
Estimating the Direct Effect between Dietary Macronutrients and Cardiometabolic Disease, Accounting for Mediation by Adiposity and Physical Activity

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Exposure, Mediator and Outcome Measures
2.3. Statistical Analysis
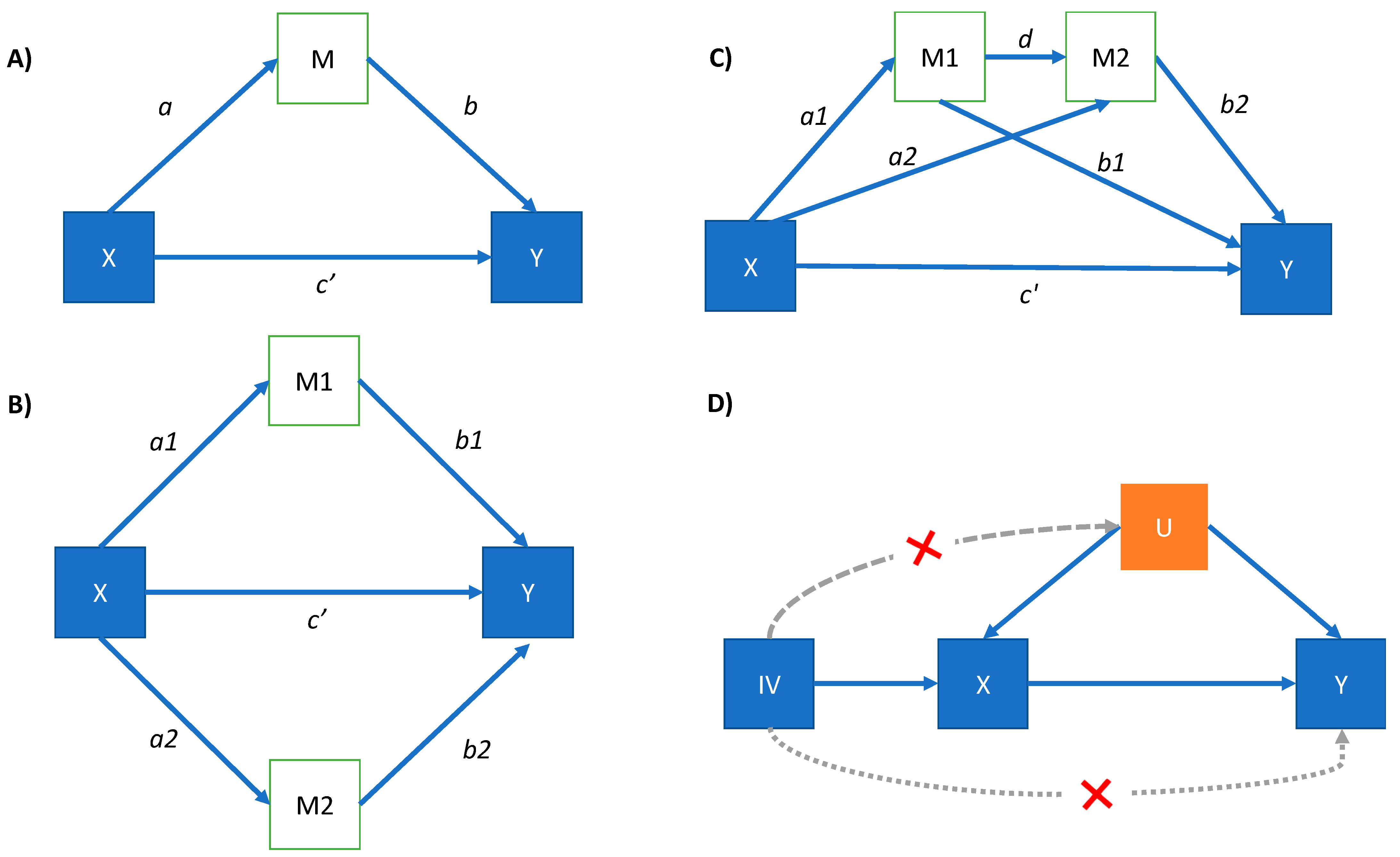
2.3.1. Mediation Analysis
2.3.2. Two-Sample Mendelian Randomization and Bayesian Colocalization
3. Results
3.1. Mediation Analysis
3.2. MR Causal Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Mozaffarian, D. Diverging global trends in heart disease and type 2 diabetes: The role of carbohydrates and saturated fats. Lancet Diabetes Endocrinol. 2015, 3, 586–588. [Google Scholar] [CrossRef]
- Afshin, A.; Sur, P.J.; Fay, K.A.; Cornaby, L.; Ferrara, G.; Salama, J.S.; Mullany, E.C.; Abate, K.H.; Abbafati, C.; Abebe, Z.; et al. Health effects of dietary risks in 195 countries, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [Google Scholar] [CrossRef]
- Cornelis, M.C.; Flint, A.; Field, A.E.; Kraft, P.; Han, J.; Rimm, E.B.; van Dam, R.M. A genome-wide investigation of food addiction. Obesity 2016, 24, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- McRae, J.F.; Jaeger, S.R.; Bava, C.M.; Beresford, M.K.; Hunter, D.; Jia, Y.; Chheang, S.L.; Jin, D.; Peng, M.; Gamble, J.C.; et al. Identification of regions associated with variation in sensitivity to food-related odors in the human genome. Curr. Biol. 2013, 23, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Meddens, S.F.W.; de Vlaming, R.; Bowers, P.; Burik, C.A.P.; Linner, R.K.; Lee, C.; Okbay, A.; Turley, P.; Rietveld, C.A.; Fontana, M.A.; et al. Genomic analysis of diet composition finds novel loci and associations with health and lifestyle. Mol. Psychiatry 2021, 26, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Hwang, L.D.; Lin, C.; Gharahkhani, P.; Cuellar-Partida, G.; Ong, J.S.; An, J.; Gordon, S.D.; Zhu, G.; MacGregor, S.; Lawlor, D.A.; et al. New insight into human sweet taste: A genome-wide association study of the perception and intake of sweet substances. Am. J. Clin. Nutr. 2019, 109, 1724–1737. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Esberg, A.; Haworth, S.; Holgerson, P.L.; Johansson, I. Allelic Variation in Taste Genes Is Associated with Taste and Diet Preferences and Dental Caries. Nutrients 2019, 11, 1491. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Group, D.P.P.R. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- Pan, X.-R.; Li, G.-w.; Hu, Y.-H.; Wang, J.-X.; Yang, W.-Y.; An, Z.-X.; Hu, Z.-X.; Xiao, J.-Z.; Cao, H.-B.; Liu, P.-A.J.D.c. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance: The Da Qing IGT and Diabetes Study. Diabetes Care 1997, 20, 537–544. [Google Scholar] [CrossRef]
- Wang, D.D.; Hu, F.B. Precision nutrition for prevention and management of type 2 diabetes. Lancet Diabetes Endocrinol. 2018, 6, 416–426. [Google Scholar] [CrossRef]
- Norberg, M.; Wall, S.; Boman, K.; Weinehall, L. The Vasterbotten Intervention Programme: Background, design and implications. Glob. Health Action 2010, 3, 6343. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Hallmans, G.; Wikman, A.; Biessy, C.; Riboli, E.; Kaaks, R. Validation and calibration of food-frequency questionnaire measurements in the Northern Sweden Health and Disease cohort. Public Health Nutr. 2002, 5, 487–496. [Google Scholar] [CrossRef]
- Ramne, S.; Alves Dias, J.; Gonzalez-Padilla, E.; Olsson, K.; Lindahl, B.; Engstrom, G.; Ericson, U.; Johansson, I.; Sonestedt, E. Association between added sugar intake and mortality is nonlinear and dependent on sugar source in 2 Swedish population-based prospective cohorts. Am. J. Clin. Nutr. 2019, 109, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I. Validity of a short questionnaire to assess physical activity in 10 European countries. Eur. J. Epidemiol. 2012, 27, 15–25. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Ng, N.; Carlberg, B.; Weinehall, L.; Norberg, M. Trends of blood pressure levels and management in Vasterbotten County, Sweden, during 1990-2010. Glob. Health Action 2012, 5, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Province, M.A.; Coon, H.; Hunt, S.C.; Eckfeldt, J.H.; Arnett, D.K.; Heiss, G.; Lewis, C.E.; Ellison, R.C.; Rao, D.C.; et al. An investigation of the effects of lipid-lowering medications: Genome-wide linkage analysis of lipids in the HyperGEN study. BMC Genet. 2007, 8, 60. [Google Scholar] [CrossRef]
- Imai, K.; Keele, L.; Tingley, D. A general approach to causal mediation analysis. Psychol. Methods 2010, 15, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.F. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression-Based Approach; Guilford Publications: New York, NY, USA, 2017. [Google Scholar]
- Tingley, D.; Yamamoto, T.; Hirose, K.; Keele, L.; Imai, K. Mediation: R Package for Causal Mediation Analysis. J. Stat. Softw. 2014, 59. [Google Scholar] [CrossRef]
- Leth-Steensen, C.; Gallitto, E. Testing Mediation in Structural Equation Modeling: The Effectiveness of the Test of Joint Significance. Educ. Psychol. Meas. 2016, 76, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Willett, W.C.; Howe, G.R.; Kushi, L.H. Adjustment for total energy intake in epidemiologic studies. Am. J. Clin. Nutr. 1997, 65, 1220S–1228S; discussion 1229S–1231S. [Google Scholar] [CrossRef] [PubMed]
- Forastiere, L.; Mattei, A.; Ding, P. Principal ignorability in mediation analysis: Through and beyond sequential ignorability. Biometrika 2018, 105, 979–986. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Davey Smith, G.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef]
- Richardson, T.G.; Sanderson, E.; Palmer, T.M.; Ala-Korpela, M.; Ference, B.A.; Davey Smith, G.; Holmes, M.V. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003062. [Google Scholar] [CrossRef]
- Borges, M.C.; Schmidt, A.F.; Jefferis, B.; Wannamethee, S.G.; Lawlor, D.A.; Kivimaki, M.; Kumari, M.; Gaunt, T.R.; Ben-Shlomo, Y.; Tillin, T.; et al. Circulating Fatty Acids and Risk of Coronary Heart Disease and Stroke: Individual Participant Data Meta-Analysis in Up to 16 126 Participants. J. Am. Heart Assoc. 2020, 9, e013131. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Davies, N.M.; Thompson, S.G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 2016, 40, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.N.; Staley, J.R.; Breen, P.G.; Sun, B.B.; Kirk, P.D.W.; Burgess, S.; Howson, J.M.M. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 2021, 12, 764. [Google Scholar] [CrossRef] [PubMed]
- Rosseel, Y. Lavaan: An R package for structural equation modeling and more. Version 0.5–12 (BETA). J. Stat. Softw. 2012, 48, 1–36. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Yavorska, O.O.; Burgess, S. MendelianRandomization: An R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 2017, 46, 1734–1739. [Google Scholar] [CrossRef]
- Wallace, C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet 2020, 16, e1008720. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.-R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, G.; Shi, D.X.; Maydeu-Olivares, A. Chi-square Difference Tests for Comparing Nested Models: An Evaluation with Non-normal Data. Struct. Equ. Model.-A Multidiscip. J. 2020, 27, 908–917. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed]
- McRae, M.P. Dietary Fiber Intake and Type 2 Diabetes Mellitus: An Umbrella Review of Meta-analyses. J. Chiropr. Med. 2018, 17, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445. [Google Scholar] [CrossRef]
- Bernstein, A.M.; Titgemeier, B.; Kirkpatrick, K.; Golubic, M.; Roizen, M.F. Major cereal grain fibers and psyllium in relation to cardiovascular health. Nutrients 2013, 5, 1471–1487. [Google Scholar] [CrossRef] [PubMed]
- Runchey, S.S.; Valsta, L.M.; Schwarz, Y.; Wang, C.; Song, X.; Lampe, J.W.; Neuhouser, M.L. Effect of low- and high-glycemic load on circulating incretins in a randomized clinical trial. Metabolism 2013, 62, 188–195. [Google Scholar] [CrossRef]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef]
- Hindy, G.; Sonestedt, E.; Ericson, U.; Jing, X.J.; Zhou, Y.; Hansson, O.; Renstrom, E.; Wirfalt, E.; Orho-Melander, M. Role of TCF7L2 risk variant and dietary fibre intake on incident type 2 diabetes. Diabetologia 2012, 55, 2646–2654. [Google Scholar] [CrossRef]
- Grau, K.; Cauchi, S.; Holst, C.; Astrup, A.; Martinez, J.A.; Saris, W.H.; Blaak, E.E.; Oppert, J.M.; Arner, P.; Rossner, S.; et al. TCF7L2 rs7903146-macronutrient interaction in obese individuals’ responses to a 10-wk randomized hypoenergetic diet. Am. J. Clin. Nutr. 2010, 91, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.; Boeing, H.; Fritsche, A.; Doering, F.; Joost, H.G.; Schulze, M.B. Whole-grain consumption and transcription factor-7-like 2 ( TCF7L2) rs7903146: Gene-diet interaction in modulating type 2 diabetes risk. Br. J. Nutr. 2009, 101, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjogren, M.; Ling, C.; Eriksson, K.F.; Lethagen, A.L.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Leiherer, A.; Muendlein, A.; Saely, C.H.; Fraunberger, P.; Drexel, H. Serotonin is elevated in risk-genotype carriers of TCF7L2 - rs7903146. Sci. Rep. 2019, 9, 12863. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.; Feng, Q.Q.; Ojo, O.O.; Wang, X.H. The Role of Dietary Fibre in Modulating Gut Microbiota Dysbiosis in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Nutrients 2020, 12, 3239. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.M.; Meigs, J.B.; Liu, S.; Rogers, G.; Yoshida, M.; Saltzman, E.; Jacques, P.F. Dietary carbohydrates and cardiovascular disease risk factors in the Framingham offspring cohort. J. Am. Coll. Nutr. 2009, 28, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.X.; Shen, P. Associations of dietary protein intake with all-cause, cardiovascular disease, and cancer mortality: A systematic review and meta-analysis of cohort studies. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1094–1105. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Satija, A.; Blondin, S.A.; Janiszewski, M.; Emlen, E.; O’Connor, L.E.; Campbell, W.W.; Hu, F.B.; Willett, W.C.; Stampfer, M.J. Meta-analysis of randomized controlled trials of red meat consumption in comparison with various comparison diets on cardiovascular risk factors. Circulation 2019, 139, 1828–1845. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Dashti, H.S.; Li, S.X.; Sarnowski, C.; Justice, A.E.; Graff, M.; Papoutsakis, C.; Smith, C.E.; Dedoussis, G.V.; Lemaitre, R.N.; et al. Genome-wide meta-analysis of macronutrient intake of 91,114 European ancestry participants from the cohorts for heart and aging research in genomic epidemiology consortium. Mol. Psychiatry 2018, 24, 1920–1932. [Google Scholar] [CrossRef]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C.; Jablonski, K.A.; Bayley, N.; Pollin, T.I.; De Bakker, P.I.; Shuldiner, A.; Knowler, W.C.; Nathan, D.M.; Altshuler, D. TCF7L2 Polymorphisms and Progression to Diabetes in the Diabetes Prevention Program. New Engl. J. Med. 2006, 355, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Garver, W.S.; Newman, S.B.; Gonzales-Pacheco, D.M.; Castillo, J.J.; Jelinek, D.; Heidenreich, R.A.; Orlando, R.A. The genetics of childhood obesity and interaction with dietary macronutrients. Genes Nutr. 2013, 8, 271–287. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.; Wallace, C.; Plagnol, V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IVW | MR-Egger | MR-PRESSO | MR-RAPS | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exposure | Outcome | Number of SNPs | F | β | 95% CI | p-Value | Q Statistic | p-Value | β | 95% CI | p-Value | Q Statistic | p-Value | Global Test p-Value | Distortion Test p-Value | β | βSE | p-Value | ||
| Sugar | FG | 26 | 5 | −0.09 | −0.18 | 0.01 | 0.07 | 24.48 | 0.18 | −0.11 | −0.5 | 0.29 | 0.6 | 24.39 | 0.14 | 0.17 | - | −0.08 | 0.05 | 0.15 |
| 2 h glucose | 24 | 5 | −0.07 | −0.54 | 0.41 | 0.79 | 16.02 | 0.85 | −0.93 | −2.9 | 1.05 | 0.36 | 15.24 | 0.85 | - | - | −0.04 | 0.25 | 0.86 | |
| HDL-C | 40 | 4 | −0.05 | −0.25 | 0.16 | 0.66 | 1265.66 | 3.62 × 10−240 | −0.21 | −0.89 | 0.48 | 0.55 | 1253.76 | 2.02 × 10−238 | <1 × 10−4 | 0.6 | −0.04 | 0.06 | 0.49 | |
| LDL-C | 40 | 4 | 0.43 | 0.05 | 0.82 | 0.03 | 3787.17 | - | 1.06 | −0.21 | 2.33 | 0.1 | 3695.86 | - | <1 × 10−4 | <1 × 10−4 | 0.1 | 0.08 | 0.16 | |
| TC | 40 | 4 | 0.32 | 0.004 | 0.64 | 0.05 | 673.38 | 1.29 × 10−116 | 0.88 | −0.17 | 1.91 | 0.1 | 657.34 | 6.04 × 10−114 | <1 × 10−4 | <1 × 10−4 | 0.12 | 0.09 | 0.17 | |
| TG | 40 | 4 | 0.17 | 0.02 | 0.32 | 0.03 | 619.14 | 1.65 × 10−105 | 0.32 | −0.17 | 0.82 | 0.2 | 611 | 1.87 × 10−104 | <1 × 10−4 | 9.00 × 10−4 | 0.05 | 0.05 | 0.28 | |
| T2D | 2 | 7 | 0.04 | 0.002 | 0.84 | 0.04 | 22.32 | 2.30 × 10−6 | - | - | - | - | - | - | - | - | −2.42 | 1.3 | 0.06 | |
| Stroke | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| * T2D | 2 | 6 | 3.9 | 0.02 | 969.07 | 0.02 | 0.24 | 0.63 | - | - | - | - | - | - | - | - | 1.36 | 2.91 | 0.64 | |
| CHD | 40 | 4 | 1.15 | 0.85 | 1.56 | 0.47 | 89.69 | 7.20 × 10−6 | 2.02 | 0.64 | 6.32 | 0.32 | 87.4 | 9.20 × 10−6 | <1 × 10−4 | <1 × 10−4 | 0.09 | 0.14 | 0.51 | |
| Fat | FG | 22 | 5 | 0.02 | −0.07 | 0.11 | 0.68 | 28.16 | 0.14 | −0.23 | −0.49 | 0.04 | 0.09 | 24.9 | 0.21 | 0.15 | - | 0.01 | 0.06 | 0.92 |
| 2h glucose | 22 | 5 | 0.09 | −0.43 | 0.62 | 0.73 | 18.73 | 0.6 | −0.31 | −2.27 | 1.65 | 0.76 | 18.37 | 0.56 | 0.62 | - | 0.17 | 0.29 | 0.56 | |
| HDL-C | 34 | 5 | −0.16 | −0.34 | 0.02 | 0.09 | 696.01 | 3.65 × 10−125 | −0.07 | −0.53 | 0.38 | 0.75 | 691.01 | 8.50 × 10−125 | <1 × 10−4 | <1 × 10−4 | −0.01 | 0.05 | 0.76 | |
| LDL-C | 34 | 5 | −0.38 | −0.79 | 0.04 | 0.08 | 3012.02 | - | −0.82 | −1.84 | 0.19 | 0.11 | 2940.35 | - | <1 × 10−4 | <1 × 10−4 | −0.19 | 0.09 | 0.05 | |
| TC | 34 | 5 | −0.28 | −0.62 | 0.06 | 0.11 | 550.56 | 3.74 × 10−95 | −0.62 | −1.44 | 0.21 | 0.15 | 538.59 | 2.56 × 10−93 | <1 × 10−4 | <1 × 10−4 | −0.16 | 0.1 | 0.11 | |
| TG | 34 | 5 | 0.11 | −0.22 | 0.43 | 0.51 | 2018.29 | - | −0.13 | −0.92 | 0.67 | 0.75 | 1995.28 | - | <1 × 10−4 | 3.00 × 10−4 | 0.04 | 0.06 | 0.57 | |
| T2D | 5 | 13 | 2.91 | 0.47 | 17.81 | 0.25 | 90 | - | 0.05 | 1.17 × 10−4 | 22.07 | 0.34 | 55.78 | - | 2.00 × 10−4 | <1 × 10−4 | −0.06 | 0.67 | 0.93 | |
| ** Stroke | 1 | - | 0.92 | 0.52 | 1.63 | 0.78 | - | - | - | - | - | - | - | - | - | - | −0.08 | 0.3 | 0.79 | |
| * T2D | 5 | 13 | 0.94 | 0.59 | 1.51 | 0.81 | 5.11 | 0.28 | 0.77 | 0.1 | 5.83 | 0.82 | 5.04 | 0.17 | 0.55 | - | −0.06 | 0.22 | 0.77 | |
| CHD | 31 | 5 | 0.81 | 0.58 | 1.12 | 0.29 | 81.71 | 1.10 × 10−6 | 0.69 | 0.36 | 1.32 | 0.21 | 80.83 | 9.00 × 10−7 | <1 × 10−4 | 0.82 | −0.21 | 0.14 | 0.12 | |
| Carbohydrates | FG | 28 | 5 | −0.07 | −0.17 | 0.03 | 0.16 | 44.25 | 0.02 | −0.12 | −0.59 | 0.35 | 0.61 | 44.16 | 0.01 | 0.02 | - | −0.13 | 0.06 | 0.02 |
| 2h glucose | 31 | 5 | −0.08 | −0.6 | 0.44 | 0.76 | 36.6 | 0.16 | −0.16 | −2.83 | 2.5 | 0.91 | 36.58 | 0.13 | 0.16 | - | −0.09 | 0.27 | 0.74 | |
| HDL-C | 44 | 4 | −0.12 | −0.32 | 0.09 | 0.27 | 1272.13 | 1.59 × 10−238 | −0.35 | −0.98 | 0.28 | 0.28 | 1254.88 | 1.22 × 10−235 | <1 × 10−4 | 0.7563 | −0.13 | 0.05 | 0.02 | |
| LDL-C | 44 | 4 | 0.44 | 0.05 | 0.82 | 0.03 | 3784.41 | - | 1 | −0.17 | 2.18 | 0.1 | 3698.32 | - | <1 × 10−4 | <1 × 10−4 | 0.08 | 0.07 | 0.25 | |
| TC | 45 | 4 | 0.33 | 0.03 | 0.63 | 0.03 | 652 | 3.30 × 10−109 | 0.76 | −0.19 | 1.7 | 0.12 | 638.86 | 3.95 × 10−107 | <1 × 10−4 | <1 × 10−4 | 0.11 | 0.08 | 0.16 | |
| TG | 44 | 4 | 0.19 | 0.03 | 0.34 | 0.02 | 663.34 | 4.11 × 10−112 | 0.37 | −0.1 | 0.84 | 0.13 | 653.47 | 1.06 × 10−110 | <1 × 10−4 | 0.1338 | 0.15 | 0.02 | 0 | |
| T2D | 6 | 5 | 0.1 | 0.01 | 0.71 | 0.02 | 80.3 | - | 0.19 | 6.56 × 10−5 | 560.9 | 0.69 | 79.55 | - | 2.00 × 10−4 | <1 × 10−4 | −1.68 | 0.63 | 0.01 | |
| Stroke | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| * T2D | 5 | 5 | 0.47 | 0.3 | 0.75 | 0.001 | 3.51 | 0.48 | 0.18 | 0.04 | 0.72 | 0.02 | 1.42 | 0.7 | 0.4343 | - | −0.82 | 0.29 | 0.004 | |
| CHD | 44 | 4 | 1.23 | 0.92 | 1.64 | 0.2 | 95.94 | 6.50 × 10−6 | 1.12 | 0.44 | 2.87 | 0.76 | 95.85 | 4.30 × 10−6 | <1 × 10−4 | 0.2603 | 0.17 | 0.12 | 0.16 | |
| Proteins | FG | 24 | 5 | −0.12 | −0.32 | 0.09 | 0.26 | 156.55 | - | 0.24 | −0.44 | 0.92 | 0.49 | 148.81 | - | <1 × 10−4 | 0.8797 | −0.1 | 0.08 | 0.24 |
| 2h glucose | 24 | 5 | 0.14 | −0.58 | 0.86 | 0.7 | 52.9 | 3.78 × 10−4 | −0.98 | -3.43 | 1.47 | 0.43 | 51.59 | 3.56 × 10−4 | 3.00 × 10−4 | 0.0805 | −0.07 | 0.32 | 0.83 | |
| HDL-C | 38 | 5 | −0.18 | −0.34 | −0.01 | 0.03 | 656.82 | 1.81 × 10−114 | −0.28 | −0.7 | 0.15 | 0.2 | 653.96 | 1.63 × 10−114 | <1 × 10−4 | <1 × 10−4 | −0.07 | 0.05 | 0.14 | |
| LDL-C | 38 | 5 | −0.19 | −0.36 | −0.03 | 0.02 | 564.08 | 1.75 × 10−95 | −0.47 | −0.9 | −0.05 | 0.03 | 540.64 | 2.63 × 10−91 | <1 × 10−4 | <1 × 10−4 | 0.1 | 0.06 | 0.09 | |
| TC | 38 | 5 | −0.19 | −0.41 | 0.03 | 0.09 | 275.49 | 8.63 × 10−38 | −0.53 | −1.06 | 0.01 | 0.05 | 262.63 | 8.45 × 10−36 | <1 × 10−4 | 0.0876 | −0.14 | 0.08 | 0.09 | |
| TG | 38 | 5 | 0.04 | −0.26 | 0.34 | 0.78 | 1927.21 | - | −0.37 | −1.13 | 0.38 | 0.33 | 1993.84 | - | <1 × 10−4 | 0.023 | −0.07 | 0.06 | 0.24 | |
| T2D | 4 | 6 | 1.78 | 0.03 | 105.08 | 0.78 | 219.15 | - | 0.01 | 4.81 × 10−27 | 1.57 × 1022 | 0.88 | 215.3 | - | <1 × 10−4 | - | −0.76 | 1.63 | 0.64 | |
| Stroke | 38 | 5 | 0.93 | 0.72 | 1.19 | 0.68 | 58.95 | 0.01 | 0.84 | 0.43 | 1.66 | 0.51 | 58.79 | 0.01 | 0.01 | 0.13 | −0.03 | 0.12 | 0.81 | |
| * T2D | 4 | 6 | 0.61 | 0.07 | 5.38 | 0.66 | 87.4 | - | 9.77 | 1.19 × 10−14 | 7.99 × 1015 | 0.91 | 86.4 | - | <1 × 10−4 | <1 × 10−4 | −0.44 | 1.07 | 0.68 | |
| CHD | 38 | 5 | 1.09 | 0.83 | 1.43 | 0.46 | 66.19 | 2.20 × 10−3 | 0.95 | 0.46 | 1.96 | 0.82 | 1.90 × 10−3 | - | 0.07 | 0.11 | 0.53 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomares-Millan, H.; Atabaki-Pasdar, N.; Coral, D.; Johansson, I.; Giordano, G.N.; Franks, P.W. Estimating the Direct Effect between Dietary Macronutrients and Cardiometabolic Disease, Accounting for Mediation by Adiposity and Physical Activity. Nutrients 2022, 14, 1218. https://doi.org/10.3390/nu14061218
Pomares-Millan H, Atabaki-Pasdar N, Coral D, Johansson I, Giordano GN, Franks PW. Estimating the Direct Effect between Dietary Macronutrients and Cardiometabolic Disease, Accounting for Mediation by Adiposity and Physical Activity. Nutrients. 2022; 14(6):1218. https://doi.org/10.3390/nu14061218
Chicago/Turabian StylePomares-Millan, Hugo, Naeimeh Atabaki-Pasdar, Daniel Coral, Ingegerd Johansson, Giuseppe N. Giordano, and Paul W. Franks. 2022. "Estimating the Direct Effect between Dietary Macronutrients and Cardiometabolic Disease, Accounting for Mediation by Adiposity and Physical Activity" Nutrients 14, no. 6: 1218. https://doi.org/10.3390/nu14061218
APA StylePomares-Millan, H., Atabaki-Pasdar, N., Coral, D., Johansson, I., Giordano, G. N., & Franks, P. W. (2022). Estimating the Direct Effect between Dietary Macronutrients and Cardiometabolic Disease, Accounting for Mediation by Adiposity and Physical Activity. Nutrients, 14(6), 1218. https://doi.org/10.3390/nu14061218