
Role of Prenatal Nutrition in the Development of Insulin Resistance in Children

 ,
, 
Abstract
1. Introduction
1.1. Definition
1.2. Measurement
1.3. Pathophysiology
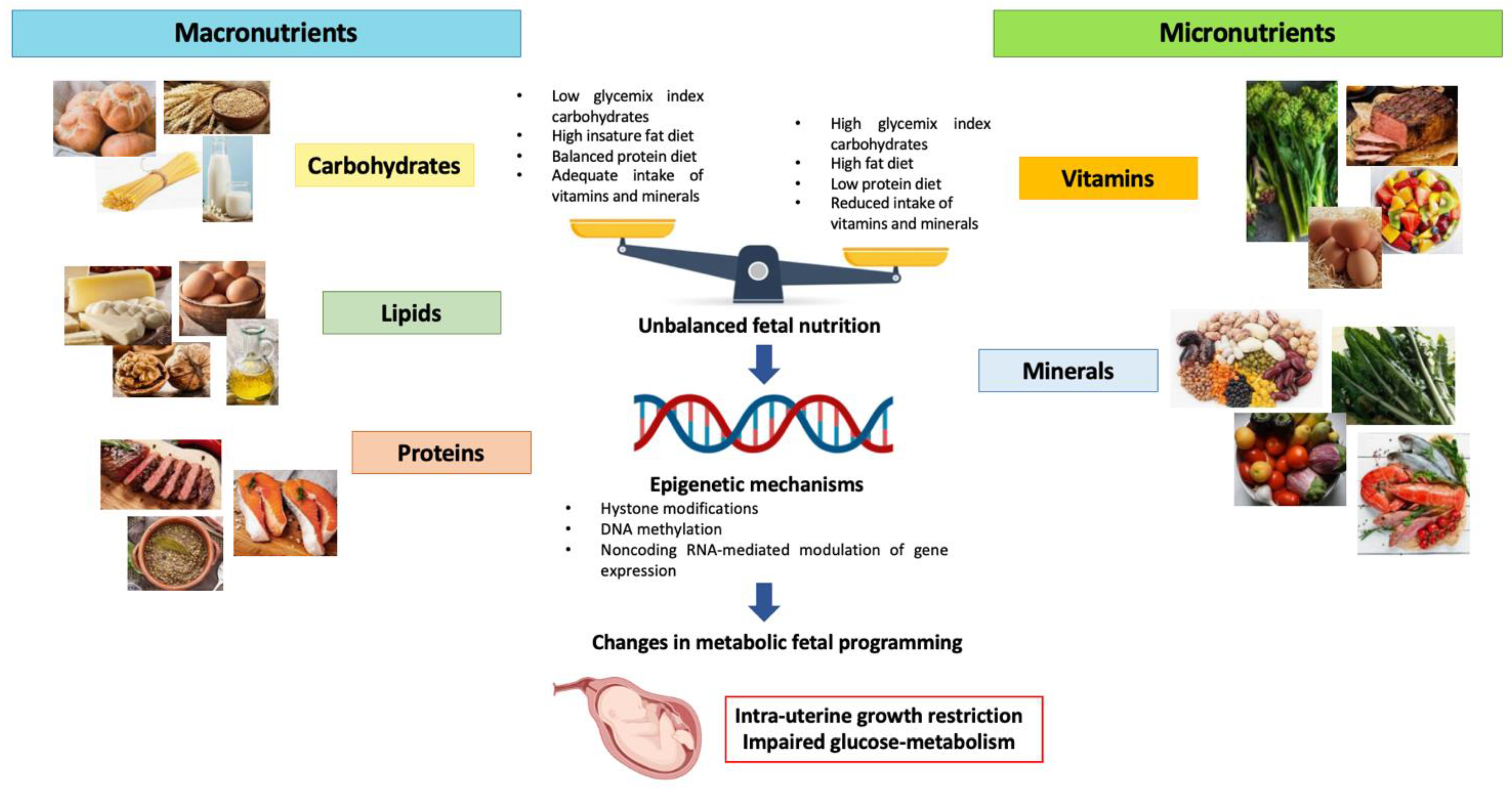
1.4. Maternal Nutrition and Intrauterine Fetal Growth
1.5. Maternal High Glycemix Index Diet
1.6. Maternal High-Fat Diet
1.7. Maternal Low-Protein Diet
1.8. Role of Micronutrients
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levy-Marchal, C.; Arslanian, S.; Cutfield, W.; Sinaiko, A.; Druet, C.; Marcovecchio, M.L.; Chiarelli, F. Insulin Resistance in Children: Consensus, Perspective, and Future Directions. J. Clin. Endocrinol. Metab. 2010, 95, 5189–5198. [Google Scholar] [CrossRef]
- Tagi, V.M.; Chiarelli, F. Obesity and insulin resistance in children. Curr. Opin. Pediatr. 2020, 32, 582–588. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Kurtoğlu, S.; Hatipoğlu, N.; Mazicioglu, M.M.; Kendirici, M.; Keskin, M.; Kondolot, M. Insulin Resistance in Obese Children and Adolescents: HOMA-IR Cut-Off Levels in the Prepubertal and Pubertal Periods—Original Article. J. Clin. Res. Pediatr. Endocrinol. 2010, 2, 100–106. [Google Scholar] [CrossRef]
- Ascaso, J.F.; Pardo, S.; Real, J.T.; Lorente, R.I.; Priego, A.; Carmena, R. Diagnosing Insulin Resistance by Simple Quantitative Methods in Subjects With Normal Glucose Metabolism. Diabetes Care 2003, 26, 3320–3325. [Google Scholar] [CrossRef]
- Brown, R.; Yanovski, J.A. Estimation of insulin sensitivity in children: Methods, measures and controversies. Pediatr. Diabetes 2014, 15, 151–161. [Google Scholar] [CrossRef][Green Version]
- Blasetti, A.; Franchini, S.; Comegna, L.; Prezioso, G.; Chiarelli, F. Role of nutrition in preventing insulin resistance in children. J. Pediatr. Endocrinol. Metab. 2016, 29, 247–257. [Google Scholar] [CrossRef]
- Tagi, V.M.; Giannini, C.; Chiarelli, F. Insulin Resistance in Children. Front Endocrinol (Lausanne). Front. Endocrinol. 2019, 10, 342. [Google Scholar] [CrossRef]
- Monetti, M.; Nagaraj, N.; Sharma, K.; Mann, M. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 2011, 8, 655–658. [Google Scholar] [CrossRef]
- Watanabe, R.M. The Genetics of Insulin Resistance: Where’s Waldo? Curr. Diabetes Rep. 2010, 10, 476–484. [Google Scholar] [CrossRef]
- Komurcu-Bayrak, E. Impact of Genetic Polymorphisms on Insulin Resistance. In Insulin Resistance; InTech: Singapore, 2012. [Google Scholar]
- Eyzaguirre, F.; Mericq, V. Insulin Resistance Markers in Children. Horm. Res. Paediatr. 2009, 71, 65–74. [Google Scholar] [CrossRef]
- Vaiserman, A.M. Early-life nutritional programming of longevity. J. Dev. Orig. Health Dis. 2014, 5, 325–338. [Google Scholar] [CrossRef]
- Bateson, P.; Gluckman, P.; Hanson, M. The biology of developmental plasticity and the Predictive Adaptive Response hypothesis. J. Physiol. 2014, 592, 2357–2368. [Google Scholar] [CrossRef]
- Darendeliler, F. IUGR: Genetic influences, metabolic problems, environmental associations/triggers, current and future management. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101260. [Google Scholar] [CrossRef]
- Vaiserman, A.; Lushchak, O. Prenatal Malnutrition-Induced Epigenetic Dysregulation as a Risk Factor for Type 2 Diabetes. Int. J. Genom. 2019, 2019, 3821409. [Google Scholar] [CrossRef]
- Hales, C.; Barker, D. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Int. J. Epidemiol. 2013, 42, 1215–1222. [Google Scholar] [CrossRef]
- Huang, L.T. Maternal and Early-Life Nutrition and Health. Int. J. Environ. Res. Public Health 2020, 17, 7982. [Google Scholar] [CrossRef]
- Marciniak, A.; Patro-Małysza, J.; Kimber-Trojnar, Ż.; Marciniak, B.; Oleszczuk, J.; Leszczyńska-Gorzelak, B. Fetal programming of the metabolic syndrome. Taiwan J. Obstet. Gynecol. 2017, 56, 133–138. [Google Scholar] [CrossRef]
- Ryznar, R.J.; Phibbs, L.; Van Winkle, L.J. Epigenetic Modifications at the Center of the Barker Hypothesis and Their Transgenerational Implications. Int. J. Environ. Res. Public Health 2021, 18, 12728. [Google Scholar] [CrossRef]
- Wells, J.C.K. The thrifty phenotype as an adaptive maternal effect. Biol. Rev. 2007, 82, 143–172. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.P. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Kislal, S.; Shook, L.L.; Edlow, A.G. Perinatal exposure to maternal obesity: Lasting cardiometabolic impact on offspring. Prenat. Diagn. 2020, 40, 1109–1125. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Garde, E.; Lopez-Bermejo, A.; de Zegher, F.; Ibañez, L. Differential DNA methylation profile in infants born small-for-gestational-age: Association with markers of adiposity and insulin resistance from birth to age 24 months. BMJ Open Diabetes Res. Care 2020, 8, e001402. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Simon, T.G.; Roelstraete, B.; Stephansson, O.; Söderling, J.; Ludvigsson, J.F. Maternal obesity increases the risk and severity of NAFLD in offspring. J. Hepatol. 2021, 75, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, Y.; Wang, L.; Yang, W.; Li, C.; Gu, P.; Xia, Y.; Yan, J.; Shen, Y.; Zhao, Q.; et al. Maternal dietary glycaemic change during gestation influences insulin-related gene methylation in the placental tissue: A genome-wide methylation analysis. Genes Nutr. 2019, 14, 17. [Google Scholar] [CrossRef]
- Jenkins avid, J.A.; Mente, A.; Bangdiwala, I.S.; Rangarajan, S.; Srichaikul, K.; Mohan, V.; Avezum, A.; Díaz, R.; Rosengren, A.; Lanas, F.; et al. Glycemic Index, Glycemic Load, and Cardiovascular Disease and Mortality. N. Engl. J. Med. 2021, 385, 378–380. [Google Scholar] [CrossRef]
- Atkinson, F.S.; Brand-Miller, J.C.; Foster-Powell, K.; Buyken, A.E.; Goletzke, J. International tables of glycemic index and glycemic load values 2021: A systematic review. Am. J. Clin. Nutr. 2021, 114, 1625–1632. [Google Scholar] [CrossRef]
- Choudhury, R.P.; Akbar, N. Beyond diabetes: A relationship between cardiovascular outcomes and glycaemic index. Cardiovasc. Res. 2021, 117, e97–e98. [Google Scholar] [CrossRef]
- Chiavaroli, L.; Lee, D.; Ahmed, A.; Cheung, A.; A Khan, T.; Blanco, S.; Mejia; Mirrahimi, A.; A Jenkins, D.J.; Livesey, G.; et al. Effect of low glycaemic index or load dietary patterns on glycaemic control and cardiometabolic risk factors in diabetes: Systematic review and meta-analysis of randomised controlled trials. BMJ 2021, 374, n1651. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.I.; E Mills, K.; Zheng, J.; Regmi, A.; Hu, S.Q.; Gou, L.; Chen, L.-L. Low-glycemic index diets as an intervention for diabetes: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2019, 110, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Maslova, E.; Hansen, S.; Grunnet, L.G.; Strøm, M.; Bjerregaard, A.A.; Hjort, L.; Kampmann, F.B.; Madsen, C.M.; Baun Thuesen, A.C.; Bech, B.H.; et al. Maternal glycemic index and glycemic load in pregnancy and offspring metabolic health in childhood and adolescence—A cohort study of 68,471 mother–offspring dyads from the Danish National Birth Cohort. Eur. J. Clin. Nutr. 2019, 73, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Sideratou, T.; Atkinson, F.; Campbell, G.J.; Petocz, P.; Bell-Anderson, K.S.; Brand-Miller, J. Glycaemic Index of Maternal Dietary Carbohydrate Differentially Alters Fto and Lep Expression in Offspring in C57BL/6 Mice. Nutrients 2018, 10, 1342. [Google Scholar] [CrossRef]
- Zhang, Y.; Chua, S. Leptin Function and Regulation. In Comprehensive Physiology; Wiley: New York, NY, USA, 2017; pp. 351–369. [Google Scholar]
- Hoggard, N.; Haggarty, P.; Thomas, L.; Lea, R. Leptin expression in placental and fetal tissues: Does leptin have a functional role? Biochem. Soc. Trans. 2001, 29, 57. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and obesity: Role and clinical implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Toh, H.; Thomson, J.A.; Jiang, P. Maternal High-Fiber Diet Protects Offspring against Type 2 Diabetes. Nutrients 2020, 13, 94. [Google Scholar] [CrossRef]
- Gawlińska, K.; Gawliński, D.; Filip, M.; Przegaliński, E. Przegaliński, E. Relationship of maternal high-fat diet during pregnancy and lactation to offspring health. Nutr. Rev. 2021, 79, 709–725. [Google Scholar] [CrossRef]
- Tain, Y.-L.; Hsu, C.-N. Maternal High-Fat Diet and Offspring Hypertension. Int. J. Mol. Sci. 2022, 23, 8179. [Google Scholar] [CrossRef]
- Saullo, C.; da Cruz, L.L.; Damasceno, D.C.; Volpato, G.T.; Sinzato, Y.K.; Karki, B.; Gallego, F.Q.; Vesentini, G. Effects of a maternal high-fat diet on adipose tissue in murine offspring: A systematic review and meta-analysis. Biochimie 2022, 201, 18–32. [Google Scholar] [CrossRef]
- Barbosa, C.M.; Figueiredo, V.P.; Barbosa, M.A.; Cardoso, L.M.; Alzamora, A.C. Maternal high-fat diet triggers metabolic syndrome disorders that are transferred to first and second offspring generations. Br. J. Nutr. 2020, 123, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xu, H.; Wu, J.; Li, J.; Zhou, Y.; Ding, Z.; Siwko, S.K.; Yuan, X.; Schalinske, K.L.; Alpini, G.; et al. Maternal high-fat diet disrupted one-carbon metabolism in offspring, contributing to nonalcoholic fatty liver disease. Liver Int. 2021, 41, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Li, J.; Xu, H.; Wang, X.; He, L.; McCauley, N.; Zhang, K.K.; Xie, L. Offspring NAFLD Liver Phospholipid Profiles are Differentially Programmed by Maternal High-fat Diet and Maternal One Carbon Supplement. J. Nutr. Biochem. 2022, 111, 109187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Wu, H.; Gao, Y.; Zheng, J.; Zhang, J. Maternal High-Fat Diet Impairs Placental Fatty Acid β-Oxidation and Metabolic Homeostasis in the Offspring. Front Nutr. 2022, 9, 849684. [Google Scholar] [CrossRef]
- Duttaroy, A.K.; Basak, S. Maternal dietary fatty acids and their roles in human placental development. Prostaglandins Leukot. Essent. Fat. Acids 2020, 155, 102080. [Google Scholar] [CrossRef]
- Lewis, R.; Desoye, G. Placental Lipid and Fatty Acid Transfer in Maternal Overnutrition. Ann. Nutr. Metab. 2017, 70, 228–231. [Google Scholar] [CrossRef]
- Perazzolo, S.; Hirschmugl, B.; Wadsack, C.; Desoye, G.; Lewis, R.M.; Sengers, B.G. The influence of placental metabolism on fatty acid transfer to the fetus. J. Lipid Res. 2017, 58, 443–454. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Cook, T.; Knipp, G.T. Effect of Placental Fatty Acid Metabolism and Regulation by Peroxisome Proliferator Activated Receptor on Pregnancy and Fetal Outcomes. J. Pharm. Sci. 2007, 96, 2582–2606. [Google Scholar] [CrossRef]
- Lewis, R.M.; Wadsack, C.; Desoye, G. Placental fatty acid transfer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 78–82. [Google Scholar] [CrossRef]
- Vipin, V.A.; Blesson, C.S.; Yallampalli, C. Maternal low protein diet and fetal programming of lean type 2 diabetes. World J. Diabetes 2022, 13, 185–202. [Google Scholar] [CrossRef]
- George, A.M.; Jacob, A.G.; Fogelfeld, L. Lean diabetes mellitus: An emerging entity in the era of obesity. World J. Diabetes 2015, 6, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Ranabir, S.; Barma, P.; Prasad, L.; Singh, T. Clinical and biochemical profile of lean type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2011, 15, S40. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.B.; Clari, R.; Vigotti, F.N.; Leone, F.; Attini, R.; Cabiddu, G.; Mauro, G.; Castelluccia, N.; Colombi, N.; Capizzi, I.; et al. Vegan-vegetarian diets in pregnancy: Danger or panacea? A systematic narrative review. BJOG Int. J. Obstet. Gynaecol. 2015, 122, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Sanders, T.A.; Obeid, O. The influence of maternal vegetarian diet on essential fatty acid status of the newborn. Eur. J. Clin. Nutr. 1994, 48, 358–368. [Google Scholar] [PubMed]
- Sebastiani, G.; Barbero, A.H.; Borràs-Novell, C.; Alsina, M.; Aldecoa-Bilbao, V.; Andreu-Fernández, V.; Tutusaus, M.P.; Martínez, S.F.; Gómez-Roig, M.D.; García-Algar, Ó. The Effects of Vegetarian and Vegan Diet during Pregnancy on the Health of Mothers and Offspring. Nutrients 2019, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, K.; Lillycrop, K.A.; Silver, M.J. Fetal programming and epigenetics. Curr. Opin. Endocr. Metab. Res. 2020, 13, 1–6. [Google Scholar] [CrossRef]
- Cox, A.R.; Beamish, C.A.; Carter, D.E.; Arany, E.J.; Hill, D.J. Cellular mechanisms underlying failed beta cell regeneration in offspring of protein-restricted pregnant mice. Exp. Biol. Med. 2013, 238, 1147–1159. [Google Scholar] [CrossRef]
- Dumortier, O.; Blondeau, B.; Duvillié, B.; Reusens, B.; Bréant, B.; Remacle, C. Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 2007, 50, 2495–2503. [Google Scholar] [CrossRef]
- Rees, W.D.; Hay, S.M.; Cruickshank, M.; Reusens, B.; Remacle, C.; Antipatis, C.; Grant, G. Maternal protein intake in the pregnant rat programs the insulin axis and body composition in the offspring. Metabolism 2006, 55, 642–649. [Google Scholar] [CrossRef]
- Sparre, T.; Reusens, B.; Cherif, H.; Larsen, M.R.; Roepstorff, P.; Fey, S.J.; Larsen, P.M.; Remacle, C.; Nerup, J. Intrauterine programming of fetal islet gene expression in rats?effects of maternal protein restriction during gestation revealed by proteome analysis. Diabetologia 2003, 46, 1497–1511. [Google Scholar] [CrossRef]
- Mateus Gonçalves, L.; Vettorazzi, J.F.; Vanzela, E.C.; Figueiredo, M.S.; Batista, T.M.; Zoppi, C.C.; Boschero, A.C.; Carneiro, E.M. Amino acid restriction increases β-cell death under challenging conditions. J. Cell Physiol. 2019, 234, 16679–16684. [Google Scholar] [CrossRef] [PubMed]
- Merezak, S.; A Hardikar, A.; Yajnik, C.S.; Remacle, C.; Reusens, B. Intrauterine low protein diet increases fetal beta-cell sensitivity to NO and IL-1 beta: The protective role of taurine. J. Endocrinol. 2001, 171, 299–308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alejandro, E.U.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet–induced microRNAs and mTOR underlie β cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Theys, N.; Bouckenooghe, T.; Ahn, M.T.; Remacle, C.; Reusens, B. Maternal low-protein diet alters pancreatic islet mitochondrial function in a sex-specific manner in the adult rat. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R1516–R1525. [Google Scholar] [CrossRef]
- Zambrano, E.; Bautista, C.J.; Deás, M.; Martínez-Samayoa, P.M.; González-Zamorano, M.; Ledesma, H.; Morales, J.; Larrea, F.; Nathanielsz, P.W. A low maternal protein diet during pregnancy and lactation has sex- and window of exposure-specific effects on offspring growth and food intake, glucose metabolism and serum leptin in the rat. J. Physiol. 2006, 571, 221–230. [Google Scholar] [CrossRef]
- Vistisen, B.; I Hellgren, L.; Vadset, T.; Scheede-Bergdahl, C.; Helge, J.W.; Dela, F.; Stallknecht, B. Effect of gender on lipid-induced insulin resistance in obese subjects. Eur. J. Endocrinol. 2008, 158, 61–68. [Google Scholar] [CrossRef]
- Varlamov, O.; Bethea, C.L.; Roberts, C.T., Jr. Sex-Specific Differences in Lipid and Glucose Metabolism. Front. Endocrinol. 2015, 5, 241. [Google Scholar] [CrossRef]
- Oreffo, R.O.; Lashbrooke, B.; I Roach, H.; Clarke, N.M.; Cooper, C. Maternal protein deficiency affects mesenchymal stem cell activity in the developing offspring. Bone 2003, 33, 100–107. [Google Scholar] [CrossRef]
- Lisboa, P.C.; Oliveira, E.; Fagundes, A.S.; Santos-Silva, A.P.; Conceição, E.S.; Passos, M.F.; Moura, E.G. Postnatal Low Protein Diet Programs Leptin Signaling in the Hypothalamic-Pituitary-Thyroid Axis and Pituitary TSH Response to Leptin in Adult Male Rats. Horm. Metab. Res. 2012, 44, 114–122. [Google Scholar] [CrossRef]
- Lisboa, P.C.; Fagundes, A.T.S.; Denolato, A.T.A.; Oliveira, E.; Bonomo, I.T.; Alves, S.B.; Curty, F.H.; Passos, M.C.F.; Moura, E. Neonatal Low-Protein Diet Changes Deiodinase Activities and Pituitary TSH Response to TRH in Adult Rats. Exp. Biol. Med. 2008, 233, 57–63. [Google Scholar] [CrossRef]
- Guzmán, C.; García-Becerra, R.; Aguilar-Medina, M.A.; Méndez, I.; Merchant-Larios, H.; Zambrano, E. Maternal Protein Restriction During Pregnancy and/or Lactation Negatively Affects Follicular Ovarian Development and Steroidogenesis in the Prepubertal Rat Offspring. Arch. Med Res. 2014, 45, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Sui, S.; Jia, Y.; He, B.; Li, R.; Li, X.; Cai, D.; Song, H.; Zhang, R.; Zhao, R. Maternal Low-protein Diet Alters Ovarian Expression of Folliculogenic and Steroidogenic Genes and Their Regulatory MicroRNAs in Neonatal Piglets. Asian-Australas. J. Anim. Sci. 2014, 27, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Hardy, G.; Wong, T.; Morrissey, H.; Anderson, C.; Moltu, S.J.; Poindexter, B.; Lapillonne, A.; Ball, P.A. Parenteral Provision of Micronutrients to Pediatric Patients: An International Expert Consensus Paper. J. Parenter. Enter. Nutr. 2020, 44, S5–S23. [Google Scholar] [CrossRef] [PubMed]
- Fall, C.H.; Yajnik, C.S.; Rao, S.; Davies, A.A.; Brown, N.; Farrant, H.J. Micronutrients and Fetal Growth. J. Nutr. 2003, 133, 1747S–1756S. [Google Scholar] [CrossRef]
- McArdle, H.J.; Ashworth, C.J. Micronutrients in fetal growth and development. Br. Med. Bull. 1999, 55, 499–510. [Google Scholar] [CrossRef]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef]
- Lyon, P.; Strippoli, V.; Fang, B.; Cimmino, L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients 2020, 12, 2867. [Google Scholar] [CrossRef]
- Rush, E.C.; Katre, P.; Yajnik, C.S. Vitamin B12: One carbon metabolism, fetal growth and programming for chronic disease. Eur. J. Clin. Nutr. 2014, 68, 2–7. [Google Scholar] [CrossRef]
- Dror, D.K.; Allen, L.H. Interventions with Vitamins B6, B12 and C in Pregnancy. Paediatr. Périnat. Epidemiol. 2012, 26, 55–74. [Google Scholar] [CrossRef]
- Molloy, A.M.; Kirke, P.N.; Brody, L.C.; Scott, J.M.; Mills, J.L. Effects of Folate and Vitamin B 12 Deficiencies During Pregnancy on Fetal, Infant, and Child Development. Food Nutr. Bull. 2008, 29 (Suppl. S1), S101–S111. [Google Scholar] [CrossRef]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle—Biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Jankovic-Karasoulos, T.; Furness, D.L.; Leemaqz, S.Y.; Dekker, G.A.; Grzeskowiak, L.E.; Grieger, J.A.; Andraweera, P.H.; McCullough, D.; McAninch, D.; McCowan, L.M.; et al. Maternal folate, one-carbon metabolism and pregnancy outcomes. Matern. Child Nutr. 2021, 17, e13064. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, U.; Katre, P.; Yajnik, C.S. Influence of Maternal Vitamin B12 and Folate on Growth and Insulin Resistance in the Offspring. In Maternal and Child Nutrition: The First 1000 Days; Karger Publishers: Basel, Switzerland, 2013; pp. 145–156. [Google Scholar]
- Sijilmassi, O. Folic acid deficiency and vision: A review. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Furness, D.; Fenech, M.; Dekker, G.; Khong, T.Y.; Roberts, C.; Hague, W. Folate, Vitamin B12, Vitamin B6 and homocysteine: Impact on pregnancy outcome. Matern. Child Nutr. 2013, 9, 155–166. [Google Scholar] [CrossRef]
- Dai, C.; Fei, Y.; Li, J.; Shi, Y.; Yang, X. A Novel Review of Homocysteine and Pregnancy Complications. BioMed Res. Int. 2021, 2021, 6652231. [Google Scholar] [CrossRef]
- Lai, J.S.; Pang, W.W.; Cai, S.; Lee, Y.S.; Chan, J.K.; Shek, L.P.; Yap, F.K.; Tan, K.H.; Godfrey, K.M.; van Dam, R.M.; et al. High folate and low vitamin B12 status during pregnancy is associated with gestational diabetes mellitus. Clin. Nutr. 2018, 37, 940–947. [Google Scholar] [CrossRef]
- Wang, Z.P.; Shang, X.X.; Zhao, Z.T. Low maternal vitamin B12 is a risk factor for neural tube defects: A meta-analysis. J. Matern. Fetal Neonatal Med. 2012, 25, 389–394. [Google Scholar] [CrossRef]
- Shub, A. Diabetes and pregnancy. Aust. N. Z. J. Obstet. Gynaecol. 2020, 60, 829–830. [Google Scholar] [CrossRef]
- Lebovitz, H. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S135–S148. [Google Scholar] [CrossRef]
- Paul, L.; Selhub, J. Interaction between excess folate and low vitamin B12 status. Mol. Asp. Med. 2017, 53, 43–47. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Vatish, M.; Alam, M.T.; Ott, S.; Kumar, S.; Saravanan, P. Low Vitamin B12 in Pregnancy Is Associated With Adipose-Derived Circulating miRs Targeting PPARγ and Insulin Resistance. J. Clin. Endocrinol. Metab. 2017, 102, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.P.; Christian, P.; Schulze, K.J.; Arguello, M.; LeClerq, S.C.; Khatry, S.K.; West, J.K.P. Low maternal vitamin B-12 status is associated with offspring insulin resistance regardless of antenatal micronutrient supplementation in rural Nepal. J. Nutr. 2011, 141, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Zhang, J.; Guan, Y.; Xing, Y. Early supplementation of folate and vitamin B12 improves insulin resistance in intrauterine growth retardation rats. Transl. Pediatr. 2022, 11, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.; Wright, C. Safety and efficacy of supplements in pregnancy. Nutr. Rev. 2020, 78, 813–826. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| INDIRECT METHODS | FORMULA |
|---|---|
| HOMA-IR | |
| QUICKI | |
| MCA | |
| FIRI | ( |
| FPI | FPI |
| ISI | |
| WBISI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasetti, A.; Quarta, A.; Guarino, M.; Cicolini, I.; Iannucci, D.; Giannini, C.; Chiarelli, F. Role of Prenatal Nutrition in the Development of Insulin Resistance in Children. Nutrients 2023, 15, 87. https://doi.org/10.3390/nu15010087
Blasetti A, Quarta A, Guarino M, Cicolini I, Iannucci D, Giannini C, Chiarelli F. Role of Prenatal Nutrition in the Development of Insulin Resistance in Children. Nutrients. 2023; 15(1):87. https://doi.org/10.3390/nu15010087
Chicago/Turabian StyleBlasetti, Annalisa, Alessia Quarta, Miriana Guarino, Ilenia Cicolini, Daniela Iannucci, Cosimo Giannini, and Francesco Chiarelli. 2023. "Role of Prenatal Nutrition in the Development of Insulin Resistance in Children" Nutrients 15, no. 1: 87. https://doi.org/10.3390/nu15010087
APA StyleBlasetti, A., Quarta, A., Guarino, M., Cicolini, I., Iannucci, D., Giannini, C., & Chiarelli, F. (2023). Role of Prenatal Nutrition in the Development of Insulin Resistance in Children. Nutrients, 15(1), 87. https://doi.org/10.3390/nu15010087