M.; Goedegebure, A.; Ikram, M.K.; Kavousi, M.; Kieboom, B.C.; Klaver, C.C.; de Knegt, R.J.; Luik, A.I.; et al. Objectives, design and main findings until 2020 from the Rotterdam Study. Eur. J. Epidemiol. 2020, 35, 483–517. [Google Scholar] [CrossRef] [PubMed]
- Feunekes, G.I.J.; Vanstaveren, W.A.; Devries, J.H.M.; Burema, J.; Hautvast, J.G.A.J. Relative and Biomarker-Based Validity of a Food-Frequency Questionnaire Estimating Intake of Fats and Cholesterol. Am. J. Clin. Nutr. 1993, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Goldbohm, R.A.; Vandenbrandt, P.A.; Brants, H.A.; van’t Veer, P.; Al, M.; Sturmans, F.; Hermus, R.J. Validation of a Dietary Questionnaire Used in a Large-Scale Prospective Cohort Study on Diet and Cancer. Eur. J. Clin. Nutr. 1994, 48, 253–265. [Google Scholar] [PubMed]
- Scheijen, J.L.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef]
- Hull, G.L.J.; Woodside, J.V.; Ames, J.M.; Cuskelly, G.J. N-epsilon-(carboxymethyl)lysine content of foods commonly consumed in a Western style diet. Food Chem. 2012, 131, 170–174. [Google Scholar] [CrossRef]
- Chen, J.; Waqas, K.; Tan, R.C.; Voortman, T.; Ikram, M.A.; Nijsten, T.E.; De Groot, L.C.; Uitterlinden, A.G.; Zillikens, M.C. The association between dietary and skin advanced glycation end products: The Rotterdam Study. Am. J. Clin. Nutr. 2020, 112, 129–137. [Google Scholar] [CrossRef]
- Willett, W.C.; Howe, G.R.; Kushi, L.H. Adjustment for total energy intake in epidemiologic studies. Am. J. Clin. Nutr. 1997, 65 (Suppl. 4), 1220S–1228S, discussion 1229S–1231S. [Google Scholar] [CrossRef]
- Voortman, T.; Kiefte-de Jong, J.C.; Ikram, M.A.; Stricker, B.H.; van Rooij, F.J.; Lahousse, L.; Tiemeier, H.; Brusselle, G.G.; Franco, O.H.; Schoufour, J.D. Adherence to the 2015 Dutch dietary guidelines and risk of non-communicable diseases and mortality in the Rotterdam Study. Eur. J. Epidemiol. 2017, 32, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; van der Duin, D.; Campos-Obando, N.; Ikram, M.A.; Nijsten, T.E.; Uitterlinden, A.G.; Zillikens, M.C. Serum 25-hydroxyvitamin D-3 is associated with advanced glycation end products (AGEs) measured as skin autofluorescence: The Rotterdam Study. Eur. J. Epidemiol. 2019, 34, 67–77. [Google Scholar] [CrossRef]
- Radjabzadeh, D.; Boer, C.G.; Beth, S.A.; van der Wal, P.; Kiefte-De Jong, J.C.; Jansen, M.A.; Konstantinov, S.R.; Peppelenbosch, M.P.; Hays, J.P.; Jaddoe, V.W.; et al. Diversity, compositional and functional differences between gut microbiota of children and adults. Sci. Rep. 2020, 10, 1040. [Google Scholar] [CrossRef]
- Schmieder, R.; Lim, Y.W.; Rohwer, F.; Edwards, R. TagCleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinform. 2010, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Li, J.; Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity Sep. 2005, 95, 221–227. [Google Scholar] [CrossRef]
- Vegan: Community Ecology Package. R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 23 May 2023).
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Mills, D.J.; Tuohy, K.M.; Booth, J.; Buck, M.; Crabbe, M.J.; Gibson, G.R.; Ames, J.M. Dietary glycated protein modulates the colonic microbiota towards a more detrimental composition in ulcerative colitis patients and non-ulcerative colitis subjects. J. Appl. Microbiol. 2008, 105, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Yao, Y.; Dong, S.; Jin, S.; Xiao, H.; Wu, H.; Zeng, M. Chemical characterization of the glycated myofibrillar proteins from grass carp (Ctenopharyngodon idella) and their impacts on the human gut microbiota in vitro fermentation. Food Funct. 2017, 8, 1184–1194. [Google Scholar] [CrossRef]
- Zhao, Q.; Ou, J.; Huang, C.; Qiu, R.; Wang, Y.; Liu, F.; Zheng, J.; Ou, S. Absorption of 1-Dicysteinethioacetal-5-Hydroxymethylfurfural in Rats and Its Effect on Oxidative Stress and Gut Microbiota. J. Agr. Food Chem. 2018, 66, 11451–11458. [Google Scholar] [CrossRef]
- Qu, W.; Nie, C.; Zhao, J.; Ou, X.; Zhang, Y.; Yang, S.; Bai, X.; Wang, Y.; Wang, J.; Li, J. Microbiome-Metabolomics Analysis of the Impacts of Long-Term Dietary Advanced-Glycation-End-Product Consumption on C57BL/6 Mouse Fecal Microbiota and Metabolites. J. Agr. Food Chem. 2018, 66, 8864–8875. [Google Scholar] [CrossRef]
- Dong, L.; Li, Y.; Chen, Q.; Liu, Y.; Qiao, Z.; Sang, S.; Zhang, J.; Zhan, S.; Wu, Z.; Liu, L. Research advances of advanced glycation end products in milk and dairy products: Formation, determination, control strategy and immunometabolism via gut microbiota. Food Chem. 2023, 417, 135861. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Pastoriza de la Cueva, S.; Peinado, M.J.; Rufian-Henares, J.A.; Navarro, M.P.; Rubio, L.A. Modifications in bacterial groups and short chain fatty acid production in the gut of healthy adult rats after long-term consumption of dietary Maillard reaction products. Food Res. Int. 2017, 100 Pt 1, 134–142. [Google Scholar] [CrossRef]
- Swiatecka, D.; Narbad, A.; Ridgway, K.P.; Kostyra, H. The study on the impact of glycated pea proteins on human intestinal bacteria. Int. J. Food Microbiol. 2011, 145, 267–272. [Google Scholar]
- van Dongen, K.C.; Linkens, A.M.; Wetzels, S.M.; Wouters, K.; Vanmierlo, T.; van de Waarenburg, M.P.; Scheijen, J.L.; de Vos, W.M.; Belzer, C.; Schalkwijk, C.G. Dietary advanced glycation endproducts (AGEs) increase their concentration in plasma and tissues, result in inflammation and modulate gut microbial composition in mice; evidence for reversibility. Food Res. Int. 2021, 147, 110547. [Google Scholar] [CrossRef] [PubMed]
- Linkens, A.M.; van Best, N.; Niessen, P.M.; Wijckmans, N.E.; de Goei, E.E.; Scheijen, J.L.; van Dongen, M.C.; van Gool, C.C.; de Vos, W.M.; Houben, A.J.; et al. A 4-Week Diet Low or High in Advanced Glycation Endproducts Has Limited Impact on Gut Microbial Composition in Abdominally Obese Individuals: The deAGEing Trial. Int. J. Mol. Sci. 2022, 23, 5328. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.P.N.; Troise, A.D.; Fogliano, V.; de Vos, W.M. Anaerobic Degradation of N-epsilon-Carboxymethyllysine, a Major Glycation End-Product, by Human Intestinal Bacteria. J. Agric. Food Chem. 2019, 67, 6594–6602. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, D. Thermal processing of food reduces gut microbiota diversity of the host and triggers adaptation of the microbiota: Evidence from two vertebrates. Microbiome 2018, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Janssens, Y.; Nielandt, J.; Bronselaer, A.; Debunne, N.; Verbeke, F.; Wynendaele, E.; Van Immerseel, F.; Vandewynckel, Y.P.; De Tré, G.; De Spiegeleer, B. Disbiome database: Linking the microbiome to disease. BMC Microbiol. 2018, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Bailén, M.; Bressa, C.; Martínez-López, S.; González-Soltero, R.; Montalvo Lominchar, M.G.; San Juan, C.; Larrosa, M. Microbiota Features Associated With a High-Fat/Low-Fiber Diet in Healthy Adults. Front. Nutr. 2020, 7, 583608. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Chinda, D.; Nakaji, S.; Fukuda, S.; Sakamoto, J.; Shimoyama, T.; Nakamura, T.; Fujisawa, T.; Terada, A.; Sugawara, K. The fermentation of different dietary fibers is associated with fecal clostridia levels in men. J. Nutr. 2004, 134, 1881–1886. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell. Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Jones, R.B.; Alderete, T.L.; Kim, J.S.; Millstein, J.; Gilliland, F.D.; Goran, M.I. High intake of dietary fructose in overweight/obese teenagers associated with depletion of Eubacterium and Streptococcus in gut microbiome. Gut Microbes 2019, 10, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, A.; Martínez, M.Á.; Babio, N.; Konstanti, P.; Tinahones, F.J.; Vioque, J.; Corella, D.; Fitó, M.; Vidal, J.; Moreno-Indias, I.; et al. Association between ultra-processed food consumption and gut microbiota in senior subjects with overweight/obesity and metabolic syndrome. Original Research. Front. Nutr. 2022, 9, 976547. [Google Scholar] [CrossRef] [PubMed]
- Mossad, O.; Batut, B.; Yilmaz, B.; Dokalis, N.; Mezö, C.; Nent, E.; Nabavi, L.S.; Mayer, M.; Maron, F.J.M.; Buescher, J.M.; et al. Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N6-carboxymethyllysine. Nat. Neurosci. 2022, 25, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.R.; Wesener, D.A.; Cheng, J.; Houston-Ludlam, A.N.; Beller, Z.W.; Hibberd, M.C.; Giannone, R.J.; Peters, S.L.; Hettich, R.L.; Leyn, S.A.; et al. Bioremediation of a Common Product of Food Processing by a Human Gut Bacterium. Cell. Host Microbe 2019, 26, 463–477.e8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristic | Total | Tertile Groups of Dietary CML Intake | ||
|---|---|---|---|---|
| Low CML | Medium CML | High CML | ||
| N | 1052 | 351 | 350 | 351 |
| Age, y | 62.5 ± 5.5 | 63.0 ± 5.4 | 62.6 ± 5.6 | 62.0 ± 5.4 |
| Sex (female) | 623 (59) | 187 (53) | 206 (59) | 230 (66) |
| Smoking status | ||||
| Never smoker | 348 (33) | 99 (28) | 125 (36) | 124 (36) |
| Ex-smoker | 536 (51) | 187 (53) | 171 (49) | 178 (51) |
| Current smoker | 163 (16) | 64 (18) | 52 (15) | 47 (13) |
| Physical activity | 50.8 (22.1, 88.4) | 46.5 (21.2, 82.8) | 52.5 (22.5, 87.3) | 52.9 (23.6, 92.8) |
| Alcohol consumption, gram/day | 8.6 (1.6, 8.6) | 8.6 (3.7, 15.0) | 8.6 (1.6, 8.6) | 6.4 (1.6, 8.6) |
| BMI, kg/m2 | 27.3 ± 4.5 | 27.2 ± 4.3 | 27.2 ± 4.2 | 27.5 ± 4.9 |
| Diabetes | 107 (10) | 36 (10) | 32 (9) | 39 (11) |
| Use of PPI, n (%) | 140 (13) | 53 (15) | 42 (12) | 45 (13) |
| Antibiotic usage | ||||
| No (n, %) | 861 (82) | 281 (80) | 289 (83) | 291 (83) |
| Within 1 m prior to collection (n, %) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 1 m–3 m prior to collection (n, %) | 67 (6) | 25 (7) | 21 (6) | 21 (6) |
| 3 m–1 y prior to collection (n, %) | 124 (12) | 45 (13) | 40 (11) | 39 (11) |
| Diet quality score | 7 (6, 8) | 7 (6, 8) | 7 (6, 8) | 7 (6, 9) |
| Energy intake, kCal/day | 2235 (1860, 2718) | 2247 (1855, 2765) | 2152 (1776, 2599) | 2302 (1953, 2752) |
| Protein intake, g/day | 89 ± 26 | 85 ± 26 | 86 ± 24 | 96 ± 26 |
| Fat, g/day | 79 (61, 101) | 77 (58, 98) | 77 (59, 96) | 82 (67, 105) |
| Carbohydrate intake, g/day | 261 ± 86 | 262 ± 90.50 | 251 ± 87 | 270 ± 81 |
| CML intake, mg/day | 2.5 ± 0.9 | 1.64 ± 0.46 | 2.40 ± 0.19 | 3.49 ± 0.76 |
| MGH1 intake, mg/day | 29.5 ± 8.1 | 24.85 ± 6.09 | 29.64 ± 5.67 | 33.93 ± 9.24 |
| CEL intake, mg/day | 2.50 ± 0.9 | 1.98 ± 0.64 | 2.50 ± 0.61 | 3.01 ± 0.95 |
| Microbial diversity | ||||
| Shannon Index | 3.99 ± 0.44 | 4.02 ± 0.40 | 3.98 ± 0.42 | 3.97 ± 0.49 |
| Inverse Simpson Index | 32.4 ± 14.55 | 33.1 ± 14.1 | 31.5 ± 14.4 | 32.6 ± 15.1 |
| Number of observed ASVs | 159 ± 56.5 | 161 ± 60 | 158 ± 56 | 157 ± 54 |
| Time in mail | 1 (1, 2) | 1 (1, 2) | 1 (1, 2) | 1 (1, 2) |
| Season of sample production | ||||
| Spring (n, %) | 261 (25) | 77 (22) | 87 (25) | 97 (28) |
| Summer (n, %) | 189 (18) | 67 (19) | 57 (16) | 65 (19) |
| Autumn (n, %) | 319 (30) | 100 (28) | 114 (33) | 105 (30) |
| Winter (n, %) | 283 (27) | 107 (30) | 92 (26) | 84 (24) |
| Number of reads | 27,201 (18,243, 34,394) | 28,442 (18,737, 34,609) | 26,276 (17,673, 34,990) | 26,910 (19,291, 33,312) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Radjabzadeh, D.; Medina-Gomez, C.; Voortman, T.; van Meurs, J.B.J.; Ikram, M.A.; Uitterlinden, A.G.; Kraaij, R.; Zillikens, M.C. Advanced Glycation End Products (AGEs) in Diet and Skin in Relation to Stool Microbiota: The Rotterdam Study. Nutrients 2023, 15, 2567. https://doi.org/10.3390/nu15112567
Chen J, Radjabzadeh D, Medina-Gomez C, Voortman T, van Meurs JBJ, Ikram MA, Uitterlinden AG, Kraaij R, Zillikens MC. Advanced Glycation End Products (AGEs) in Diet and Skin in Relation to Stool Microbiota: The Rotterdam Study. Nutrients. 2023; 15(11):2567. https://doi.org/10.3390/nu15112567
Chicago/Turabian StyleChen, Jinluan, Djawad Radjabzadeh, Carolina Medina-Gomez, Trudy Voortman, Joyce B. J. van Meurs, M. Arfan Ikram, André G. Uitterlinden, Robert Kraaij, and M. Carola Zillikens. 2023. "Advanced Glycation End Products (AGEs) in Diet and Skin in Relation to Stool Microbiota: The Rotterdam Study" Nutrients 15, no. 11: 2567. https://doi.org/10.3390/nu15112567
APA StyleChen, J., Radjabzadeh, D., Medina-Gomez, C., Voortman, T., van Meurs, J. B. J., Ikram, M. A., Uitterlinden, A. G., Kraaij, R., & Zillikens, M. C. (2023). Advanced Glycation End Products (AGEs) in Diet and Skin in Relation to Stool Microbiota: The Rotterdam Study. Nutrients, 15(11), 2567. https://doi.org/10.3390/nu15112567