
Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
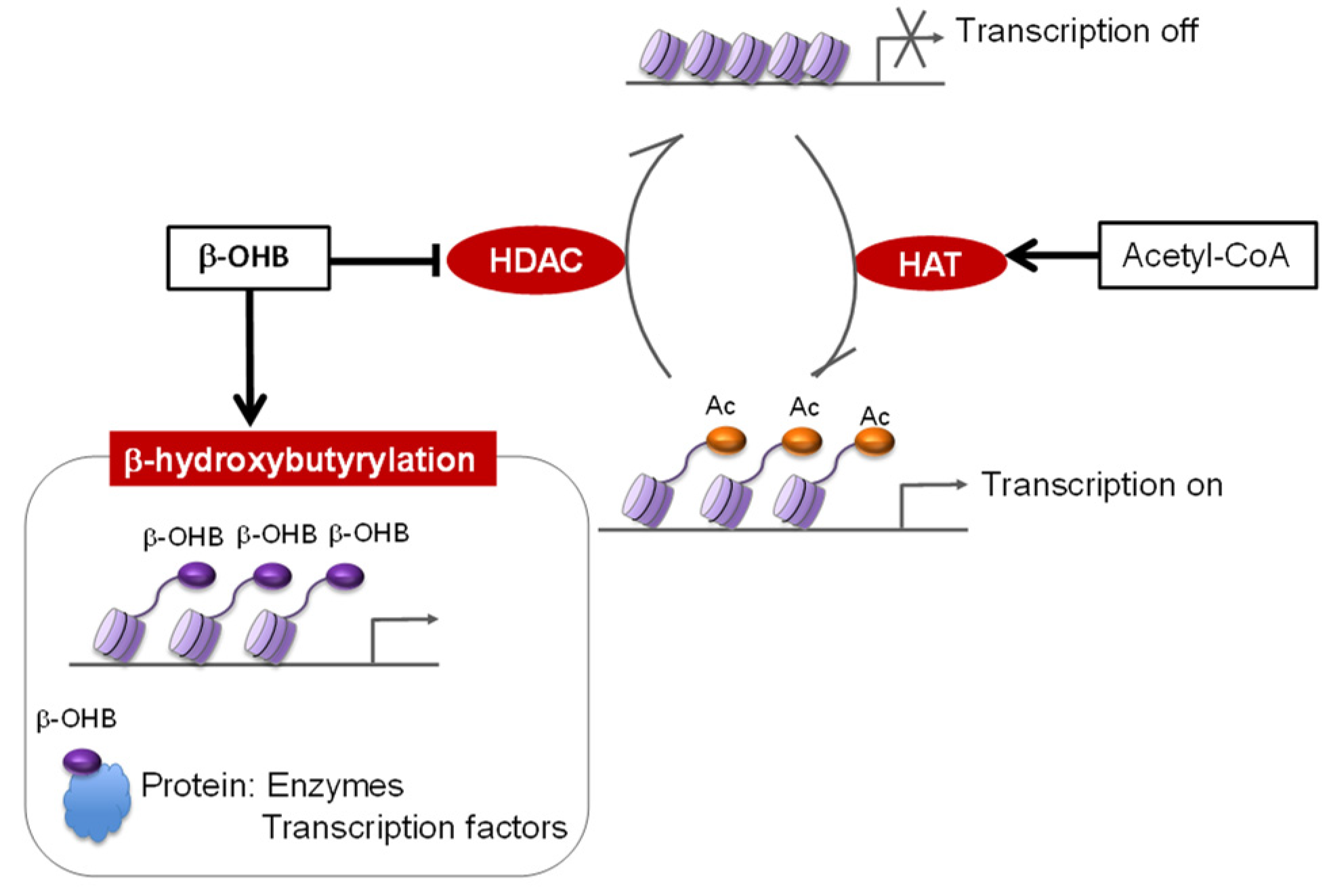
2. A Ketogenic Diet as an Epigenetic Modifier
3. Ketone Bodies as Endogenous Ligands for G-Protein-Coupled Receptors
4. Physiological Impact of a Ketogenic Diet
4.1. Nervous System
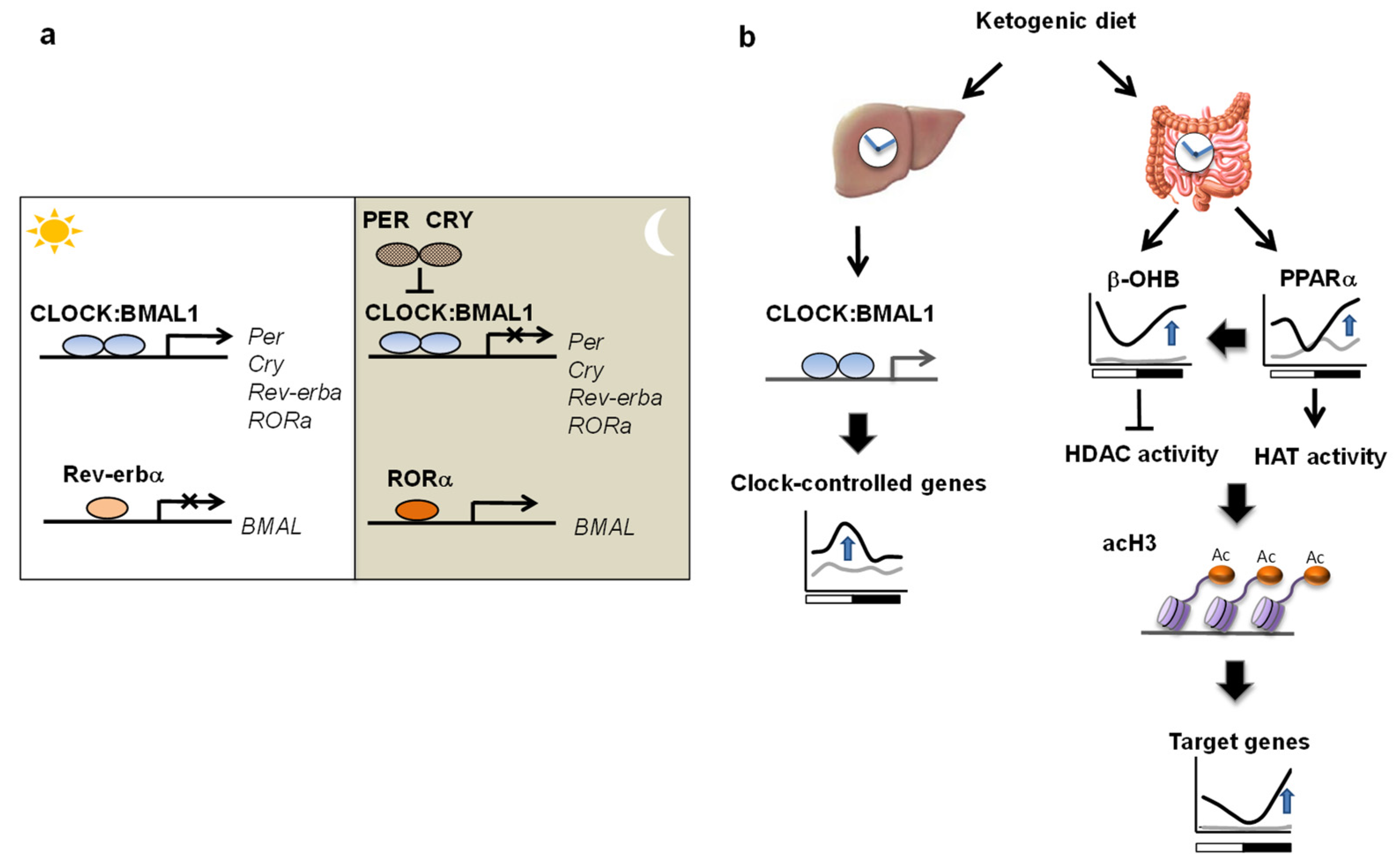
4.2. Circadian Clock
4.3. Metabolism
4.4. Immune System
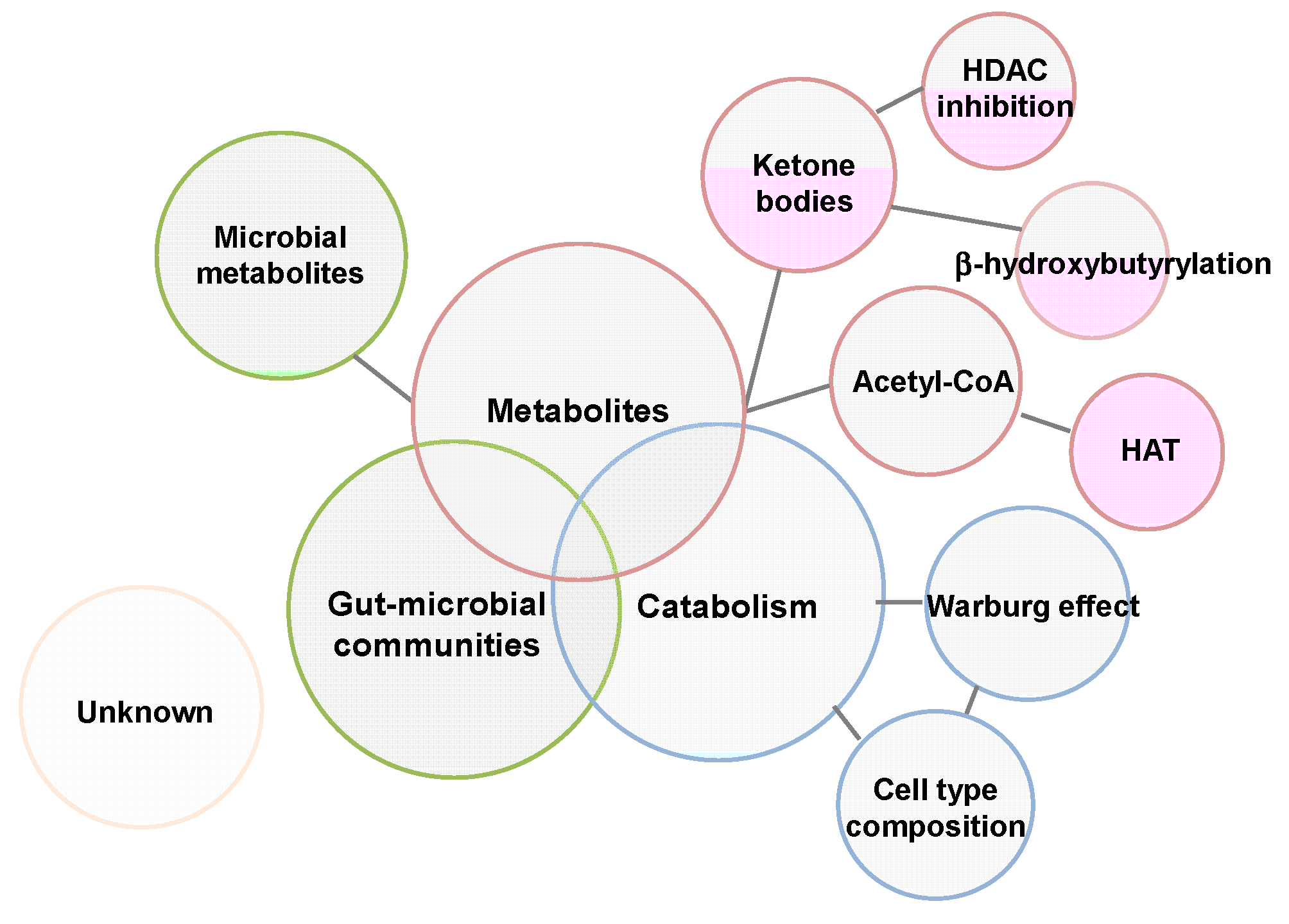
5. Influence of a Ketogenic Diet on Intestinal Microenvironment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hugo, S.E.; Cruz-Garcia, L.; Karanth, S.; Anderson, R.M.; Stainier, D.Y.; Schlegel, A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 2012, 26, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukao, T.; Song, X.Q.; Mitchell, G.A.; Yamaguchi, S.; Sukegawa, K.; Orii, T.; Kondo, N. Enzymes of ketone body utilization in human tissues: Protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr. Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Yang, H.; Kong, X.; Wang, K.; Mao, X.; Yan, X.; Wang, Y.; Liu, S.; Zhang, X.; Li, J.; et al. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E287–E295. [Google Scholar] [CrossRef]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurle, J.B.; Philp, N.J. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ishibashi, J.; Trefely, S.; Shao, M.; Cowan, A.J.; Sakers, A.; Lim, H.W.; O’Connor, S.; Doan, M.T.; Cohen, P.; et al. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019, 30, 174–189.e5. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Weber, W.A.; Schwaiger, M.; Avril, N. Quantitative assessment of tumor metabolism using FDG-PET imaging. Nucl. Med. Biol. 2000, 27, 683–687. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma Utilizes Fatty Acids and Ketone Bodies for Growth Allowing Progression during Ketogenic Diet Therapy. iScience 2020, 23, 101453. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Loscher, W.; Rho, J.M. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med. 2016, 6, a022780. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- Pougovkina, O.; te Brinke, H.; Ofman, R.; van Cruchten, A.G.; Kulik, W.; Wanders, R.J.; Houten, S.M.; de Boer, V.C. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef] [Green Version]
- Dieterich, I.A.; Lawton, A.J.; Peng, Y.; Yu, Q.; Rhoads, T.W.; Overmyer, K.A.; Cui, Y.; Armstrong, E.A.; Howell, P.R.; Burhans, M.S.; et al. Acetyl-CoA flux regulates the proteome and acetyl-proteome to maintain intracellular metabolic crosstalk. Nat. Commun. 2019, 10, 3929. [Google Scholar] [CrossRef]
- Arima, Y.; Nakagawa, Y.; Takeo, T.; Ishida, T.; Yamada, T.; Hino, S.; Nakao, M.; Hanada, S.; Umemoto, T.; Suda, T.; et al. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat. Metab. 2021, 3, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Candido, E.P.; Reeves, R.; Davie, J.R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978, 14, 105–113. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131.e15. [Google Scholar] [CrossRef] [Green Version]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, K.; Ma, J.; Zhou, L.; Liu, J.; Zeng, L.; Zhu, L.; Xu, P.; Chen, J.; Wei, K.; et al. Ketogenesis-generated beta-hydroxybutyrate is an epigenetic regulator of CD8(+) T-cell memory development. Nat. Cell Biol. 2020, 22, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Greco, C.M.; Huang, H.; Kim, J.K.; Fribourgh, J.L.; Crosby, P.; Mathur, L.; Ren, X.; Partch, C.L.; Jang, C.; et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine beta-hydroxybutyrylation. Cell Rep. 2021, 36, 109487. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [Green Version]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Ohue-Kitano, R.; Mukouyama, H.; Nishida, A.; Watanabe, K.; Igarashi, M.; Irie, J.; Tsujimoto, G.; Satoh-Asahara, N.; Itoh, H.; et al. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 23813–23821. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, S.; Le Roy, T.; Furgiuele, S.; Coste, V.; Bindels, L.B.; Leyrolle, Q.; Neyrinck, A.M.; Quoilin, C.; Amadieu, C.; Petit, G.; et al. Gut Microbiota-Induced Changes in beta-Hydroxybutyrate Metabolism Are Linked to Altered Sociability and Depression in Alcohol Use Disorder. Cell Rep. 2020, 33, 108238. [Google Scholar] [CrossRef]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillere, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Lutas, A.; Yellen, G. The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Vining, E.P.; Freeman, J.M.; Ballaban-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D.L.; Pyzik, P.L.; Freeman, J.M. The ketogenic diet: Seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J. Child Neurol. 2000, 15, 787–790. [Google Scholar] [CrossRef]
- D’Andrea Meira, I.; Romao, T.T.; Pires do Prado, H.J.; Kruger, L.T.; Pires, M.E.P.; da Conceicao, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasior, M.; French, A.; Joy, M.T.; Tang, R.S.; Hartman, A.L.; Rogawski, M.A. The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia 2007, 48, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, H.M. Factors influencing experimentally produced convulsions. Arch. Neurol. Psychiatry 1933, 29, 148. [Google Scholar] [CrossRef]
- Rho, J.M.; Anderson, G.D.; Donevan, S.D.; White, H.S. Acetoacetate, acetone, and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia 2002, 43, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Thio, L.L.; Wong, M.; Yamada, K.A. Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 2000, 54, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef] [Green Version]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Lazarow, A.; Luhovyy, B.; Wehrli, S.; Nissim, I. Response of brain amino acid metabolism to ketosis. Neurochem. Int. 2005, 47, 119–128. [Google Scholar] [CrossRef]
- Wang, Z.J.; Bergqvist, C.; Hunter, J.V.; Jin, D.; Wang, D.J.; Wehrli, S.; Zimmerman, R.A. In vivo measurement of brain metabolites using two-dimensional double-quantum MR spectroscopy—Exploration of GABA levels in a ketogenic diet. Magn. Reson. Med. 2003, 49, 615–619. [Google Scholar] [CrossRef]
- Dahlin, M.; Elfving, A.; Ungerstedt, U.; Amark, P. The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res. 2005, 64, 115–125. [Google Scholar] [CrossRef]
- Calderon, N.; Betancourt, L.; Hernandez, L.; Rada, P. A ketogenic diet modifies glutamate, gamma-aminobutyric acid and agmatine levels in the hippocampus of rats: A microdialysis study. Neurosci. Lett. 2017, 642, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, J.; Li, Y.Q.; Yao, S.; Wu, C.H.; Wang, Y.; Gao, F.; Xu, M.D.; Huang, G.B.; Zhao, C.Q.; et al. Neuregulin 1/ErbB4 signaling contributes to the anti-epileptic effects of the ketogenic diet. Cell Biosci. 2021, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Granados-Rojas, L.; Jeronimo-Cruz, K.; Juarez-Zepeda, T.E.; Tapia-Rodriguez, M.; Tovar, A.R.; Rodriguez-Jurado, R.; Carmona-Aparicio, L.; Cardenas-Rodriguez, N.; Coballase-Urrutia, E.; Ruiz-Garcia, M.; et al. Ketogenic Diet Provided during Three Months Increases KCC2 Expression but Not NKCC1 in the Rat Dentate Gyrus. Front. Neurosci. 2020, 14, 673. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lira, G.; Mendoza-Torreblanca, J.G.; Granados-Rojas, L. Ketogenic diet does not change NKCC1 and KCC2 expression in rat hippocampus. Epilepsy Res. 2011, 96, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ding, Y.; Ding, X.Y.; Liu, Z.R.; Shen, C.H.; Jin, B.; Guo, Y.; Wang, S.; Ding, M.P. Effectiveness of ketogenic diet in pentylenetetrazol-induced and kindling rats as well as its potential mechanisms. Neurosci. Lett. 2016, 614, 1–6. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J. Clin. Invest. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.; Qiu, X.; Lai, W.; Li, W.; Zhang, L.; Zhu, X.; He, S.; Duan, J.; Chen, L. Altered composition of the gut microbiome in patients with drug-resistant epilepsy. Epilepsy Res. 2018, 147, 102–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef]
- Lindefeldt, M.; Eng, A.; Darban, H.; Bjerkner, A.; Zetterstrom, C.K.; Allander, T.; Andersson, B.; Borenstein, E.; Dahlin, M.; Prast-Nielsen, S. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Liu, X.; Chen, C.; Lin, J.; Li, A.; Guo, K.; An, D.; Zhou, D.; Hong, Z. Alteration of Gut Microbiota in Patients with Epilepsy and the Potential Index as a Biomarker. Front. Microbiol. 2020, 11, 517797. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic diet in neuromuscular and neurodegenerative diseases. BioMed Res. Int. 2014, 2014, 474296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraeuter, A.K.; Phillips, R.; Sarnyai, Z. Ketogenic therapy in neurodegenerative and psychiatric disorders: From mice to men. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 101, 109913. [Google Scholar] [CrossRef]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, C.; Wu, J.; Liu, P.; Deng, X.; Zhang, Y.; Peng, B.; Zhu, Y. Ketogenic diet ameliorates cognitive impairment and neuroinflammation in a mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2021, 00, 1–13. [Google Scholar] [CrossRef]
- Beckett, T.L.; Studzinski, C.M.; Keller, J.N.; Paul Murphy, M.; Niedowicz, D.M. A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1505, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Park, S.; Zhang, T.; Wu, X.; Yi Qiu, J. Ketone production by ketogenic diet and by intermittent fasting has different effects on the gut microbiota and disease progression in an Alzheimer’s disease rat model. J. Clin. Biochem. Nutr. 2020, 67, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Koppel, S.J.; Pei, D.; Wilkins, H.M.; Weidling, I.W.; Wang, X.; Menta, B.W.; Perez-Ortiz, J.; Kalani, A.; Manley, S.; Novikova, L.; et al. A ketogenic diet differentially affects neuron and astrocyte transcription. J. Neurochem. 2021, 157, 1930–1945. [Google Scholar] [CrossRef]
- Gzielo, K.; Soltys, Z.; Rajfur, Z.; Setkowicz, Z.K. The Impact of the Ketogenic Diet on Glial Cells Morphology. A Quantitative Morphological Analysis. Neuroscience 2019, 413, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Fortier, M.; Rheault, F.; Edde, M.; Croteau, E.; Castellano, C.A.; Langlois, F.; St-Pierre, V.; Cuenoud, B.; Bocti, C.; et al. A ketogenic supplement improves white matter energy supply and processing speed in mild cognitive impairment. Alzheimers Dement. 2021, 7, e12217. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Brandt, J.; Buchholz, A.; Henry-Barron, B.; Vizthum, D.; Avramopoulos, D.; Cervenka, M.C. Preliminary Report on the Feasibility and Efficacy of the Modified Atkins Diet for Treatment of Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 969–981. [Google Scholar] [CrossRef]
- Li, Q.; Liang, J.; Fu, N.; Han, Y.; Qin, J. A Ketogenic Diet and the Treatment of Autism Spectrum Disorder. Front. Pediatr. 2021, 9, 650624. [Google Scholar] [CrossRef]
- Phelps, J.R.; Siemers, S.V.; El-Mallakh, R.S. The ketogenic diet for type II bipolar disorder. Neurocase 2013, 19, 423–426. [Google Scholar] [CrossRef]
- Caminha, M.C.; Moreira, A.B.; Matheus, F.C.; Rieger, D.K.; Moreira, J.D.; Dalmarco, E.M.; Demarchi, I.G.; Lin, K. Efficacy and tolerability of the ketogenic diet and its variations for preventing migraine in adolescents and adults: A systematic review. Nutr. Rev. 2021, nuab080. [Google Scholar] [CrossRef]
- Palmer, C.M.; Gilbert-Jaramillo, J.; Westman, E.C. The ketogenic diet and remission of psychotic symptoms in schizophrenia: Two case studies. Schizophr. Res. 2019, 208, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Bernal, I.; Becerril-Perez, F.; Aguilar-Arnal, L. Circadian rhythms in the three-dimensional genome: Implications of chromatin interactions for cyclic transcription. Clin. Epigenet. 2019, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokkan, K.A.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the circadian clock in the liver by feeding. Science 2001, 291, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel-Mahan, K.L.; Patel, V.R.; de Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.A.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the circadian clock by nutritional challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Tognini, P.; Liu, Y.; Eckel-Mahan, K.L.; Baldi, P.; Sassone-Corsi, P. Gut microbiota directs PPARgamma-driven reprogramming of the liver circadian clock by nutritional challenge. EMBO Rep. 2016, 17, 1292–1303. [Google Scholar] [CrossRef] [Green Version]
- Chavan, R.; Feillet, C.; Costa, S.S.; Delorme, J.E.; Okabe, T.; Ripperger, J.A.; Albrecht, U. Liver-derived ketone bodies are necessary for food anticipation. Nat. Commun. 2016, 7, 10580. [Google Scholar] [CrossRef] [Green Version]
- Oishi, K.; Uchida, D.; Ohkura, N.; Doi, R.; Ishida, N.; Kadota, K.; Horie, S. Ketogenic diet disrupts the circadian clock and increases hypofibrinolytic risk by inducing expression of plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1571–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Yamamoto, S.; Uchida, D.; Doi, R. Ketogenic diet and fasting induce the expression of cold-inducible RNA-binding protein with time-dependent hypothermia in the mouse liver. FEBS Open Bio 2013, 3, 192–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genzer, Y.; Dadon, M.; Burg, C.; Chapnik, N.; Froy, O. Ketogenic diet delays the phase of circadian rhythms and does not affect AMP-activated protein kinase (AMPK) in mouse liver. Mol. Cell. Endocrinol. 2015, 417, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Roberson, R.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Kennedy, A.R.; Adams, A.C.; Pissios, P.; Maratos-Flier, E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1197–E1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Jurczak, M.J.; Lee, H.Y.; Birkenfeld, A.L.; Frederick, D.W.; Zhang, D.; Zhang, X.M.; Samuel, V.T.; Shulman, G.I. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E808–E815. [Google Scholar] [CrossRef] [PubMed]
- Grandl, G.; Straub, L.; Rudigier, C.; Arnold, M.; Wueest, S.; Konrad, D.; Wolfrum, C. Short-term feeding of a ketogenic diet induces more severe hepatic insulin resistance than an obesogenic high-fat diet. J. Physiol. 2018, 596, 4597–4609. [Google Scholar] [CrossRef]
- Choi, Y.J.; Jeon, S.M.; Shin, S. Impact of a Ketogenic Diet on Metabolic Parameters in Patients with Obesity or Overweight and with or without Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Nutrients 2020, 12, 2005. [Google Scholar] [CrossRef]
- Badman, M.K.; Koester, A.; Flier, J.S.; Kharitonenkov, A.; Maratos-Flier, E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 2009, 150, 4931–4940. [Google Scholar] [CrossRef] [Green Version]
- Douris, N.; Desai, B.N.; Fisher, F.M.; Cisu, T.; Fowler, A.J.; Zarebidaki, E.; Nguyen, N.L.T.; Morgan, D.A.; Bartness, T.J.; Rahmouni, K.; et al. Beta-adrenergic receptors are critical for weight loss but not for other metabolic adaptations to the consumption of a ketogenic diet in male mice. Mol. Metab. 2017, 6, 854–862. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Shchukina, I.; Asher, J.L.; Sidorov, S.; Artyomov, M.N.; Dixit, V.D. Ketogenesis activates metabolically protective gammadelta T cells in visceral adipose tissue. Nat. Metab. 2020, 2, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I.; Blau, J.E.; Rother, K.I. SGLT2 Inhibitors May Predispose to Ketoacidosis. J. Clin. Endocrinol. Metab. 2015, 100, 2849–2852. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e6. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. beta-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef]
- Tate, M.D.; Mansell, A. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Koutnik, A.P.; Goldberg, E.L.; Upadhyay, V.; Turnbaugh, P.J.; Verdin, E.; Newman, J.C. Investigating Ketone Bodies as Immunometabolic Countermeasures against Respiratory Viral Infections. Med. 2020, 1, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Neudorf, H.; Durrer, C.; Myette-Cote, E.; Makins, C.; O’Malley, T.; Little, J.P. Oral Ketone Supplementation Acutely Increases Markers of NLRP3 Inflammasome Activation in Human Monocytes. Mol. Nutr. Food Res. 2019, 63, e1801171. [Google Scholar] [CrossRef] [PubMed]
- Neudorf, H.; Myette-Cote, E.; Little, J.P. The Impact of Acute Ingestion of a Ketone Monoester Drink on LPS-Stimulated NLRP3 Activation in Humans with Obesity. Nutrients 2020, 12, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, E.L.; Molony, R.D.; Kudo, E.; Sidorov, S.; Kong, Y.; Dixit, V.D.; Iwasaki, A. Ketogenic diet activates protective gammadelta T cell responses against influenza virus infection. Sci. Immunol. 2019, 4, eaav2026. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Tognini, P. The Circadian Clock as an Essential Molecular Link between Host Physiology and Microorganisms. Front. Cell. Infect. Microbiol. 2019, 9, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275.e16. [Google Scholar] [CrossRef]
- Ni, F.F.; Li, C.R.; Liao, J.X.; Wang, G.B.; Lin, S.F.; Xia, Y.; Wen, J.L. The effects of ketogenic diet on the Th17/Treg cells imbalance in patients with intractable childhood epilepsy. Seizure 2016, 38, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism 2016, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Zhou, Q.; Qiu, C.Z.; Dai, W.K.; Wang, H.P.; Li, Y.H.; Liao, J.X.; Lu, X.G.; Lin, S.F.; Ye, J.H.; et al. Ketogenic diet poses a significant effect on imbalanced gut microbiota in infants with refractory epilepsy. World J. Gastroenterol. 2017, 23, 6164–6171. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, M.; Prast-Nielsen, S. The gut microbiome and epilepsy. EBioMedicine 2019, 44, 741–746. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, M.; Tognini, P. Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet. Nutrients 2022, 14, 782. https://doi.org/10.3390/nu14040782
Murakami M, Tognini P. Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet. Nutrients. 2022; 14(4):782. https://doi.org/10.3390/nu14040782
Chicago/Turabian StyleMurakami, Mari, and Paola Tognini. 2022. "Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet" Nutrients 14, no. 4: 782. https://doi.org/10.3390/nu14040782
APA StyleMurakami, M., & Tognini, P. (2022). Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet. Nutrients, 14(4), 782. https://doi.org/10.3390/nu14040782