
Dysmetabolism and Neurodegeneration: Trick or Treat?

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
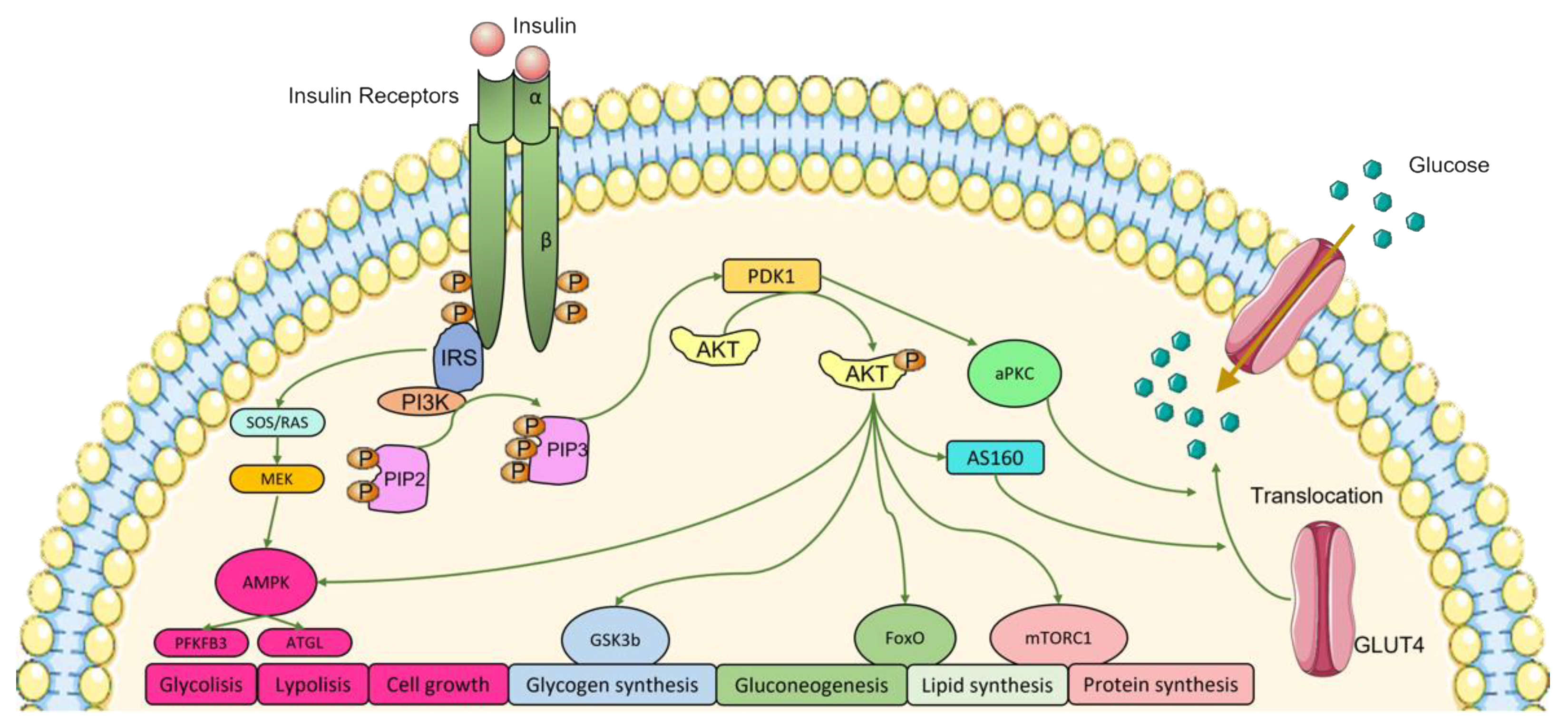
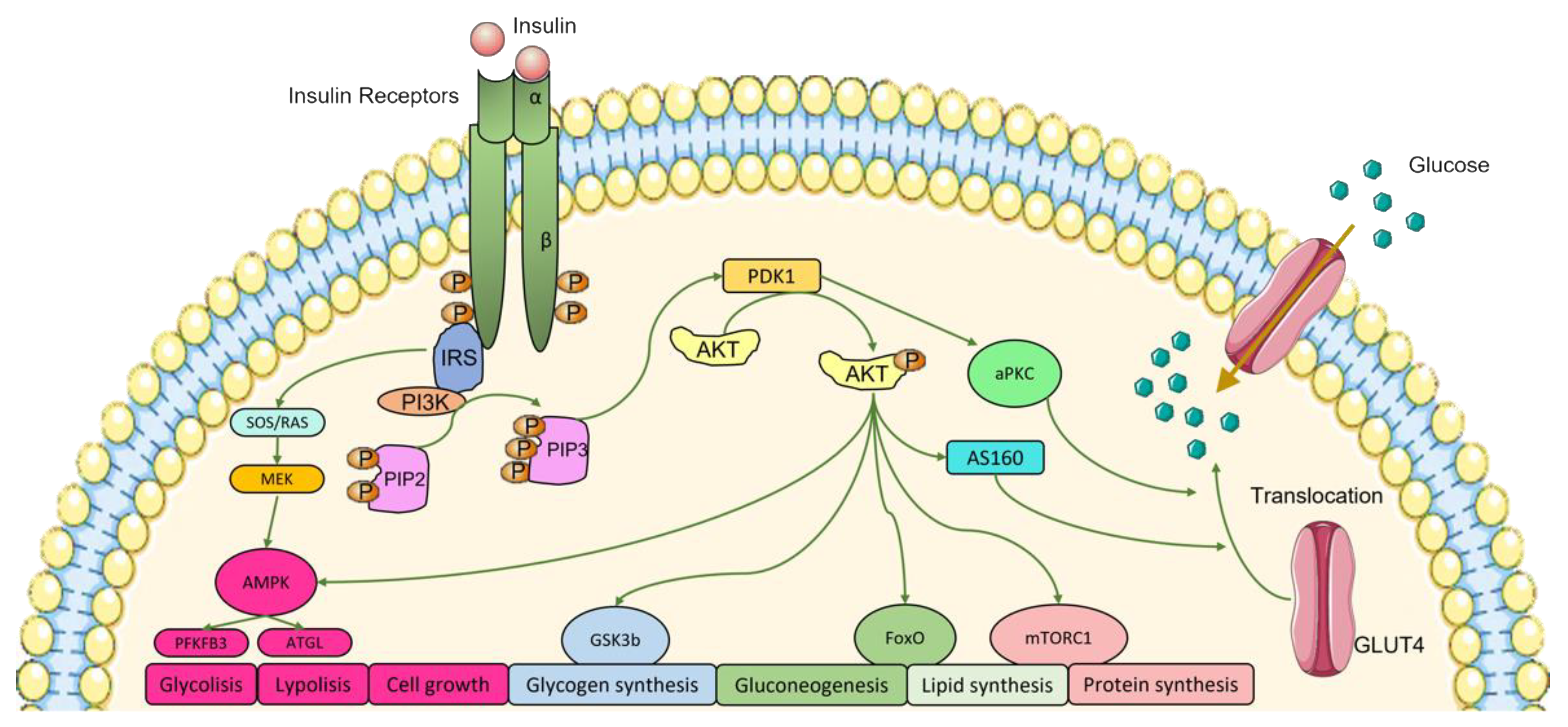
2. Insulin and Insulin Signaling Pathways
3. Metabolic Syndrome: Insulin Resistance and Diabetes
Impact of Hypercaloric Diets on Insulin Resistance and Metabolic Syndrome
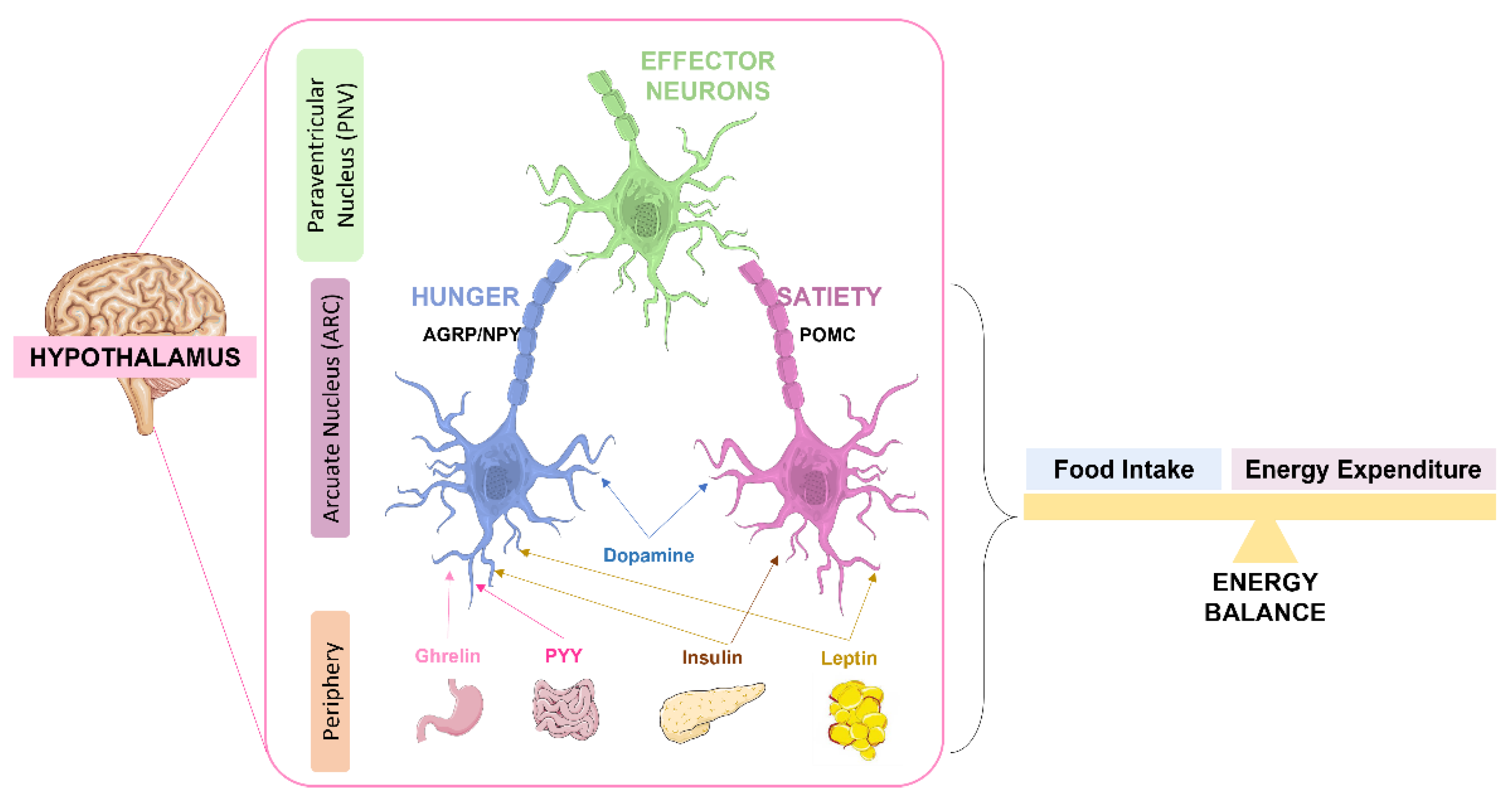
4. The Role of Insulin in the Brain
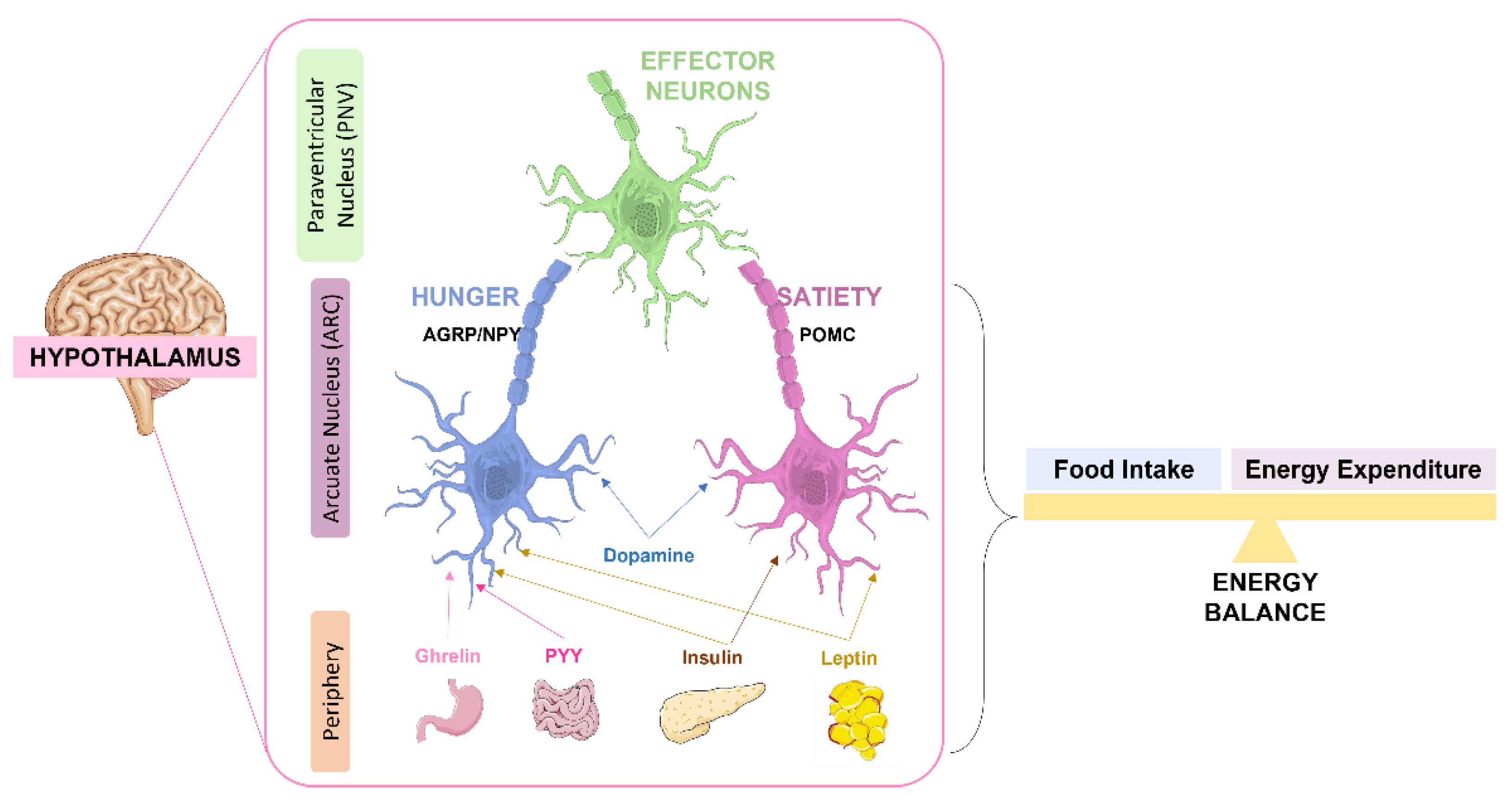
4.1. Role of Insulin in Brain Glucose Metabolism and Feeding
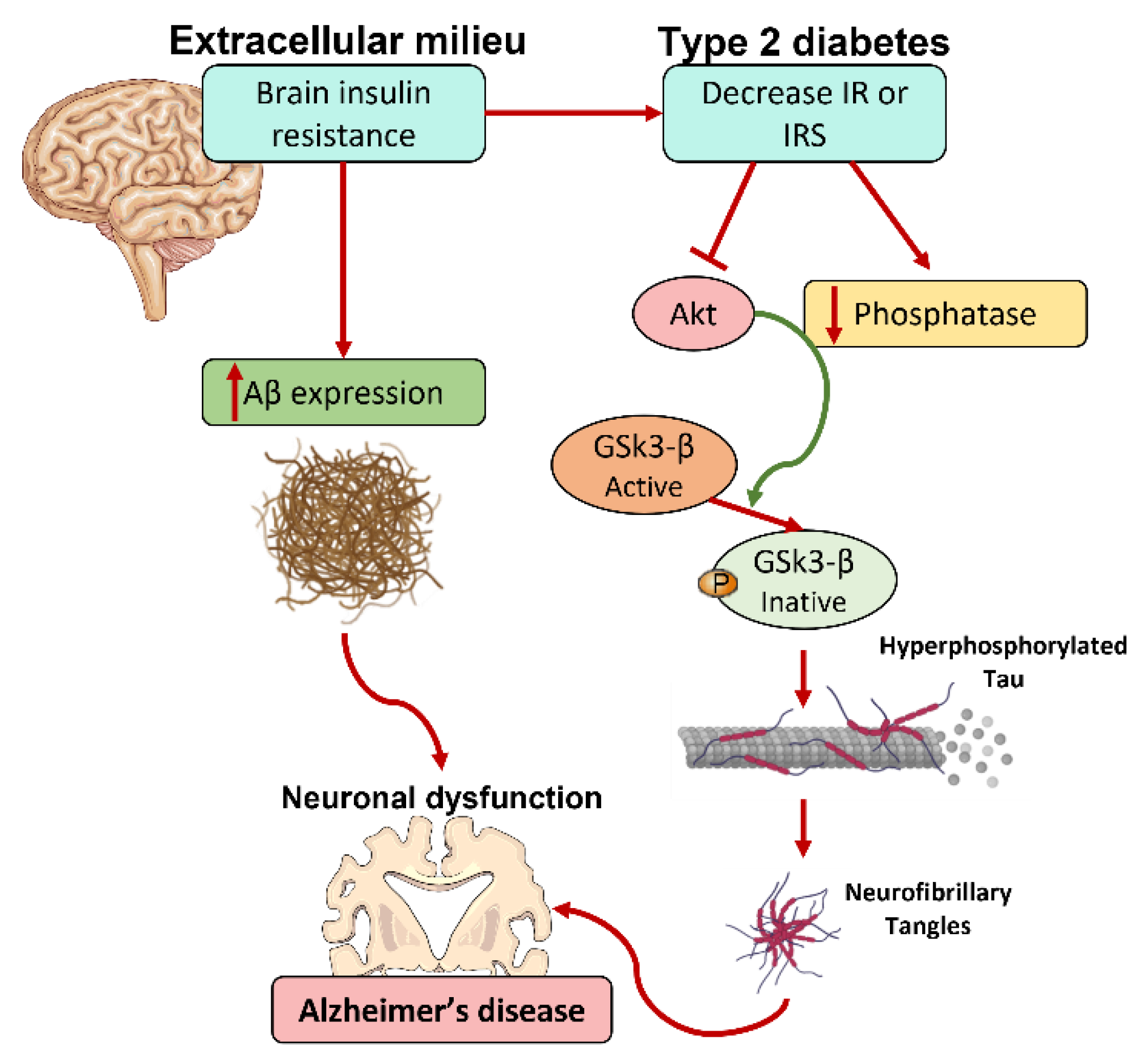
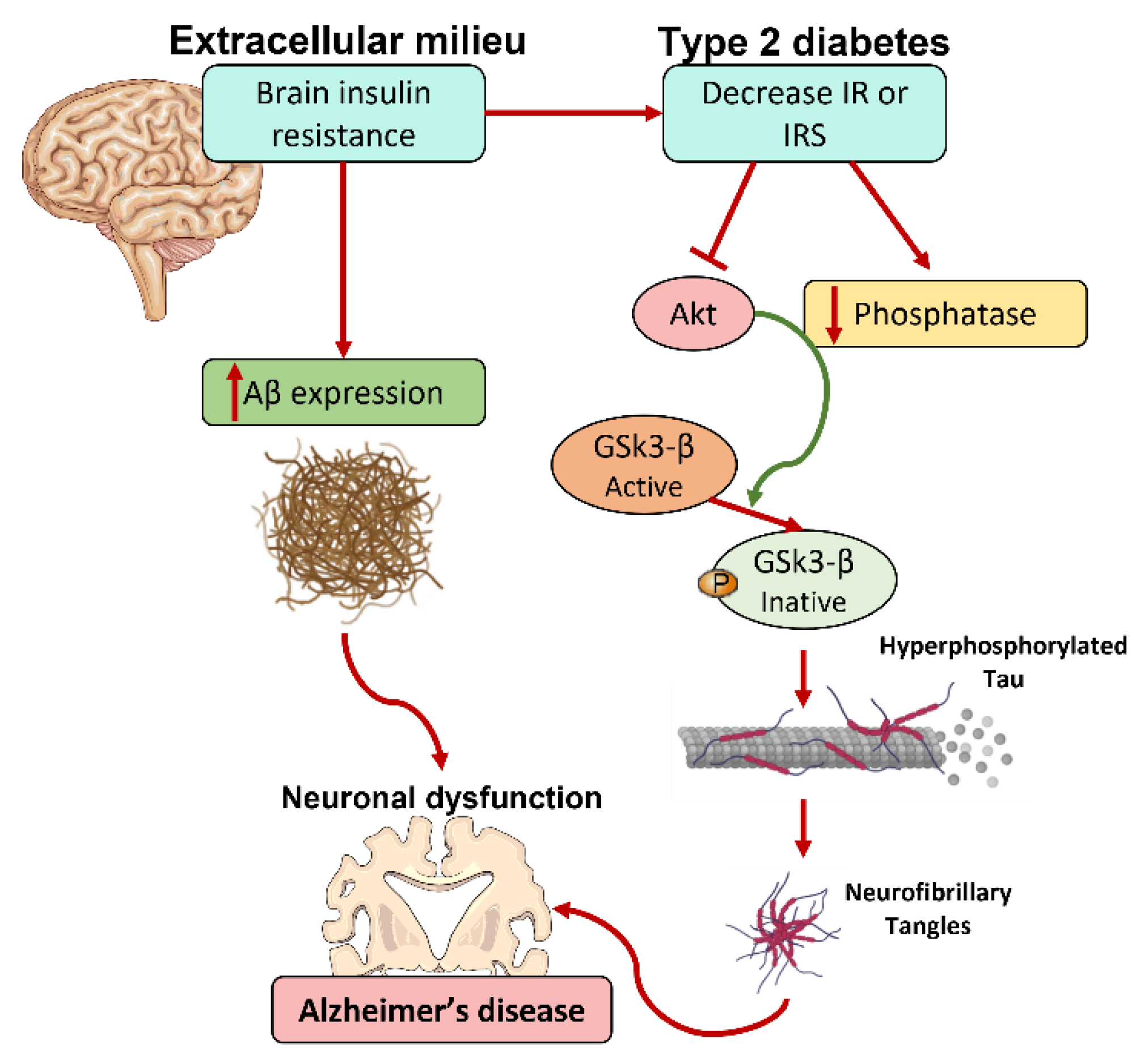
4.2. Insulin and Cognitive Function
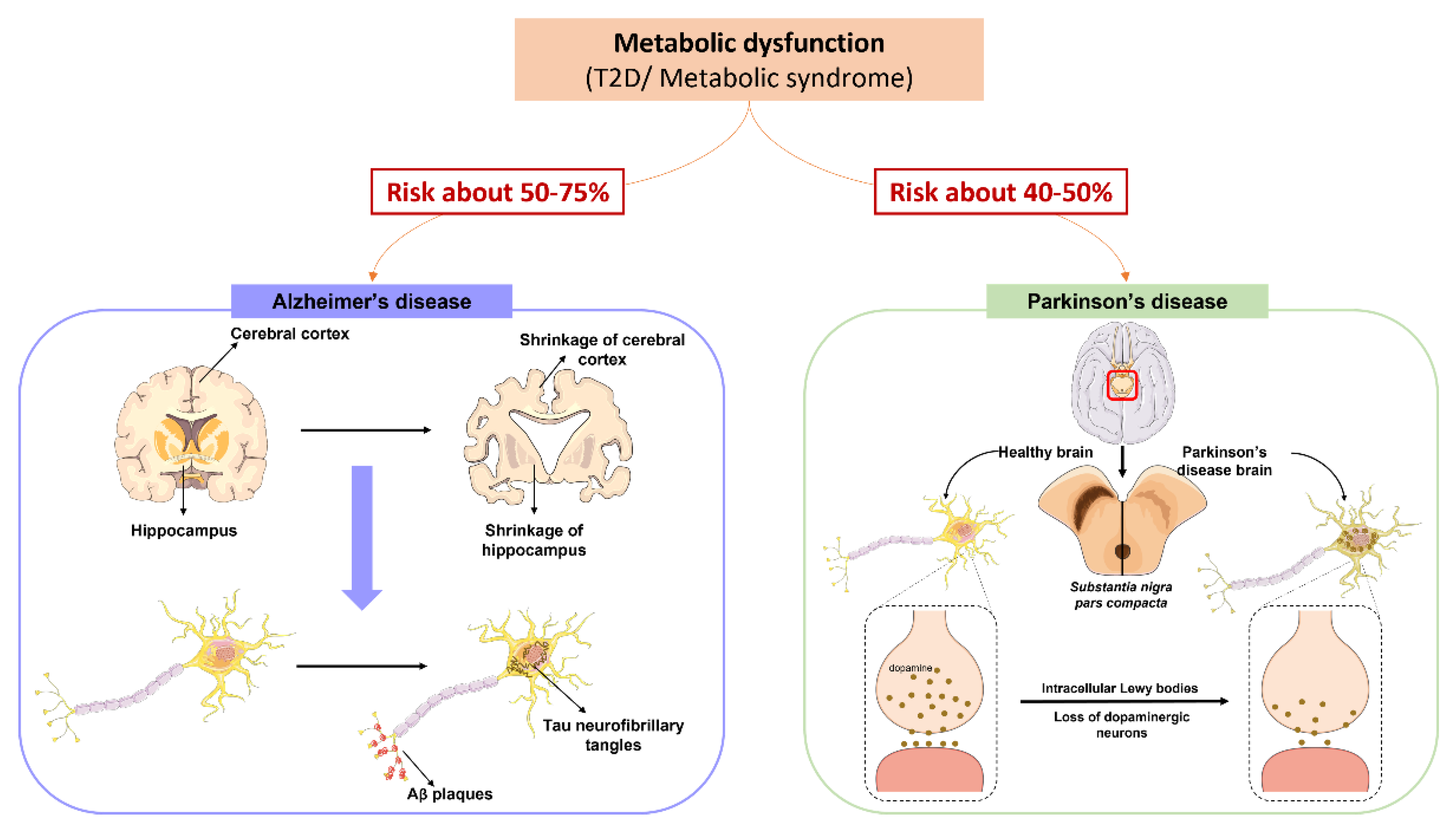
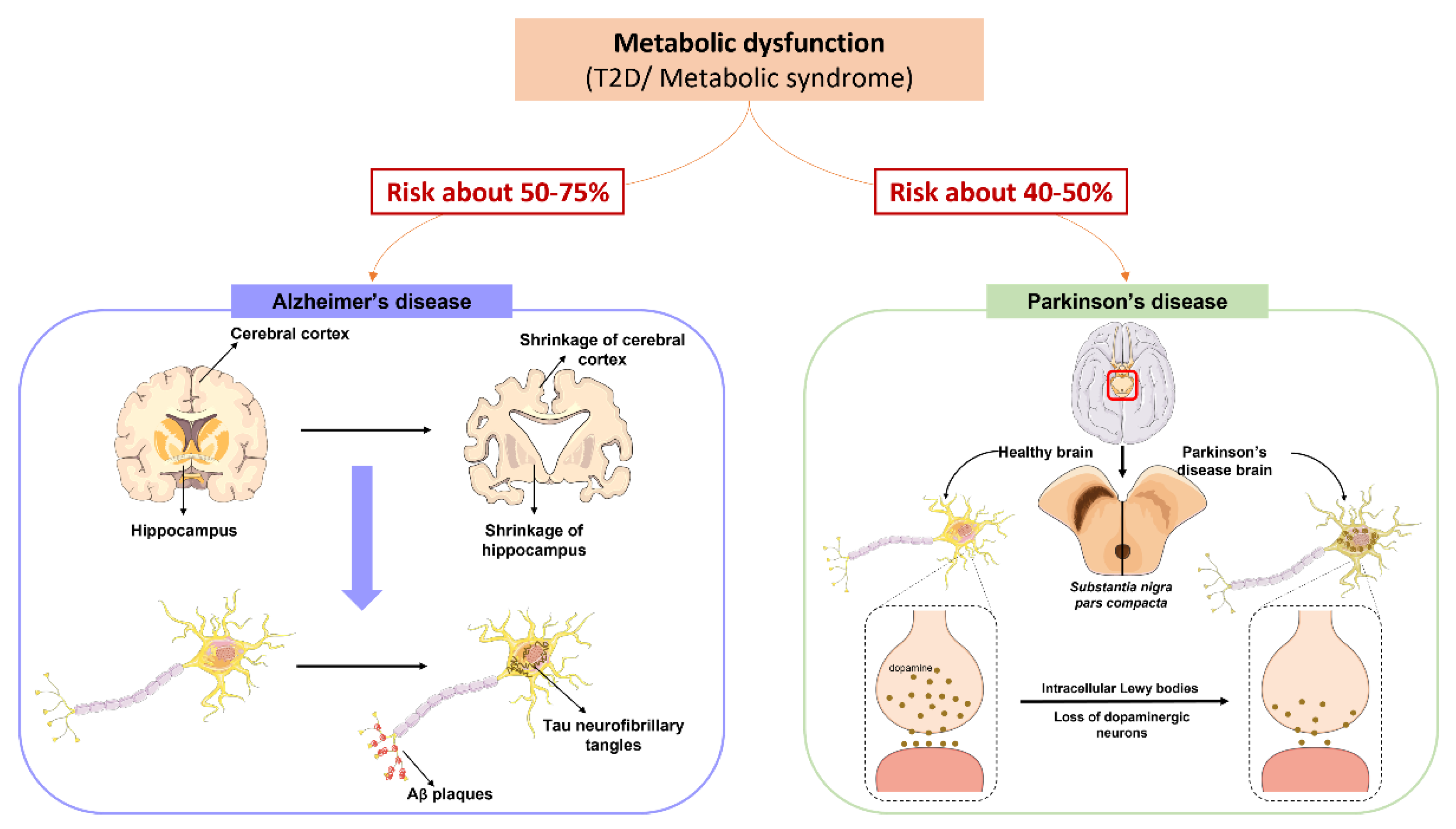
5. Metabolic Syndrome and the Neurodegenerative Process
5.1. The Impact of Diet in Neurodegeneration: Evidence from Animal Models
5.1.1. Parkinson’s Disease Animal Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Diet Regiment | Rodent Model | Outcomes |
|---|---|---|---|
| Choi et al. [181] | 8 weeks of HF diet | MPTP-lesioned PD-like mice | Severe decrease in the levels of striatal dopamine and of nigral microtubule-associated protein 2, manganese superoxide dismutase, TH. Elevated striatal nNOS phosphorylation and dopamine turnover. |
| Bousquet et al. [182] | 8 weeks of HF diet | MPTP-lesioned PD-like mice | Decreased levels of striatal TH and dopamine, exacerbated MPTP-induced dopaminergic degeneration. |
| Sharma and Taliyan [183] | 8 weeks of HF diet | 6-OHDA-induced PD-like rats | Decreased levels of striatal dopamine, motor abnormalities, exacerbated 6-OHDA mediated neurotoxicity. |
| Morris et al. [184] | 5 weeks of HF diet | 6-OHDA-induced PD-like rats | Peripheral dysmetabolic features, increased dopamine depletion and oxidative stress in the substantia nigra and the striatum, without locomotor dysfunction. |
| Ma et al. [185] | 3 month of HF diet, followed by 3 months of a low-fat diet | 6-OHDA-induced PD-like rats | Reversed peripheral dysmetabolism and mitochondrial and proteasomal function in the striatum, although without altering nigrostriatal vulnerability. |
| Rotermund et al. [191] | HF diet from 5 weeks old onward throughout their lifespan | Mutant A30P aSyn transgenic mice | Accelerated onset of brainstem aSyn pathology and lethal locomotor features. |
| Hong et al. [192] | 2 weeks of HF diet | MitoPark transgenic mice | Increased SNCA expression (coding for aSyn) in the dopaminergic neurons of both the WT and MitoPark mice; enhanced dopaminergic degeneration in the MitoPark mice. |
| Morris et al. 2011 [188] | 12 weeks of HF diet | WT rats | Attenuated dopamine release and clearance and increased iron deposition in the substantia nigra. |
| Jang et al. [189] | 13 weeks of HF diet | WT mice | Decreased in movement accompanied by abnormal motor behavior. Decreased levels of TH in the substantia nigra and striatum. |
| Kao et al. [193] | 5 months of HF diet | WT mice | Dopaminergic neurons degeneration and reduced dopaminergic neuroplasticity in the substantia nigra. |
| Bittencourt et al. [180] | 25 weeks of HF diet | WT rats | Reduced levels of TH through metabolic dysfunction, neuroinflammation and oxidative stress, associated with impaired locomotor activity, and anxiety-related behaviors, without changes in motor coordination or memory. No differences in the levels of aSyn. |
5.1.2. Alzheimer’s Disease Animal Models
5.2. Sex Differences in the Link Dysmetabolism-Neurodegeneration
6. Regulation of Metabolic Function as a Prevention of Neurodegeneration
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepúlveda, J.; Murray, C. The state of global health in 2014. Science 2014, 345, 1275–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the Metabolic Syndrome in the United States, 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef]
- Hinnouho, G.-M.; Czernichow, S.; Dugravot, A.; Nabi, H.; Brunner, E.; Kivimaki, M.; Singh-Manoux, A. Metabolically healthy obesity and the risk of cardiovascular disease and type 2 diabetes: The Whitehall II cohort study. Eur. Heart J. 2014, 36, 551–559. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef]
- Deng, Y.; Li, B.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. Am. J. Pathol. 2009, 175, 2089–2098. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Li, H.; Yan, H.; Zhang, P.; Chang, L.; Li, T. Risk of Parkinson Disease in Diabetes Mellitus: An Updated Meta-Analysis of Population-Based Cohort Studies. Medicine 2016, 95, e3549. [Google Scholar] [CrossRef]
- Podolsky, S.; Leopold, N.A.; Sax, D.S. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet 1972, 299, 1356–1358. [Google Scholar] [CrossRef]
- Hegele, R.A.; Maltman, G.M. Insulin’s centenary: The birth of an idea. Lancet Diabetes Endocrinol. 2020, 8, 971–977. [Google Scholar] [CrossRef]
- Kahn, C.R.; White, M.F. The insulin receptor and the molecular mechanism of insulin action. J. Clin. Investig. 1988, 82, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell. Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Hermida, M.A.; Kumar, J.D.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Inoue, H.; Ravnskjaer, K.; Viste, K.; Miller, N.; Liu, Y.; Hedrick, S.; Vera, L.; Montminy, M. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 3087–3092. [Google Scholar] [CrossRef] [Green Version]
- Eldin, W.S.; Emara, M.; Shoker, A. Prediabetes: A must to recognise disease state. Int. J. Clin. Pract. 2008, 62, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IDFD Atlas. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.; Gavin, J.R.; Aguilar, R.B. The Time Is Right for a New Classification System for Diabetes: Rationale and Implications of the β-Cell–Centric Classification Schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2013, 383, 1084–1094. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2016, 66, 241–255. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Classification of Diabetes Mellitus; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Lovejoy, J.C. The influence of dietary fat on insulin resistance. Curr. Diabetes Rep. 2002, 2, 435–440. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwater, M.L.; Fletcher, P.C.; Ziauddeen, H. Sugar addiction: The state of the science. Eur. J. Nutr. 2016, 55 (Suppl. S2), 55–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, B.; Sacramento, J.F.; Ribeiro, M.J.; Prego, C.S.; Correia, M.C.; Coelho, J.C.; Cunha-Guimaraes, J.P.; Rodrigues, T.; Martins, I.B.; Guarino, M.P.; et al. Evaluating the Impact of Different Hypercaloric Diets on Weight Gain, Insulin Resistance, Glucose Intolerance, and its Comorbidities in Rats. Nutrients 2019, 11, 1197. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- He, Q.; Gao, Z.; Yin, J.; Zhang, J.; Yun, Z.; Ye, J. Regulation of HIF-1(alpha) activity in adipose tissue by obesity-associated factors: Adipogenesis, insulin, and hypoxia. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E877–E885. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, K.; Sarkadi-Nagy, E.; Duncan, R.E.; Ahmadian, M.; Sul, H.S. Regulation of Triglyceride Metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol. Liver Physiol. 2007, 293, G1–G4. [Google Scholar]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Halbleib, M.; Ahmad, F.; Manganiello, V.C.; Greenberg, A.S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 2002, 51, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, C.R.; Zehra, A.; Ramirez, V.; Wiers, C.E.; Volkow, N.D.; Wang, G.J. Impact of sugar on the body, brain, and behavior. Front. Biosci. 2018, 23, 2255–2266. [Google Scholar]
- Greenberg, D.; St Peter, J.V. Sugars and Sweet Taste: Addictive or Rewarding? Int. J. Environ. Res. Public Health 2021, 18, 9791. [Google Scholar] [CrossRef]
- Alam, Y.H.; Kim, R.; Jang, C. Metabolism and Health Impacts of Dietary Sugars. J. Lipid Atheroscler. 2022, 11, 20–38. [Google Scholar] [CrossRef]
- Jalal, D.I.; Smits, G.; Johnson, R.J.; Chonchol, M. Increased Fructose Associates with Elevated Blood Pressure. J. Am. Soc. Nephrol. 2010, 21, 1543–1549. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; O’Keefe, J.H.; Lucan, S.C. Added fructose: A principal driver of type 2 diabetes mellitus and its consequences. Mayo Clin. Proc. 2015, 90, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Malik, V.S.; Hu, F.B. Fructose and Cardiometabolic Health: What the Evidence From Sugar-Sweetened Beverages Tells Us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Madero, M.; Arriaga, J.C.; Jalal, D.; Rivard, C.; McFann, K.; Pérez-Méndez, O.; Vázquez, A.; Ruiz, A.; Lanaspa, M.A.; Jimenez, C.R.; et al. The effect of two energy-restricted diets, a low-fructose diet versus a moderate natural fructose diet, on weight loss and metabolic syndrome parameters: A randomized controlled trial. Metabolism 2011, 60, 1551–1559. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children with Obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Monterosso, J.R.; Sarpelleh, K.; Page, K.A. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc. Natl. Acad. Sci. USA 2015, 112, 6509–6514. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014, 6, 3117–3129. [Google Scholar] [CrossRef] [Green Version]
- Rizkalla, S.W. Health implications of fructose consumption: A review of recent data. Nutr. Metab. 2010, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Benito, M. Tissue specificity on insulin action and resistance: Past to recent mechanisms. Acta Physiol. 2011, 201, 297–312. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef]
- Wallum, B.J.; Taborsky, G.J.; Porte, D.; Figlewicz, D.P.; Jacobson, L.; Beard, J.C.; Ward, W.K.; Dorsa, D. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man. J. Clin. Endocrinol. Metab. 1987, 64, 190–194. [Google Scholar] [CrossRef]
- Bromander, S.; Anckarsäter, R.; Ahren, B.; Kristiansson, M.; Blennow, K.; Holmäng, A.; Zetterberg, H.; Anckarsäter, H.; Wass, C.E. Cerebrospinal fluid insulin during non-neurological surgery. J. Neural Transm. 2010, 117, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Figlewicz, D.P.; Baskin, D.G.; Woods, S.C.; Porte, D. Insulin in the brain: A hormonal regulator of energy balance. Endocr. Rev. 1992, 13, 387–414. [Google Scholar]
- Coker, G.T.; Studelska, D.; Harmon, S.; Burke, W.; O’Malley, K.L. Analysis of tyrosine hydroxylase and insulin transcripts in human neuroendocrine tissues. Mol. Brain Res. 1990, 8, 93–98. [Google Scholar] [CrossRef]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Aβ oligomers. Mol. Biol. Cell 2017, 28, 2623–2636. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Kagalwala, M.N.; Onuma, Y.; Ito, Y.; Warashina, M.; Terashima, K.; Sanosaka, T.; Nakashima, K.; Gage, F.H.; Asashima, M. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol. Med. 2011, 3, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Koarashi, K.; Kawabe, K.; Itakura, M.; Nakajima, H.; Moriyama, M.; Nakamura, Y. Insulin expression in cultured astrocytes and the decrease by amyloid β. Neurochem. Int. 2018, 119, 171–177. [Google Scholar] [CrossRef]
- Dakic, T.B.; Jevdjovic, T.; Peric, M.I.; Bjelobaba, I.; Markelić, M.; Milutinovic, B.S.; Lakic, I.V.; Jasnic, N.; Djordjevic, J.D.; Vujovic, P.Z. Short-term fasting promotes insulin expression in rat hypothalamus. Eur. J. Neurosci. 2017, 46, 1730–1737. [Google Scholar] [CrossRef]
- Molnár, G.; Faragó, N.; Kocsis, K.; Rózsa, M.; Lovas, S.; Boldog, E.; Báldi, R.; Csajbók, É.; Gardi, J.; Puskás, L.G.; et al. GABAergic Neurogliaform Cells Represent Local Sources of Insulin in the Cerebral Cortex. J. Neurosci. 2014, 34, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.-P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018, 24, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Vigneri, P.; Pennisi, M.G.; Malandrino, P.; Squatrito, S.; Pezzino, V.; Vigneri, R. Biological Effects of Insulin and Its Analogs on Cancer Cells with Different Insulin Family Receptor Expression. J. Cell. Physiol. 2014, 229, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Pandini, G.; Le Moli, R.; Squatrito, S.; Vigneri, R. Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia 2010, 53, 1743–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre-Eugene, C.; Pagesy, P.; Nguyen, T.T.; Neuillé, M.; Tschank, G.; Tennagels, N.; Hampe, C.; Issad, T. Effect of Insulin Analogues on Insulin/IGF1 Hybrid Receptors: Increased Activation by Glargine but Not by Its Metabolites M1 and M2. PLoS ONE 2012, 7, e41992. [Google Scholar] [CrossRef] [Green Version]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Wu, Z.; Farrell, R.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e3. [Google Scholar] [CrossRef]
- Uemura, E.; Greenlee, H.W. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 2006, 198, 48–53. [Google Scholar] [CrossRef]
- Heidenreich, K.A.; Gilmore, P.R.; Garvey, W.T. Glucose transport in primary cultured neurons. J. Neurosci. Res. 1989, 22, 397–407. [Google Scholar] [CrossRef]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 1999, 57, 693–705. [Google Scholar] [CrossRef]
- Duelli, R.; Kuschinsky, W. Brain Glucose Transporters: Relationship to Local Energy Demand. Physiology 2001, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Morikawa, Y.; Tamura, S.; Doi, A.; Nanjo, K.; Senba, E. Subcellular localization of glucose transporter 4 in the hypothalamic arcuate nucleus of ob/ob mice under basal conditions. Brain Res. 2005, 1049, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- McEwen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Goodner, C.J.; Hom, F.G.; Berrie, M.A. Investigation of the Effect of Insulin upon Regional Brain Glucose Metabolism in the Rat in Vivo. Endocrinology 1980, 107, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Hom, F.G.; Goodner, C.J.; Berrie, M.A. A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes 1984, 33, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Lesniak, M.A.; Hill, J.M.; Kiess, W.; Rojeski, M.; Pert, C.B.; Roth, J. Receptors for Insulin-like Growth Factors I and II: Autoradiographic Localization in Rat Brain and Comparison to Receptors for Insulin. Endocrinology 1988, 123, 2089–2099. [Google Scholar] [CrossRef]
- Khanh, D.V.; Choi, Y.H.; Moh, S.H.; Kinyua, A.W.; Kim, K.W. Leptin and insulin signaling in dopaminergic neurons: Relationship between energy balance and reward system. Front. Psychol. 2014, 5, 846. [Google Scholar] [CrossRef] [Green Version]
- Könner, A.C.; Hess, S.; Tovar, S.; Mesaros, A.; Sánchez-Lasheras, C.; Evers, N.; Verhagen, L.A.; Brönneke, H.S.; Kleinridders, A.; Hampel, B.; et al. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011, 13, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Heni, M.; Kullmann, S.; Ketterer, C.; Guthoff, M.; Linder, K.; Wagner, R.; Stingl, K.T.; Veit, R.; Staiger, H.; Häring, H.U.; et al. Nasal insulin changes peripheral insulin sensitivity simultaneously with altered activity in homeostatic and reward-related human brain regions. Diabetologia 2012, 55, 1773–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heni, M.; Wagner, R.; Kullmann, S.; Veit, R.; Mat Husin, H.; Linder, K.; Benkendorff, C.; Peter, A.; Stefan, N.; Häring, H.U.; et al. Central insulin administration improves whole-body insulin sensitivity via hypothalamus and parasympathetic outputs in men. Diabetes 2014, 63, 4083–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauda, E.B.; Conde, S.; Bassi, M.; Zoccal, D.B.; Colombari, D.S.A.; Colombari, E.; Despotovic, N. Leptin: Master Regulator of Biological Functions that Affects Breathing. Compr. Physiol. 2020, 10, 1047–1083. [Google Scholar] [PubMed]
- Kleinridders, A.; Könner, A.C.; Bruning, J.C. CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 2009, 9, 794–804. [Google Scholar] [CrossRef]
- Könner, A.C.; Klöckener, T.; Brüning, J.C. Control of energy homeostasis by insulin and leptin: Targeting the arcuate nucleus and beyond. Physiol. Behav. 2009, 97, 632–638. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Reagan, L.P. Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology 2018, 136, 182–191. [Google Scholar] [CrossRef]
- De Felice, F.G.; Benedict, C. A Key Role of Insulin Receptors in Memory. Diabetes 2015, 64, 3653–3655. [Google Scholar] [CrossRef] [Green Version]
- Dhikav, V.; Anand, K.S. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef]
- Tyng, C.M.; Amin, H.U.; Saad, M.N.M.; Malik, A.S. The Influences of Emotion on Learning and Memory. Front. Psychol. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Mizuseki, K.; Royer, S.; Diba, K.; Buzsáki, G. Activity dynamics and behavioral correlates of CA3 and CA1 hippocampal pyramidal neurons. Hippocampus 2012, 22, 1659–1680. [Google Scholar] [CrossRef] [Green Version]
- Alkadhi, K.A. Cellular and Molecular Differences between Area CA1 and the Dentate Gyrus of the Hippocampus. Mol. Neurobiol. 2019, 56, 6566–6580. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Miles, R. The CA3 region of the hippocampus: How is it? What is it for? How does it do it? Front. Cell Neurosci. 2015, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunsaker, M.R.; Lee, B.; Kesner, R.P. Evaluating the temporal context of episodic memory: The role of CA3 and CA1. Behav. Brain Res. 2008, 188, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoge, J.; Kesner, R.P. Role of CA3 and CA1 subregions of the dorsal hippocampus on temporal processing of objects. Neurobiol. Learn. Mem. 2007, 88, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahata, Y.; Yasuda, R. Plasticity of Spine Structure: Local Signaling, Translation and Cytoskeletal Reorganization. Front. Synaptic Neurosci. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porte, D.; Baskin, D.G., Jr.; Schwartz, M.W. Insulin signaling in the central nervous system: A critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes 2005, 54, 1264–1276. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-C.; Huang, C.-C.; Hsu, K.-S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Van der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef]
- Choi, J.; Ko, J.; Racz, B.; Burette, A.; Lee, J.R.; Kim, S.; Na, M.; Lee, H.W.; Kim, K.; Weinberg, R.J.; et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J. Neurosci. 2005, 25, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Christie, J.; Wenthold, R.J.; Monaghan, D.T. Insulin Causes a Transient Tyrosine Phosphorylation of NR2A and NR2B NMDA Receptor Subunits in Rat Hippocampus. J. Neurochem. 2001, 72, 1523–1528. [Google Scholar] [CrossRef]
- Liu, L.; Brown, J.C.; Webster, W.W.; Morrisett, R.A.; Monaghan, D.T. Insulin potentiates N-methyl-d-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neurosci. Lett. 1995, 192, 5–8. [Google Scholar] [CrossRef]
- Martín, E.D.; Sánchez-Perez, A.; Trejo, J.L.; Martin-Aldana, J.A.; Jaimez, M.C.; Pons, S.; Umanzor, C.A.; Menes, L.; White, M.F.; Burks, D.J. IRS-2 Deficiency Impairs NMDA Receptor-Dependent Long-term Potentiation. Cereb. Cortex 2011, 22, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Dong, Z.; Bagot, R.C.; Howland, J.G.; Phillips, A.G.; Wong, T.P.; Wang, Y.T. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc. Natl. Acad. Sci. USA 2010, 107, 16697–16702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euston, D.R.; Gruber, A.; McNaughton, B.L. The Role of Medial Prefrontal Cortex in Memory and Decision Making. Neuron 2012, 76, 1057–1070. [Google Scholar] [CrossRef] [Green Version]
- Chao, O.Y.; Silva, M.A.D.S.; Yang, Y.-M.; Huston, J.P. The medial prefrontal cortex-hippocampus circuit that integrates information of object, place and time to construct episodic memory in rodents: Behavioral, anatomical and neurochemical properties. Neurosci. Biobehav. Rev. 2020, 113, 373–407. [Google Scholar] [CrossRef]
- Niblock, M.M.; Brunso-Bechtold, J.K.; Riddle, D.R. Insulin-like growth factor I stimulates dendritic growth in primary soma-tosensory cortex. J. Neurosci. 2000, 20, 4165–4176. [Google Scholar] [CrossRef]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Häring, H.-U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef]
- Taouis, M.; Torres-Aleman, I. Editorial: Insulin and the Brain. Front. Endocrinol. 2019, 10, 299. [Google Scholar] [CrossRef]
- Heni, M.; Schöpfer, P.; Peter, A.; Sartorius, T.; Fritsche, A.; Synofzik, M.; Häring, H.-U.; Maetzler, W.; Hennige, A.M. Evidence for altered transport of insulin across the blood–brain barrier in insulin-resistant humans. Geol. Rundsch. 2013, 51, 679–681. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain insulin resistance impairs hippocampal plasticity. Vitam. Horm. 2020, 114, 281–306. [Google Scholar]
- García-Cáceres, C.; Lechuga-Sancho, A.; Argente, J.; Frago, L.M.; Chowen, J.A. Death of Hypothalamic Astrocytes in Poorly Controlled Diabetic Rats is Associated with Nuclear Translocation of Apoptosis Inducing Factor. J. Neuroendocr. 2008, 20, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodl, C.T.; Seaquist, E.R. Cognitive Dysfunction and Diabetes Mellitus. Endocr. Rev. 2008, 29, 494–511. [Google Scholar] [CrossRef] [PubMed]
- Fadel, J.R.; Reagan, L.P. Stop signs in hippocampal insulin signaling: The role of insulin resistance in structural, functional and behavioral deficits. Curr. Opin. Behav. Sci. 2015, 9, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.-R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Ramakers, G.M.; Gispen, W.H.; Biessels, G.J.; Al Ansari, A. Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp. Brain Res. 2013, 226, 45–51. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Dai, C.L.; Liang, Z.; Run, X.; Iqbal, K.; Liu, F.; Gong, C.X. Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Aβ level and microglia activation in the brains of 3xTg-AD mice. Exp. Neurol. 2014, 261, 610–619. [Google Scholar] [CrossRef]
- Yang, Y.W.; Hsieh, T.F.; Li, C.I.; Liu, C.S.; Lin, W.Y.; Chiang, J.H.; Li, T.C.; Lin, C.C. Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine 2017, 96, e5921. [Google Scholar] [CrossRef]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Puschmann, A.; Bhidayasiri, R.; Weiner, W.J. Synucleinopathies from bench to bedside. Park. Relat. Disord. 2012, 18 (Suppl. S1), S24–S27. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinson’s Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Eberstein, J.A. Metabolic syndrome and the role of dietary lifestyles in Alzheimer’s disease. J. Neurochem. 2008, 106, 1503–1514. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Helkala, E.; Tuomilehto, J.; Nissinen, A.; Soininen, H.; Kivipelto, M. Fat intake at midlife and cognitive impairment later in life: A population-based CAIDE study. Int. J. Geriatr. Psychiatry 2008, 23, 741–747. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Lourenco, M.V. Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Wilson, R.S. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 2004, 62, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Nyaradi, A.; Foster, J.K.; Hickling, S.; Li, J.; Ambrosini, G.; Jacques, A.; Oddy, W.H. Prospective associations between dietary patterns and cognitive performance during adolescence. J. Child Psychol. Psychiatry 2014, 55, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Øverby, N.C.; Lüdemann, E.; Høigaard, R. Self-repor.rted learning difficulties and dietary intake in Norwegian adolescents. Scand. J. Public Health 2013, 41, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, Q.; Jackson, J.C.; Zhang, J. Overweight is associated with decreased cognitive functioning among school-age children and adolescents. Obesity 2008, 16, 1809–1815. [Google Scholar] [CrossRef]
- Holloway, C.J.; E Cochlin, L.; Emmanuel, Y.; Murray, A.; Codreanu, I.; Edwards, L.M.; Szmigielski, C.; Tyler, D.J.; Knight, N.S.; Saxby, B.K.; et al. A high-fat diet impairs cardiac high-energy phosphate metabolism and cognitive function in healthy human subjects. Am. J. Clin. Nutr. 2011, 93, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Edwards, L.M.; Murray, A.J.; Holloway, C.J.; Carter, E.E.; Kemp, G.J.; Codreanu, I.; Brooker, H.; Tyler, D.J.; Robbins, P.A.; Clarke, K. Short-term consumption of a high-fat diet impairs whole-body efficiency and cognitive function in sedentary men. FASEB J. 2010, 25, 1088–1096. [Google Scholar] [CrossRef]
- Luchsinger, J.; Tang, M.-X.; Shea, S.; Mayeux, R. Caloric Intake and the Risk of Alzheimer Disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef]
- Brands, A.M.; Biessels, G.J.; de Haan, E.H.; Kappelle, L.J.; Kessels, R.P. The effects of type 1 diabetes on cognitive performance: A meta-analysis. Diabetes Care 2005, 28, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; Kappelle, L.J. Increased risk of Alzheimer’s disease in Type II diabetes: Insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 2005, 33 Pt 5, 1041–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilliox, L.A.; Chadrasekaran, K.; Kwan, J.Y.; Russell, J.W. Diabetes and Cognitive Impairment. Curr. Diabetes Rep. 2016, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Kuo, C.L.; Chang, Y.H.; Lu, C.L.; Martini, S.; Hou, W.H. Association between trajectory of severe hypoglycemia and dementia in patients with type 2 diabetes: A population-based study. J. Epidemiol. 2021, JE20200518. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Spires-Jones, T.; Pooler, A.M.; Lechuga-Sancho, A.; Bacskai, B.J.; Garcia-Alloza, M. Progressive Neuronal Pathology and Synaptic Loss Induced by Prediabetes and Type 2 Diabetes in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2016, 54, 3428–3438. [Google Scholar] [CrossRef]
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Cardiovasc. Med. 2006, 2, 159–166. [Google Scholar] [CrossRef]
- Watson, G.; Craft, S. Modulation of memory by insulin and glucose: Neuropsychological observations in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 97–113. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Abbott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed] [Green Version]
- Qu, Z.-S.; Li, L.; Sun, X.-J.; Zhao, Y.-W.; Zhang, J.; Geng, Z.; Fu, J.-L.; Ren, Q.-G. Glycogen Synthase Kinase-3 Regulates Production of Amyloid-βPeptides and Tau Phosphorylation in Diabetic Rat Brain. Sci. World J. 2014, 2014, 878123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Deng, Y.; Zhang, B.; Gong, C.-X. Deregulation of brain insulin signaling in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-Fat Diet Induces Hepatic Insulin Resistance and Impairment of Synaptic Plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 Diabetes and the Risk of Parkinson’s Disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.A.; Potashkin, J.A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 2013, 19, 176–186. [Google Scholar] [CrossRef]
- Lu, M.; Hu, G. Targeting metabolic inflammation in Parkinson’s disease: Implications for prospective therapeutic strategies. Clin. Exp. Pharmacol. Physiol. 2011, 39, 577–585. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Nelson, J.S.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Popper, J.S.; Petrovitch, H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology 2002, 59, 1051–1057. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145, 98–120. [Google Scholar] [CrossRef]
- Gentier, I.; D’Hondt, E.; Shultz, S.; Deforche, B.; Augustijn, M.; Hoorne, S.; Verlaecke, K.; De Bourdeaudhuij, I.; Lenoir, M. Fine and gross motor skills differ between healthy-weight and obese children. Res. Dev. Disabil. 2013, 34, 4043–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krombholz, H. Motor and Cognitive Performance of Overweight Preschool Children. Percept. Mot. Skills 2013, 116, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Mond, J.M.; Stich, H.; Hay, P.; Kraemer, A.; Baune, B.T. Associations between obesity and developmental functioning in pre-school children: A population-based study. Int. J. Obes. 2007, 31, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.; Veneri, D.; Decker, R.; Gannotti, M. Weight Status and Gross Motor Skill in Kindergarten Children. Pediatr. Phys. Ther. 2012, 24, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, A.A.; Desha, L.; Ziviani, J.; Griffiths, L.; Heaslop, A.; Khan, A.; Leong, G. Fundamental movement skills and self-concept of children who are overweight. Pediatr. Obes. 2011, 6, e464–e471. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Okely, A.; Caputi, P.; Cliff, D. Perceived and actual competence among overweight and non-overweight children. J. Sci. Med. Sport 2010, 13, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Slining, M.; Adair, L.S.; Goldman, B.D.; Borja, J.B.; Bentley, M. Infant Overweight Is Associated with Delayed Motor Development. J. Pediatr. 2010, 157, 20–25.e1. [Google Scholar] [CrossRef] [Green Version]
- Bittencourt, A.; Brum, P.O.; Ribeiro, C.T.; Gasparotto, J.; Bortolin, R.C.; de Vargas, A.R.; Heimfarth, L.; de Almeida, R.F.; Moreira, J.C.F.; de Oliveira, J.; et al. High fat diet-induced obesity causes a reduction in brain tyrosine hydroxylase levels and non-motor features in rats through metabolic dysfunction, neuroinflammation and oxidative stress. Nutr. Neurosci. 2020, 1831261. [Google Scholar] [CrossRef]
- Choi, J.Y.; Jang, E.H.; Park, C.S.; Kang, J.H. Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic. Biol. Med. 2005, 38, 806–816. [Google Scholar] [CrossRef]
- Bousquet, M.; St-Amour, I.; Vandal, M.; Julien, P.; Cicchetti, F.; Calon, F. High-fat diet exacerbates MPTP-induced dopaminergic degeneration in mice. Neurobiol. Dis. 2012, 45, 529–538. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R. High fat diet feeding induced insulin resistance exacerbates 6-OHDA mediated neurotoxicity and behavioral abnormalities in rats. Behav. Brain Res. 2018, 351, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Shuler, J.M.; Raider, K.D.; Rogers, R.S.; Wheatley, J.L.; Geiger, P.C.; Stanford, J.A. Effects of discontinuing a high-fat diet on mitochondrial proteins and 6-hydroxydopamine-induced dopamine depletion in rats. Brain Res. 2015, 1613, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotermund, C.; Truckenmüller, F.M.; Schell, H.; Kahle, P.J. Diet-induced obesity accelerates the onset of terminal phenotypes in α-synuclein transgenic mice. J. Neurochem. 2014, 131, 848–858. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Park. Relat. Disord. 2009, 15, S185–S188. [Google Scholar] [CrossRef]
- Morris, J.; Bomhoff, G.; Gorres, B.; Davis, V.; Kim, J.; Lee, P.-P.; Brooks, W.; Gerhardt, G.; Geiger, P.; Stanford, J. Insulin resistance impairs nigrostriatal dopamine function. Exp. Neurol. 2011, 231, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Lee, M.J.; Han, J.; Kim, S.J.; Ryu, I.; Ju, X.; Ryu, M.J.; Chung, W.; Oh, E.; Kweon, G.R.; et al. A High-fat Diet Induces a Loss of Midbrain Dopaminergic Neuronal Function That Underlies Motor Abnormalities. Exp. Neurobiol. 2017, 26, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-C.; Wei, W.-Y.; Tsai, K.-J.; Wang, L.-C. High Fat Diet Suppresses Peroxisome Proliferator-Activated Receptors and Reduces Dopaminergic Neurons in the Substantia Nigra. Int. J. Mol. Sci. 2019, 21, 207. [Google Scholar] [CrossRef] [Green Version]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Hong, C.T.; Chen, K.Y.; Wang, W.; Chiu, J.Y.; Wu, D.; Chao, T.Y.; Hu, C.J.; Chau, K.D.; Bamodu, O.A. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-F.; Kao, L.-T.; Shih, J.-H.; Kao, H.-H.; Chou, Y.-C.; Li, I.-H.; Kao, S. Pioglitazone use and Parkinson’s disease: A retrospective cohort study in Taiwan. BMJ Open 2018, 8, e023302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sah, S.K.; Lee, C.; Jang, J.-H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valladolid-Acebes, I.; Fole, A.; Martín, M.; Morales, L.; Victoria Cano, M.; Ruiz-Gayo, M.; Del Olmo, N. Spatial memory impairment and changes in hippocampal morphology are triggered by high-fat diets in adolescent mice. Is there a role of leptin? Neurobiol. Learn. Mem. 2013, 106, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ledreux, A.; Wang, X.; Schultzberg, M.; Granholm, A.-C.; Freeman, L.R. Detrimental effects of a high fat/high cholesterol diet on memory and hippocampal markers in aged rats. Behav. Brain Res. 2016, 312, 294–304. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Pallàs, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of β-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef]
- Tran, D.M.; Westbrook, R.F. A high-fat high-sugar diet-induced impairment in place-recognition memory is reversible and training-dependent. Appetite 2017, 110, 61–71. [Google Scholar] [CrossRef]
- Spencer, S.J.; D’Angelo, H.; Soch, A.; Watkins, L.R.; Maier, S.F.; Barrientos, R.M. High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol. Aging 2017, 58, 88–101. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Fu, Z.; Wu, J.; Nesil, T.; Li, M.D.; Aylor, K.W.; Liu, Z. Long-term high-fat diet induces hippocampal microvascular insulin resistance and cognitive dysfunction. Am. J. Physiol. Metab. 2017, 312, E89–E97. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, G.; Zizza, M.; Di Vito, A.; Alò, R.; Mele, M.; Bruno, R.; Barni, T.; Facciolo, R.M.; Canonaco, M. Reduced learning and memory performances in high-fat treated hamsters related to brain neurotensin receptor1 expression variations. Behav. Brain Res. 2018, 347, 227–233. [Google Scholar] [CrossRef]
- Abbott, K.N.; Arnott, C.K.; Westbrook, R.F.; Tran, D.M. The effect of high fat, high sugar, and combined high fat-high sugar diets on spatial learning and memory in rodents: A meta-analysis. Neurosci. Biobehav. Rev. 2019, 107, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Wolf, P.A.; Beiser, A.; Au, R.; McNulty, K.; White, R.; D’Agostino, R.B. Lifetime risk of dementia and Alzheimer’s disease. The impact of mortality on risk estimates in the Framingham Study. Neurology 1997, 49, 1498–1504. [Google Scholar] [CrossRef]
- Plassman, B.L.; Langa, K.M.; McCammon, R.J.; Fisher, G.G.; Potter, G.G.; Burke, J.R.; Steffens, D.C.; Foster, N.L.; Giordani, B.; Unverzagt, F.W.; et al. Incidence of dementia and cognitive impairment, not dementia in the United States. Ann. Neurol. 2011, 70, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, K.; Laws, K.R.; Gale, T.M.; Kondel, T.K. Greater cognitive deterioration in women than men with Alzheimer’s disease: A meta analysis. J. Clin. Exp. Neuropsychol. 2012, 34, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease—The gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.; Launer, L.J.; Dewey, M.E.; Letenneur, L.; Ott, A.; Copeland, J.R.M.; Dartigues, J.-F.; Kragh-Sorensen, P.; Baldereschi, M.; Brayne, C.; et al. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. Neurology 1999, 53, 1992. [Google Scholar] [CrossRef]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.F.; Ahmad, A.; Bhat, S.H.; Abu-Duhier, F.M.; Barreto, G.E.; Ashraf, G.M. Impact of sex differences and gender specificity on behavioral characteristics and pathophysiology of neurodegenerative disorders. Neurosci. Biobehav. Rev. 2019, 102, 95–105. [Google Scholar] [CrossRef]
- Gillies, G.E.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrinol. 2014, 35, 370–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solla, P.; Cannas, A.; Ibba, F.C.; Loi, F.; Corona, M.; Orofino, G.; Marrosu, M.G.; Marrosu, F. Gender differences in motor and non-motor symptoms among Sardinian patients with Parkinson’s disease. J. Neurol. Sci. 2012, 323, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.; Peterson, B.J.; Ahlskog, J.; Schaid, D.J.; A Rocca, W. Risk tables for parkinsonism and Parkinson’s disease. J. Clin. Epidemiol. 2002, 55, 25–31. [Google Scholar] [CrossRef]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Gaignard, P.; Liere, P.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 406. [Google Scholar] [CrossRef]
- Gaignard, P.; Fréchou, M.; Liere, P.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Sex differences in brain mitochondrial metabolism: Influence of endogenous steroids and stroke. J. Neuroendocr. 2018, 30, e12497. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Q.; Terry, R.; Brown, T.R.; Russo, J. Regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity associated with increased exposure to endocrine disruptors. Biochim. Biophys. Acta 2009, 1793, 1128–1143. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.M.; Jones, T.H. Testosterone: A metabolic hormone in health and disease. J. Endocrinol. 2013, 217, R25–R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of Estrogen and Other Sex Hormones in Brain Aging. Neuroprotection and DNA Repair. Front. Aging Neurosci. 2017, 9, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, A.N.; Siddiqui, N.; Khan, R.A.; Kalam, A.; Jabir, N.R.; Kamal, M.A.; Firoz, C.K.; Tabrez, S. Neuroprotective Role of Steroidal Sex Hormones: An Overview. CNS Neurosci. Ther. 2016, 22, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, R.D.; Voskuhl, R.R. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front. Neuroendocrinol. 2012, 33, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghava, N.; Das, B.C.; Ray, S.K. Neuroprotective effects of estrogen in CNS injuries: Insights from animal models. Neurosci. Neuroecon. 2017, 6, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, M.; Weill-Engerer, S.; Liere, P.; Robert, F.; Franklin, R.; Garcia-Segura, L.; Lambert, J.; Mayo, W.; Melcangi, C.R.; Parducz, A.; et al. Steroid hormones and neurosteroids in normal and pathological aging of the nervous system. Prog. Neurobiol. 2003, 71, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Wahlqvist, M.L.; Lee, M.-S.; Hsu, C.-C.; Chuang, S.-Y.; Lee, J.-T.; Tsai, H.-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Park. Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef]
- Miranda, H.V.; El-Agnaf, O.M.A.; Outeiro, T.F. Glycation in Parkinson’s disease and Alzheimer’s disease. Mov. Disord. 2016, 31, 782–790. [Google Scholar] [CrossRef]
- Patil, S.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Koehler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiho, T.; Kato, M.; Usui, S.; Hirano, K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin. Chim. Acta 2005, 358, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin inhibition of glycation processes. Diabetes Metab. 2003, 29, 6S95–6S103. [Google Scholar] [CrossRef]
- Kender, Z.; Fleming, T.; Kopf, S.; Torzsa, P.; Grolmusz, V.; Herzig, S.; Schleicher, E.; Rácz, K.; Reismann, P.; Nawroth, P. Effect of Metformin on Methylglyoxal Metabolism in Patients with Type 2 Diabetes. Exp. Clin. Endocrinol. Diabetes 2014, 122, 316–319. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal to Form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain. Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Cheng, Y.-C.; Chen, S.-J.; Yen, C.-H.; Huang, R.-N. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef]
- Perez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Dulovic, M.; Jovanovic, M.; Xilouri, M.; Stefanis, L.; Harhaji-Trajkovic, L.; Stevovic, T.K.; Paunovic, V.; Ardah, M.T.; El-Agnaf, O.M.; Kostic, V.; et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuro-Psychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.K.; Park, H.Y.; Go, J.; Choi, D.H.; Kim, Y.H.; Hwang, J.H.; Noh, J.R.; Lee, T.G.; Lee, C.H.; Kim, K.S. Metformin Inhibits the Development of L-DOPA-Induced Dyskinesia in a Murine Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 5715–5726. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-Term Metformin Usage and Cognitive Function among Older Adults with Diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: A population-based case-control study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef] [Green Version]
- Kuan, Y.C.; Huang, K.W.; Lin, C.L.; Hu, C.J.; Kao, C.H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 77–83. [Google Scholar] [CrossRef]
- Ping, F.; Jiang, N.; Li, Y. Association between metformin and neurodegenerative diseases of observational studies: Systematic review and meta-analysis. BMJ Open Diabetes Res. Care 2020, 8, e001370. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association Between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 678649. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.; Sun, W.; Li, J. Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS ONE 2015, 10, e0123864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaf, N.; El-Shamarka, M.E.; Salem, N.A.; Khadrawy, Y.A.; El Sayed, N.S. Neuroprotective effect of PPAR α and γ agonists in a mouse model of amyloidogenesis through modulation of the Wnt/β catenin pathway via targeting α- and β-secretases. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 97, 109793. [Google Scholar] [CrossRef] [PubMed]
- Searcy, J.L.; Phelps, J.T.; Pancani, T.; Kadish, I.; Popovic, J.; Anderson, K.L.; Beckett, T.L.; Murphy, M.P.; Chen, K.-C.; Blalock, E.M.; et al. Long-Term Pioglitazone Treatment Improves Learning and Attenuates Pathological Markers in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 30, 943–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, L.; Simón, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; López de Maturana, R.; García-Osta, A.; Ricobaraza, A.; Pérez-Mediavilla, A.; Del Río, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-H.; Chang, P.-C.; Wey, S.-P.; Chen, P.-M.; Chen, C.; Chan, M.-H. Therapeutic effects of honokiol on motor impairment in hemiparkinsonian mice are associated with reversing neurodegeneration and targeting PPARγ regulation. Biomed. Pharmacother. 2018, 108, 254–262. [Google Scholar] [CrossRef]
- Machado, M.M.F.; Bassani, T.B.; Cóppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Andreatini, R.; Vital, M. PPAR-γ agonist pioglitazone reduces microglial proliferation and NF-κB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol. Rep. 2019, 71, 556–564. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Rahimmi, A.; Moloudi, M.R.; Maccarone, R.; Corbo, M.; Izadpanah, E.; Feligioni, M. Effect of lobeglitazone on motor function in rat model of Parkinson’s disease with diabetes co-morbidity. Brain Res. Bull. 2021, 173, 184–192. [Google Scholar] [CrossRef]
- Das, N.R.; Vaidya, B.; Khare, P.; Bishnoi, M.; Sharma, S.S. Combination of Peroxisome Proliferator-activated Receptor γ (PPARγ) Agonist and PPAR Gamma Co-Activator 1α (PGC-1α) Activator Ameliorates Cognitive Deficits, Oxidative Stress, and Inflammation in Rodent Model of Parkinson’s Disease. Curr. Neurovasc. Res. 2021, 18, 497–507. [Google Scholar] [CrossRef]
- Schintu, N.; Frau, L.; Ibba, M.; Caboni, P.; Garau, A.; Carboni, E.; Carta, A.R. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 954–963. [Google Scholar] [CrossRef]
- Breidert, T.; Callebert, J.; Heneka, M.T.; Landreth, G.; Launay, J.M.; Hirsch, E.C. Protective action of the peroxisome proliferator-activated receptor-γ agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Tyson, T.; George, S.; Hildebrandt, E.N.; Steiner, J.A.; Madaj, Z.; Schulz, E.; Machiela, E.; McDonald, W.G.; Escobar Galvis, M.L.; et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci. Transl. Med. 2016, 8, 368ra174. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Rosenblat, J.D.; Brietzke, E.; Park, C.; Lee, Y.; Musial, N.; Pan, Z.; Mansur, R.B.; McIntyre, R.S. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2018, 20, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; West, E.; Nolan, W.; McHale-Owen, H.; Williams, A.; Bate, C. Glimepiride protects neurons against amyloid-β-induced synapse damage. Neuropharmacology 2016, 101, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.C.; Wahlqvist, M.L.; Lee, M.S.; Tsai, H.N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J. Alzheimer’s Dis. 2011, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Tat, V.; Forest, C.P. The role of SGLT2 inhibitors in managing type 2 diabetes. J. Am. Acad. Physician Assist. 2018, 31, 35–40. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef] [Green Version]
- Naznin, F.; Sakoda, H.; Okada, T.; Tsubouchi, H.; Waise, T.Z.; Arakawa, K.; Nakazato, M. Canagliflozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced inflammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur. J. Pharmacol. 2017, 794, 37–44. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Chen, S.; Peng, P.; Gu, Z.; Yu, J.; Zhao, G.; Deng, Y. Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3β. Biochem. Biophys. Res. Commun. 2019, 511, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; E Silva, N.M.L.; Brito-Moreira, J.; Boehnke, S.; Winterborn, A.; Coe, B.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The Diabetes Drug Liraglutide Prevents Degenerative Processes in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Labandeira, C.M.; Fraga-Bau, A.; Arias Ron, D.; Muñoz, A.; Alonso-Losada, G.; Koukoulis, A.; Romero-Lopez, J.; Rodriguez-Perez, A.I. Diabetes, insulin and new therapeutic strategies for Parkinson’s disease: Focus on glucagon-like peptide-1 receptor agonists. Front. Neuroendocrinol. 2021, 62, 100914. [Google Scholar] [CrossRef]
- Labandeira, C.; Fraga-Bau, A.; Ron, D.A.; Alvarez-Rodriguez, E.; Vicente-Alba, P.; Lago-Garma, J.; Rodriguez-Perez, A. Parkinson’s disease and diabetes mellitus: Common mechanisms and treatment repurposing. Neural Regen. Res. 2022, 17, 1652. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [Green Version]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Kahan, J.; Ell, P.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Motor and Cognitive Advantages Persist 12 Months after Exenatide Exposure in Parkinson’s Disease. J. Park. Dis. 2014, 4, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Wu, S.-L.; Chen, T.-C.; Chuang, C.-S. Antidiabetic Agents for Treatment of Parkinson’s Disease: A Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 4805. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Dong, Y.; Chen, J.; Guan, T.; Cao, B.; Zhang, Y.; Qi, Y.; Guan, Z.; Wang, Y. Liraglutide Regulates Mitochondrial Quality Control System through PGC-1α in a Mouse Model of Parkinson’s Disease. Neurotox. Res. 2022, 40, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Zhang, Y.; Chen, J.; Wu, P.; Dong, Y.; Wang, Y. Neuroprotective effects of liraglutide against inflammation through the AMPK/NF-κB pathway in a mouse model of Parkinson’s disease. Metab. Brain Dis. 2022, 37, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Hölscher, C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides 2018, 71, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Hölscher, C. Semaglutide is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinson’s Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Muthureddy Nataraj, S.K.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1773–1784. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, M.; Sun, J.; Guo, A.; Fernando, R.L.; Chen, Y.; Peng, P.; Zhao, G.; Deng, Y. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 2019, 157, 107668. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Li, C.; Li, M.; Ma, L. Sitagliptin rescues memory deficits in Parkinsonian rats via upregulating BDNF to prevent neuron and dendritic spine loss. Neurol. Res. 2018, 40, 736–743. [Google Scholar] [CrossRef]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef]
- Zhou, B.; Zissimopoulos, J.; Nadeem, H.; Crane, M.A.; Goldman, D.; Romley, J.A. Association between exenatide use and incidence of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12139. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.J.; Sacramento, J.F.; Gonzalez, C.; Guarino, M.P.; Monteiro, E.C.; Conde, S.V. Carotid Body Denervation Prevents the Development of Insulin Resistance and Hypertension Induced by Hypercaloric Diets. Diabetes 2013, 62, 2905–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.J.; Sacramento, J.F.; Gallego-Martin, T.; Olea, E.; Melo, B.F.; Guarino, M.P.; Yubero, S.; Obeso, A.; Conde, S.V. High fat diet blunts the effects of leptin on ventilation and on carotid body activity. J. Physiol. 2018, 596, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Sacramento, J.F.; Ribeiro, M.J.; Rodrigues, T.; Olea, E.; Melo, B.F.; Guarino, M.P.; Fonseca-Pinto, R.; Ferreira, C.R.; Coelho, J.; Obeso, A.; et al. Functional abolition of carotid body activity restores insulin action and glucose homeostasis in rats: Key roles for visceral adipose tissue and the liver. Diabetologia 2016, 60, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, C.; López-López, J.; Obeso, A.; Pérez-García, M.T.; Rocher, A. Cellular mechanisms of oxygen chemoreception in the carotid body. Respir. Physiol. 1995, 102, 137–147. [Google Scholar] [CrossRef]
- Nurse, C.A. Synaptic and paracrine mechanisms at carotid body arterial chemoreceptors. J. Physiol. 2014, 592, 3419–3426. [Google Scholar] [CrossRef]
- Zera, T.; Moraes, D.J.A.; Da Silva, M.P.; Fisher, J.P.; Paton, J.F.R. The Logic of Carotid Body Connectivity to the Brain. Physiology 2019, 34, 264–282. [Google Scholar] [CrossRef]
- Sacramento, J.F.; Chew, D.J.; Melo, B.; Donegá, M.; Dopson, W.; Guarino, M.P.; Robinson, A.; Prieto-Lloret, J.; Patel, S.; Holinski, B.J.; et al. Bioelectronic modulation of carotid sinus nerve activity in the rat: A potential therapeutic approach for type 2 diabetes. Diabetologia 2018, 61, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Guimaraes, J.P.; Guarino, M.P.; Timóteo, A.T.; Caires, I.; Sacramento, J.F.; Ribeiro, M.J.; Selas, M.; Santiago, J.C.P.; Carmo, M.; Conde, S.V. Carotid body chemosensitivity: Early biomarker of dysmetabolism in humans. Eur. J. Endocrinol. 2020, 182, 549–557. [Google Scholar] [CrossRef]
- Pauza, A.G.; Thakkar, P.; Tasic, T.; Felippe, I.; Bishop, P.; Greenwood, M.P.; Rysevaite-Kyguoliene, K.; Ast, J.; Broichhagen, J.; Hodson, D.J.; et al. GLP1R Attenuates Sympathetic Response to High Glucose via Carotid Body Inhibition. Circ. Res. 2022, 130, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Cracchiolo, M.; Sacramento, J.F.; Mazzoni, A.; Panarese, A.; Carpaneto, J.; Conde, S.V.; Micera, S. Decoding Neural Metabolic Markers from the Carotid Sinus Nerve in a Type 2 Diabetes Model. IEEE Trans. Neural Syst. Rehabil. Eng. 2019, 27, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
| Study | Diet Regiment | Rodent Model | Outcomes |
|---|---|---|---|
| Velazquez et al. [194] | NC diet | Tg2576 and 3xTg-AD transgenic AD-like mice | Central insulin dysregulation and energy dyshomeostasis, developed before peripheral insulin resistance. |
| Sah et al. [195] | 16 weeks of HF diet | 3xTg-AD transgenic AD-like mice | Enhanced memory impairment, without alteration in the levels of Aβ and phosphorylation of tau in the cortical region. Increased neuronal oxidative stress and apoptosis. |
| Thériault et al. [196] | 4 months of a HF diet | APPswe/PS1 transgenic AD-like mice | Accelerated age-associated cognitive decline without affecting parenchymal Aβ. Loss of synaptic plasticity and exacerbated systemic inflammation and oxidative stress. |
| Valladolid-Acebes et al. [197] | 8 weeks of HF diet | WT mice | Spatial memory impairment and changes in hippocampal morphology, accompanied by an increase of dendritic spine density in CA1 pyramidal neurons that correlated with the upregulation of neural cell adhesion molecule (NCAM) in this area and a desensitization of the Akt pathway coupled to hippocampal leptin receptors. |
| Ledreux et al. [198] | 6 months of HF or high cholesterol diet | WT rats | Memory impairment, neurodegeneration in the hippocampus, increased activation of microglia and abnormal phosphorylation of Tau. |
| Busquets et al. [199] | 15 months of a HF diet | WT mice | Long-term exposure to HF diet favors the appearance of Aβ depositions in the brain, thought increased inflammation leading to a decrease in the neuronal precursor cells, and dysregulation in normal autophagy and apoptosis. |
| Tran and Westbrook [200] | HFHSu and NC diets | WT rats | Impairment in place-recognition memory, that is reversible and training-dependent. |
| Spencer et al. [201] | 3 days of HF diet | WT rats | Impaired long-term contextual (hippocampal-dependent) and auditory-cued fear (amygdalar-dependent) memory in aged, but not young adult rats. Increased activation of microglia. |
| Kothari et al. [202] | 14 weeks of HFHSu diet | WT mice | Induced brain insulin resistance, accompanied by inflammatory and stress responses as well as by increased Aβ deposition and neurofibrillary tangle formation, and decreased synaptic plasticity and cognitive impairment. |
| Fu et al. [203] | 6 months of HF diet | WT rats | Induced hippocampal microvascular insulin resistance and cognitive dysfunction. |
| Fazzari et al. [204] | 12 weeks of HF diet | WT hamsters | Reduced locomotor activities such as exploratory bouts, rearing and grooming behaviors, cognitive and memory impairment. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capucho, A.M.; Chegão, A.; Martins, F.O.; Vicente Miranda, H.; Conde, S.V. Dysmetabolism and Neurodegeneration: Trick or Treat? Nutrients 2022, 14, 1425. https://doi.org/10.3390/nu14071425
Capucho AM, Chegão A, Martins FO, Vicente Miranda H, Conde SV. Dysmetabolism and Neurodegeneration: Trick or Treat? Nutrients. 2022; 14(7):1425. https://doi.org/10.3390/nu14071425
Chicago/Turabian StyleCapucho, Adriana M., Ana Chegão, Fátima O. Martins, Hugo Vicente Miranda, and Sílvia V. Conde. 2022. "Dysmetabolism and Neurodegeneration: Trick or Treat?" Nutrients 14, no. 7: 1425. https://doi.org/10.3390/nu14071425
APA StyleCapucho, A. M., Chegão, A., Martins, F. O., Vicente Miranda, H., & Conde, S. V. (2022). Dysmetabolism and Neurodegeneration: Trick or Treat? Nutrients, 14(7), 1425. https://doi.org/10.3390/nu14071425