

Annona muricate Extract Supplementation Contributes to Improve Aberrant Multi-Organ Energy Metabolism via Muscle–Brain Connectivity in Diabetic Mice


Abstract
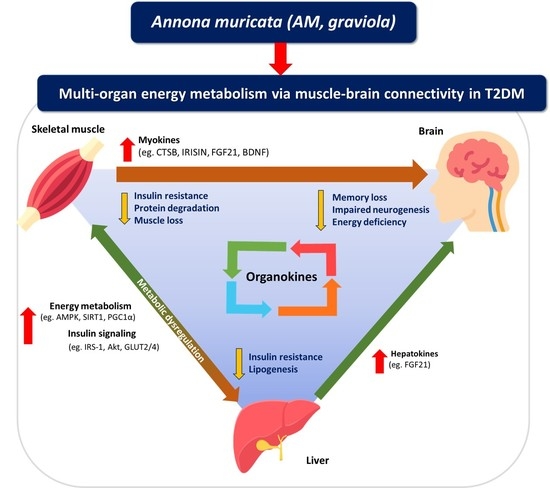
1. Introduction
2. Materials and Methods
2.1. Annona Muricata (AM) Extract
2.2. Animals and Experimental Design
2.3. Measurement of Body Composition
2.4. Hemoglobin A1c (% HbA1c) and Insulin Level
2.5. Homeostasis Model Assessment of IR (HOMA-IR) Level
2.6. Histological Analysis
2.7. Protein Extraction and Western Blot Analysis
2.8. Statistical Analysis
3. Results
3.1. Effects of AME Treatment on Body Composition, Food Intake, and Glycemic Regulation in T2DM Mice
3.2. Effects of AME Treatment on Skeletal Muscle and Brain Morphology in T2DM Mice
3.3. Effects of AME Treatment on Insulin-Signaling-Related Markers in T2DM Mice
3.4. Effects of AME Treatment on Energy Metabolism-Related Markers in T2DM Mice
3.5. Effects of AME Treatment on Hepatokines and Myokines Associated with Neuroprotection in T2DM Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gregory, J.W. Metabolic disorders. Endocr. Dev. 2009, 15, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.C.; Chen, W.L.; Wu, L.W.; Chang, Y.W.; Kao, T.W. Sarcopenia and cognitive impairment: A systematic review and meta-analysis. Clin. Nutr. 2020, 39, 2695–2701. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Fukunishi, S.; Asai, A.; Yokohama, K.; Ohama, H.; Nishiguchi, S.; Higuchi, K. Sarcopenia, frailty and type 2 diabetes mellitus (Review). Mol. Med. Rep. 2021, 24, 854. [Google Scholar] [CrossRef]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, M.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-Driven Neuroinflammation Compromises BBB Leading to Memory Loss in Both Diabetes Mellitus (DM) Type 1 and Type 2 Mouse Models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Moheet, A.; Mangia, S.; Seaquist, E.R. Impact of diabetes on cognitive function and brain structure. Ann. N. Y. Acad. Sci. 2015, 1353, 60–71. [Google Scholar] [CrossRef]
- Low, S.; Goh, K.S.; Ng, T.P.; Moh, A.; Ang, S.F.; Khoo, J.; Ang, K.; Yap, P.; Cheong, C.Y.; Tang, W.E.; et al. Decline in skeletal muscle mass is associated with cognitive decline in type 2 diabetes mellitus. J. Diabetes Complicat. 2022, 36, 108258. [Google Scholar] [CrossRef]
- Tsugawa, A.; Ogawa, Y.; Takenoshita, N.; Kaneko, Y.; Hatanaka, H.; Jaime, E.; Fukasawa, R.; Hanyu, H. Decreased Muscle Strength and Quality in Diabetes-Related Dementia. Dement. Geriatr. Cogn. Dis. Extra. 2017, 7, 454–462. [Google Scholar] [CrossRef]
- Low, S.; Ng, T.P.; Lim, C.L.; Moh, A.; Ang, S.F.; Wang, J.; Goh, K.S.; Ang, K.; Tang, W.E.; Kwan, P.Y.; et al. Association between Lower Extremity Skeletal Muscle Mass and Impaired Cognitive Function in Type 2 Diabetes. Sci. Rep. 2020, 10, 2956. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Lim, G.; Lee, H.; Lim, Y. Potential Effects of Resistant Exercise on Cognitive and Muscle Functions Mediated by Myokines in Sarcopenic Obese Mice. Biomedicines 2022, 10, 2529. [Google Scholar] [CrossRef]
- Lee, H.; Lim, Y. The Potential Role of Myokines/Hepatokines in the Progression of Neuronal Damage in Streptozotocin and High-Fat Diet-Induced Type 2 Diabetes Mellitus Mice. Biomedicines 2022, 10, 1521. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. Springerplus 2014, 3, 2. [Google Scholar] [CrossRef]
- Jiang, Y.; Lin, L.; Liu, N.; Wang, Q.; Yuan, J.; Li, Y.; Chung, K.K.; Guo, S.; Yu, Z.; Wang, X. FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation. Int. J. Mol. Sci. 2020, 21, 824. [Google Scholar] [CrossRef]
- Villegas, R.; Williams, S.M.; Gao, Y.T.; Long, J.; Shi, J.; Cai, H.; Li, H.; Chen, C.C.; Tai, E.S.; Hu, F.; et al. Genetic variation in the peroxisome proliferator-activated receptor (PPAR) and peroxisome proliferator-activated receptor gamma co-activator 1 (PGC1) gene families and type 2 diabetes. Ann. Hum. Genet. 2014, 78, 23–32. [Google Scholar] [CrossRef]
- Goto, T.; Hirata, M.; Aoki, Y.; Iwase, M.; Takahashi, H.; Kim, M.; Li, Y.; Jheng, H.F.; Nomura, W.; Takahashi, N.; et al. The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. J. Biol. Chem. 2017, 292, 9175–9190. [Google Scholar] [CrossRef]
- Al Yaad, K.M.; Elsaid, S.F.G.; Abdraboh, M.E.; Al-Doaiss, A.A. Effect of Graviola (Annona Muricata l.) and Ginger (Zingiber Officinale Roscoe) on Diabetes Mellitus Induced in Male Wistar Albino Rats. Folia. Biol. 2019, 65, 275–284. [Google Scholar]
- Sasso, S.; Sampaio, E.; Souza, P.C.; Santana, L.F.; Cardoso, C.A.L.; Alves, F.M.; Portugal, L.C.; Faria, B.B.; Silva, A.F.; Motta-Castro, A.R.; et al. Use of an Extract of Annona muricata Linn to Prevent High-Fat Diet Induced Metabolic Disorders in C57BL/6 Mice. Nutrients 2019, 11, 1509. [Google Scholar] [CrossRef]
- Olas, B. The Antioxidant Potential of Graviola and Its Potential Medicinal Application. Nutrients 2023, 15, 402. [Google Scholar] [CrossRef]
- Son, Y.; Lee, H.; Son, S.Y.; Lee, C.H.; Kim, S.Y.; Lim, Y. Ameliorative Effect of Annona muricata (Graviola) Extract on Hyperglycemia Induced Hepatic Damage in Type 2 Diabetic Mice. Antioxidants 2021, 10, 1546. [Google Scholar] [CrossRef]
- Alsenosy, A.A.; El-Far, A.H.; Sadek, K.M.; Ibrahim, S.A.; Atta, M.S.; Sayed-Ahmed, A.; Al Jaouni, S.K.; Mousa, S.A. Graviola (Annona muricata) attenuates behavioural alterations and testicular oxidative stress induced by streptozotocin in diabetic rats. PLoS ONE 2019, 14, e0222410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lv, X.Y.; Li, J.; Xu, Z.G.; Chen, L. The characterization of high-fat diet and multiple low-dose streptozotocin induced type 2 diabetes rat model. Exp. Diabetes. Res. 2008, 2008, 704045. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Beck, C.A.; Robinson, D.; Ye, J.; Mills, B.; Gerber, S.A.E.; Schwarz, M.; Linehan, D. Dual Energy X-ray Absorptiometry (DEXA) as a longitudinal outcome measure of cancer-related muscle wasting in mice. PLoS ONE 2020, 15, e0230695. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, S.Y.; Lim, Y. Lespedeza bicolor extract supplementation reduced hyperglycemia-induced skeletal muscle damage by regulation of AMPK/SIRT/PGC1α-related energy metabolism in type 2 diabetic mice. Nutr. Res. 2023, 110, 1–13. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, H.; Kim, S.Y.; Lim, Y. Effects of Lespedeza Bicolor Extract on Regulation of AMPK Associated Hepatic Lipid Metabolism in Type 2 Diabetic Mice. Antioxidants 2019, 8, 599. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Zanatta, L.; Rosso, A.; Folador, P.; Figueiredo, M.S.; Pizzolatti, M.G.; Leite, L.D.; Silva, F.R. Insulinomimetic effect of kaempferol 3-neohesperidoside on the rat soleus muscle. J. Nat. Prod. 2008, 71, 532–535. [Google Scholar] [CrossRef]
- Hu, T.; Lu, X.Y.; Shi, J.J.; Liu, X.Q.; Chen, Q.B.; Wang, Q.; Chen, Y.B.; Zhang, S.J. Quercetin protects against diabetic encephalopathy via SIRT1/NLRP3 pathway in db/db mice. J. Cell. Mol. Med. 2020, 24, 3449–3459. [Google Scholar] [CrossRef]
- Henagan, T.M.; Lenard, N.R.; Gettys, T.W.; Stewart, L.K. Dietary quercetin supplementation in mice increases skeletal muscle PGC1α expression, improves mitochondrial function and attenuates insulin resistance in a time-specific manner. PLoS ONE 2014, 9, e89365. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta. Mol. Basis. Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flie, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Prida, E.; Álvarez-Delgado, S.; Pérez-Lois, R.; Soto-Tielas, M.; Estany-Gestal, A.; Fernø, J.; Seoane, L.M.; Quiñones, M.; Al-Massadi, O. Liver Brain Interactions: Focus on FGF21 a Systematic Review. Int. J. Mol. Sci. 2022, 23, 13318. [Google Scholar] [CrossRef]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Onishi, Y.T.; Ueha, T.; Kawamoto, H.; Hara, M.; Toda, R.; Harada, M.; Minoda, M.; Kurosaka, T.; Akisue, T. Regulation of mitochondrial proliferation by PGC-1α induces cellular apoptosis in musculoskeletal malignancies. Sci. Rep. 2014, 4, 3916. [Google Scholar] [CrossRef]
- Zhang, F.; Feng, J.; Zhang, J.; Kang, X.; Qian, D. Quercetin modulates AMPK/SIRT1/NF-κB signaling to inhibit inflammatory/oxidative stress responses in diabetic high fat diet-induced atherosclerosis in the rat carotid artery. Exp. Ther. Med. 2020, 20, 280. [Google Scholar] [CrossRef]
- Seo, S.; Lee, M.S.; Chang, E.; Shin, Y.; Oh, S.; Kim, I.H.; Kim, Y. Rutin Increases Muscle Mitochondrial Biogenesis with AMPK Activation in High-Fat Diet-Induced Obese Rats. Nutrients 2015, 7, 8152–8169. [Google Scholar] [CrossRef]
- Belizário, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. Springerplus 2016, 5, 619. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing. Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef]
- Chen, W.; Wang, L.; You, W.; Shan, T. Myokines mediate the cross talk between skeletal muscle and other organs. J. Cell. Physiol. 2021, 236, 2393–2412. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell. Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell. Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, 369. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.F.; Zhang, J.; Liu, X.Y.; Fang, H.; Tian, L.B.; Zhou, D.H.; Kosten, T.R.; Zhang, X.Y. Low BDNF is associated with cognitive deficits in patients with type 2 diabetes. Psychopharmacology 2013, 227, 93–100. [Google Scholar] [CrossRef]
- Nakagawa, T.; Ono-Kishino, M.; Sugaru, E.; Yamanaka, M.; Taiji, M.; Noguchi, H. Brain-derived neurotrophic factor (BDNF) regulates glucose and energy metabolism in diabetic mice Diabetes. Metab. Res. Rev. 2002, 18, 185–191. [Google Scholar] [CrossRef]
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B Improves ß-Amyloidosis and Learning and Memory in Models of Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 340–352. [Google Scholar] [CrossRef]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.E.; Roberson, D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Smith, D.R.; Fernyhough, P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 2013, 51, 56–65. [Google Scholar] [CrossRef]
- Lucas, E.K.; Markwardt, S.J.; Gupta, S.; Meador-Woodruff, J.H.; Lin, J.D.; Overstreet-Wadiche, L.; Cowell, R.M. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J. Neurosci. 2010, 30, 7227–7235. [Google Scholar] [CrossRef]
- Roy Chowdhury, S.K.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 2012, 135, 1751–1766. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing. Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, W.; Zhao, B.; Xie, J.; Sun, Q.; Shi, X.; Yan, B.; Tian, G.; Liang, X. Quercetin Attenuates Diabetic Peripheral Neuropathy by Correcting Mitochondrial Abnormality via Activation of AMPK/PGC-1α Pathway in vivo and in vitro. Front. Neurosci. 2021, 15, 636172. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GROUP | CON | DMC | LAM | HAM |
|---|---|---|---|---|
| Body weight (g) | ||||
| Before treatment | 26.76 ± 0.50 a | 34.87 ± 0.77 b | 33.91 ± 1.22 b | 34.69 ± 0.72 b |
| After treatment | 30.40 ± 0.67 a | 40.20 ± 1.27 b | 39.82 ± 1.37 b | 40.94 ± 1.46 b |
| Gain | 3.20 ± 0.39 a | 4.66 ± 0.48 ab | 4.67 ± 0.62 ab | 5.56 ± 0.63 b |
| % Fat | 31.61 ± 2.04 a | 42.40 ± 3.7 b | 46.10 ± 2.04 b | 45.43 ± 2.77 b |
| % Lean | 65.31 ± 2.21 b | 55.16 ± 3.90 a | 51.08 ± 2.04 a | 52.24 ± 2.94 a |
| Gastrocnemius weight (g/kg BW) | 0.54 ± 0.01 b | 0.43 ± 0.01 a | 0.43 ± 0.02 a | 0.42 ± 0.02 a |
| Quadriceps weight (g/kg BW) | 0.63 ± 0.07 b | 0.43 ± 0.01 a | 0.43 ± 0.05 a | 0.48 ± 0.02 a |
| Food intake (g/day) | 3.35 ± 0.01 | 3.25 ± 0.08 | 3.41 ± 0.03 | 3.28 ± 0.07 |
| Fasting blood glucose level (mg/dl) | ||||
| 4 weeks after treatment | 163.64 ± 7.37 a | 331.18 ± 18.28 c | 269.78 ± 9.91 b | 336.56 ± 23.67 c |
| 9 weeks after treatment | 146.36 ± 4.55 a | 291.64 ± 26.97 c | 235.33 ± 7.00 b | 282.33± 20.67 bc |
| Plasma insulin (μU/mL) | 10.00 ± 1.23 a | 22.49 ± 5.36 b | 9.52 ± 2.68 a | 14.14 ± 3 ab |
| HbA1c% | 5.01 ± 0.07 a | 5.94 ± 0.23 b | 5.25 ± 0.17 a | 5.18 ± 0.21 a |
| HOMA-IR (mmol/L × μU/mL) | 3.65 ± 0.51 a | 16.01 ± 3.6 b | 5.74 ± 1.79 a | 10.08 ± 2.41 ab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kim, S.Y.; Lim, Y. Annona muricate Extract Supplementation Contributes to Improve Aberrant Multi-Organ Energy Metabolism via Muscle–Brain Connectivity in Diabetic Mice. Nutrients 2023, 15, 2559. https://doi.org/10.3390/nu15112559
Lee H, Kim SY, Lim Y. Annona muricate Extract Supplementation Contributes to Improve Aberrant Multi-Organ Energy Metabolism via Muscle–Brain Connectivity in Diabetic Mice. Nutrients. 2023; 15(11):2559. https://doi.org/10.3390/nu15112559
Chicago/Turabian StyleLee, Heaji, Sun Yeou Kim, and Yunsook Lim. 2023. "Annona muricate Extract Supplementation Contributes to Improve Aberrant Multi-Organ Energy Metabolism via Muscle–Brain Connectivity in Diabetic Mice" Nutrients 15, no. 11: 2559. https://doi.org/10.3390/nu15112559
APA StyleLee, H., Kim, S. Y., & Lim, Y. (2023). Annona muricate Extract Supplementation Contributes to Improve Aberrant Multi-Organ Energy Metabolism via Muscle–Brain Connectivity in Diabetic Mice. Nutrients, 15(11), 2559. https://doi.org/10.3390/nu15112559