
Molecular Basis of Resveratrol-Induced Resensitization of Acquired Drug-Resistant Cancer Cells



Abstract
:1. Introduction
2. In Vitro and In Vivo Activity of RES in Different Tumor Models
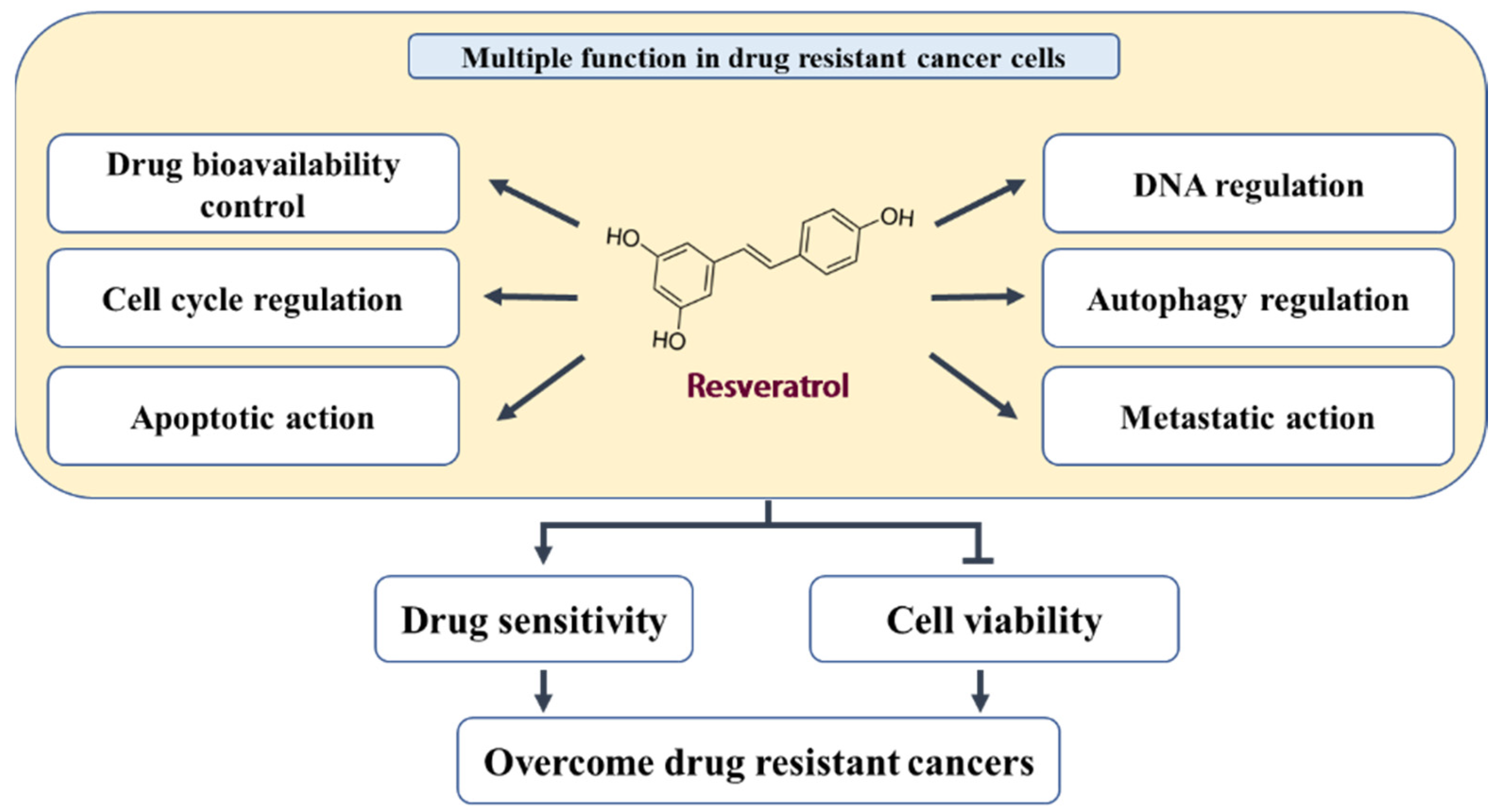
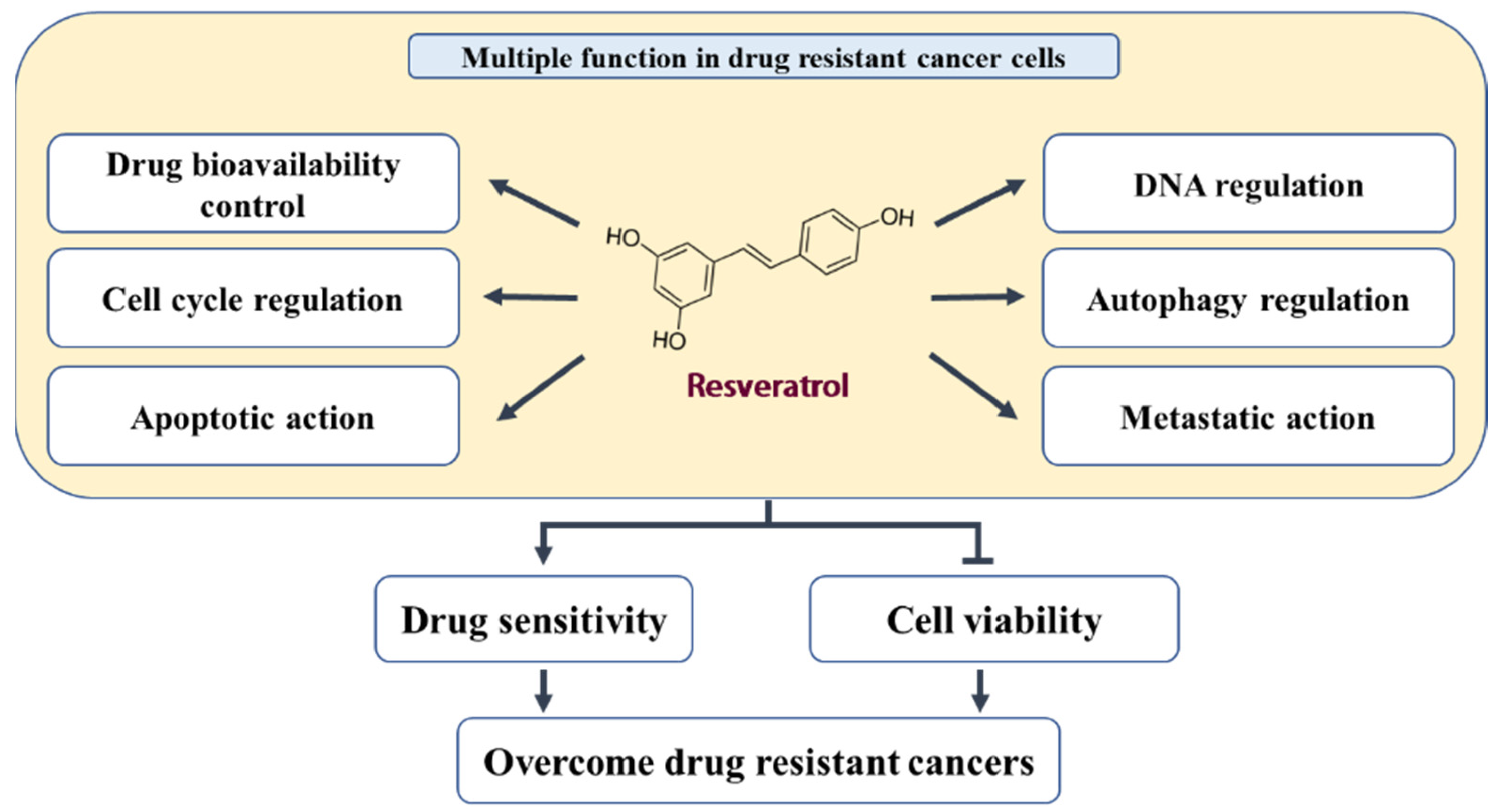
3. Biological Effects and Mechanisms of RES in Acquired Drug-Resistant Cancer Cells
3.1. Inhibition of Drug Transporters and Drug-Metabolizing Enzymes
3.1.1. P-Glycoprotein (P-gp/MDR1)
3.1.2. Multidrug Resistance-Associated Protein 1 (MRP1)
3.1.3. BCRP (ABCG2)
3.2. Promotion of DNA Damage and Inhibition of DNA Repair and Replication
3.3. Cell Cycle Regulation
3.4. Pro-Apoptotic and Antisurvival Actions
3.4.1. p53 Induction
3.4.2. Caspase Activation
3.4.3. Inhibition of ERK and AKT Pathways
3.4.4. Inhibition of NF-κB
3.4.5. Alteration of Bcl-2 Family Members
3.4.6. Prevention of Clusterin Expression
3.4.7. Other Factors
3.5. Autophagy Regulation
3.6. Inhibition of EMT and CSCs
4. Conclusions

{kind=link}
| Cancer | Selecting Drug | Resistant Cells | Assessed Parameter | Outcome Measures | Reference |
|---|---|---|---|---|---|
| Bladder | Doxorubicin | Pumc-91/ADM | ↑Topo-II, ↓Bcl-2, ↓GST, ↓LRP, ↓MRP1 | ↑S arrest, ↓Proliferation | [24] |
| Breast | Cisplatin | MCF-7R | ↑γ-H2AX, ↓Rad51 | ↓Proliferation, ↓Repair of DNA damage | [7] |
| MCF-7R | ↑AMPK, ↑Bax, ↑CHK2, ↑CK1, ↑p21, ↑PUMA, ↑p-p53 (Serine20), ↓Bcl-2, ↓p-p53 (Serine15 and 46) | ↑Apoptosis, ↓Proliferation | [47] | ||
| Doxorubicin | MCF-7-ADR | ↑Caspase-8, ↑Caspase-9, ↑miR-122-5p ↓Bcl-2, ↓CDK2, ↓CDK4, ↓CDK6 | ↑Apoptosis, ↑G1 arrest, ↓Proliferation | [46] | |
| MCF7/ADR | ↑SIRT1, ↓β-Catenin, ↓N-Cadherin, ↓Imentin | ↑Apoptosis, ↓Cell migration, ↓EMT, ↓Proliferation | [78] | ||
| MCF-7/DOX | ↑Caspase-3, ↓PI3K, ↓p-Akt/Akt, ↓p-mTOR/mTOR | ↑Apoptosis, ↓Cell migration, ↓Cell invasion, ↓Metastasis, ↓Proliferation, ↓Tumor volume | [58] | ||
| MCF-7/adr | ↓MDR1, ↓MRP1 | ↑Cellular influx, ↓Proliferation, ↓Tumor volume | [34] | ||
| Mitoxantrone | MCF/MR | - | ↑ATPase activity, ↑Cellular accumulation | [38] | |
| Paclitaxel | MDA-MB-231/PacR | ↑Caspase-7, ↓Survivin | ↑Apoptosis, ↑Senescence ↑Sub-G1 arrest, ↓Colony formation ↓Proliferation | [59] | |
| Tamoxifen | MCF-7/TR | ↑Caspase-3, ↑E-Cadherin, ↑γ-Catenin, ↓Fibronectin, ↓N-Cadherin, ↓p-Akt, ↓p-ERK, ↓p-Smad2, ↓p-Smad3, ↓TGF-β, ↓Vimentin | ↑Apoptosis, ↓EMT | [56] | |
| MCF-7 TR1 | ↑p21, ↑p53, ↓Cyclin D1, ↓ERα, ↓IRS1 | ↑G1 arrest, ↓Proliferation | [51] | ||
| Colon | 5-FU | HCT-116R | ↑Caspase-3, ↑E-Cadherin, ↓ALDH1, ↓CD133, ↓CD44, ↓CXCR4, ↓MMP-9, ↓p-NF-kB, ↓Slug, ↓Vimentin | ↑Apoptosis, ↓Cell invasion, ↓CSC, ↓EMT, ↓Metastasis | [25] |
| HCT116R | ↑Caspase-3, ↑E-Cadherin, ↓MMP-9, ↓p-IkBa, ↓p-NF-kB, ↓Slug, ↓Vimentin, | ↑Apoptosis, ↑Intracellular junction, ↓Cell invasion, ↓EMT, ↓Metastasis, ↓Proliferation | [57] | ||
| 5-FU-R | ↑APC, ↑Bax/Bcl-xL, ↑Caspase-3, ↑p21, ↑PTEN ↓CDC-2, ↓DDB2, ↓FEN-1, ↓POLH, ↓POL-β | ↑Apoptosis, ↑DNA damage, ↑Sub-G1 arrest, ↓Proliferation | [44] | ||
| Cisplatin | CIS-resistant HCT-116 CRC | - | ↑Apoptosis, ↑Cellular uptake, ↑G0 arrest, ↓Proliferation | [30] | |
| Oxaliplatin | HCT116/L-OHP | ↑AMPK, ↓MDR1, ↓p-CREB, ↓p-NF-kB | ↑Cellular uptake, ↓Proliferation | [31] | |
| Gastric | Doxorubicin | SGC7901/DOX | ↑Caspase-3, ↑Caspase-9, ↑E-Cadherin, ↑TSC1, ↑TSC2, ↓p-mTOR, ↓p-Akt, ↓β-Catenin, ↓Vimentin | ↑Apoptosis, ↓Cell migration, ↓EMT, ↓Proliferation, ↓Tumor volume | [26] |
| Leukemia | Doxorubicin | CEM/ADR5000 | - | ↑Apoptosis, ↑Cellular uptake, ↓Proliferation | [32] |
| HL-60/ADR | ↓MRP1, ↓Nrf2, ↓p-Akt, ↓PI3K | ↑Cellular uptake, ↓Proliferation | [35] | ||
| AML-2/DX300 | ↓MRP1 | ↑Apoptosis, ↑Cellular uptake, ↓Proliferation | [36] | ||
| Imatinib | IM-R K562 | ↑Atg3, ↑LC3-Ⅱ, ↑p62/SQSTM1, ↑p-AMPK, ↑p-JNK, ↓p-mTOR | ↑Autophagy, ↓Colony formation, ↓Proliferation | [70] | |
| Vincristine | HL60/VCR | - | ↑S arrest, ↑sub-G1 arrest, ↓Proliferation | [52] | |
| Lung | Cisplatin | SPC-A-1/CDDP | ↑Caspase-3, ↓Survivin | ↑Apoptosis, ↑G0-G1 arrest, ↓Proliferation, ↓Tumor volume | [16] |
| Gefitinib | PC9/G | ↑Caspase-3, ↑LC3B-II, ↑p21, ↑p53, ↓p-EGFR, ↓ABCG2, ↓CYP1A1 | ↑Apoptosis, ↑Autophagy, ↑Cellular uptake, ↑G2/M arrest, ↑Senescence | [37] | |
| Melanoma | Vemurafenib | VEM resistant | ↓p-Akt | ↓Proliferation | [61] |
| Oral | Cetuximab | SAS-R, Sa3-R, HSC-3-R | ↓Integrin β1, ↓p-ERK1/2, ↓p-FAK, ↓uPAR | ↓Proliferation, ↓Tumor volume | [60] |
| Cisplatin | CAR | ↑AIF, ↑Apaf-1, ↑Atg5, ↑Atg7, ↑Atg12, ↑Atg14, ↑Atg16L1, ↑Bad, ↑Bax, ↑Beclin-1, ↑Caspase-3, ↑Caspase-9, ↑Endo G, ↑LC3-Ⅱ, ↑p-AMPKα, ↓p-AKT, ↓p-mTOR, ↓Bcl-2, ↓p-Bad, ↓Rubicon | ↑Apoptosis, ↑Autophagy, ↓Proliferation | [63] | |
| ↓MMP-2, ↓MMP-9, ↓p-ERK, ↓p-p38 | ↓Cell migration ↓Cell invasion | [79] | |||
| Ovarian | Cisplatin | A2780CP | ↑LC3B-Ⅱ, ↓p-ERK, ↓Snail, | ↑Autophagy, ↑Cell death, ↑Senescence, ↓Cell migration, ↓EMT | [71] |
| A2780cisR | - | ↑Cellular accumulation, ↑Platinum–DNA binding, ↓Proliferation | [33] | ||
| Prostate | Docetaxel | PC3-DR | ↑Caspase-3, ↓Clusterin, ↓p-Jak, ↓p-Src, ↓p-Stat1 | ↑Apoptosis | [65] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bugde, P.; Biswas, R.; Merien, F.; Lu, J.; Liu, D.X.; Chen, M.; Zhou, S.; Li, Y. The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 2017, 21, 511–530. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.; Li, H.; Li, M.; Shu, X. Resveratrol-mediated reversal of tumor multi-drug resistance. Curr. Drug Metab. 2014, 15, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, F.U.; Sufiyan Chhipa, A.; Mishra, V.; Gupta, V.K.; Rawat, S.G.; Kumar, A.; Pathak, C. Molecular and cellular paradigms of multidrug resistance in cancer. Cancer Rep. 2020, 13, e1291. [Google Scholar] [CrossRef]
- Shnaider, P.V.; Ivanova, O.M.; Malyants, I.K.; Anufrieva, K.S.; Semenov, I.A.; Pavlyukov, M.S.; Lagarkova, M.A.; Govorun, V.M.; Shender, V.O. New Insights into Therapy-Induced Progression of Cancer. Int. J. Mol. Sci. 2020, 21, 7872. [Google Scholar] [CrossRef]
- Leon-Galicia, I.; Diaz-Chavez, J.; Albino-Sanchez, M.E.; Garcia-Villa, E.; Bermudez-Cruz, R.; Garcia-Mena, J.; Herrera, L.A.; Garcia-Carranca, A.; Gariglio, P. Resveratrol decreases Rad51 expression and sensitizes cisplatinresistant MCF7 breast cancer cells. Oncol. Rep. 2018, 39, 3025–3033. [Google Scholar] [CrossRef]
- Beaudeux, J.L.; Nivet-Antoine, V.; Giral, P. Resveratrol: A relevant pharmacological approach for the treatment of metabolic syndrome? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 729–736. [Google Scholar] [CrossRef]
- Gusman, J.; Malonne, H.; Atassi, G. A reappraisal of the potential chemopreventive and chemotherapeutic properties of resveratrol. Carcinogenesis 2001, 22, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef]
- Holmes-McNary, M.; Baldwin, A.S., Jr. Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IkappaB kinase. Cancer Res. 2000, 60, 3477–3483. [Google Scholar]
- Opipari, A.W., Jr.; Tan, L.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Bao, P.; Qi, H.; You, H. Resveratrol down-regulates survivin and induces apoptosis in human multidrug-resistant SPC-A-1/CDDP cells. Oncol. Rep. 2010, 23, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Luther, D.J.; Ohanyan, V.; Shamhart, P.E.; Hodnichak, C.M.; Sisakian, H.; Booth, T.D.; Meszaros, J.G.; Bishayee, A. Chemopreventive doses of resveratrol do not produce cardiotoxicity in a rodent model of hepatocellular carcinoma. Investig. New Drugs 2011, 29, 380–391. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kannappan, R.; Reuter, S.; Kim, J.H.; Aggarwal, B.B. Chemosensitization of tumors by resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Ozben, T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett. 2006, 580, 2903–2909. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [Green Version]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, Q.; Xie, Q.; Zhang, M. Effect and mechanism of resveratrol on drug resistance in human bladder cancer cells. Mol. Med. Rep. 2017, 15, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Shayan, P.; Goel, A.; Aggarwal, B.B.; Shakibaei, M. Resveratrol Chemosensitizes TNF-beta-Induced Survival of 5-FU-Treated Colorectal Cancer Cells. Nutrients 2018, 10, 888. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, D.; Niu, H.; Zhu, G.; Xu, Y.; Ye, D.; Li, J.; Zhang, Q. Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2017, 36, 19. [Google Scholar] [CrossRef] [Green Version]
- Kourti, M.; Vavatsi, N.; Gombakis, N.; Sidi, V.; Tzimagiorgis, G.; Papageorgiou, T.; Koliouskas, D.; Athanassiadou, F. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int. J. Hematol. 2007, 86, 166–173. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Kaur, G.; Gupta, S.K.; Singh, P.; Ali, V.; Kumar, V.; Verma, M. Drug-metabolizing enzymes: Role in drug resistance in cancer. Clin. Transl. Oncol. 2020, 22, 1667–1680. [Google Scholar] [CrossRef]
- Osman, A.M.; Al-Malki, H.S.; Al-Harthi, S.E.; El-Hanafy, A.A.; Elashmaoui, H.M.; Elshal, M.F. Modulatory role of resveratrol on cytotoxic activity of cisplatin, sensitization and modification of cisplatin resistance in colorectal cancer cells. Mol. Med. Rep. 2015, 12, 1368–1374. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, L.; Ni, Z.; Sun, J.; Gao, H.; Cheng, Z.; Xu, J.; Yin, P. Resveratrol induces AMPK-dependent MDR1 inhibition in colorectal cancer HCT116/L-OHP cells by preventing activation of NF-kappaB signaling and suppressing cAMP-responsive element transcriptional activity. Tumour Biol. 2015, 36, 9499–9510. [Google Scholar] [CrossRef] [PubMed]
- El-Readi, M.Z.; Eid, S.; Abdelghany, A.A.; Al-Amoudi, H.S.; Efferth, T.; Wink, M. Resveratrol mediated cancer cell apoptosis, and modulation of multidrug resistance proteins and metabolic enzymes. Phytomedicine 2019, 55, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Nessa, M.U.; Beale, P.; Chan, C.; Yu, J.Q.; Huq, F. Combinations of resveratrol, cisplatin and oxaliplatin applied to human ovarian cancer cells. Anticancer Res. 2012, 32, 53–59. [Google Scholar] [PubMed]
- Kim, T.H.; Shin, Y.J.; Won, A.J.; Lee, B.M.; Choi, W.S.; Jung, J.H.; Chung, H.Y.; Kim, H.S. Resveratrol enhances chemosensitivity of doxorubicin in multidrug-resistant human breast cancer cells via increased cellular influx of doxorubicin. Biochim. Biophys. Acta 2014, 1840, 615–625. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Feng, Z.; Bergan, R.; Li, B.; Qin, Y.; Zhao, L.; Zhang, Z.; Shi, M. Involvement of the PI3K/Akt/Nrf2 Signaling Pathway in Resveratrol-Mediated Reversal of Drug Resistance in HL-60/ADR Cells. Nutr. Cancer 2019, 71, 1007–1018. [Google Scholar] [CrossRef]
- Kweon, S.H.; Song, J.H.; Kim, T.S. Resveratrol-mediated reversal of doxorubicin resistance in acute myeloid leukemia cells via downregulation of MRP1 expression. Biochem. Biophys. Res. Commun. 2010, 395, 104–110. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef] [Green Version]
- Cooray, H.C.; Janvilisri, T.; van Veen, H.W.; Hladky, S.B.; Barrand, M.A. Interaction of the breast cancer resistance protein with plant polyphenols. Biochem. Biophys. Res. Commun. 2004, 317, 269–275. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Salehan, M.R.; Morse, H.R. DNA damage repair and tolerance: A role in chemotherapeutic drug resistance. Br. J. Biomed. Sci 2013, 70, 31–40. [Google Scholar] [CrossRef]
- Beck, W.T.; Danks, M.K. Mechanisms of resistance to drugs that inhibit DNA topoisomerases. Semin. Cancer Biol. 1991, 2, 235–244. [Google Scholar]
- Beck, W.T.; Danks, M.K.; Wolverton, J.S.; Granzen, B.; Chen, M.; Schmidt, C.A.; Bugg, B.Y.; Friche, E.; Suttle, D.P. Altered DNA topoisomerase II in multidrug resistance. Cytotechnology 1993, 11, 115–119. [Google Scholar] [CrossRef]
- Kellner, U.; Sehested, M.; Jensen, P.B.; Gieseler, F.; Rudolph, P. Culprit and victim—DNA topoisomerase II. Lancet Oncol. 2002, 3, 235–243. [Google Scholar] [CrossRef]
- Das, D.; Preet, R.; Mohapatra, P.; Satapathy, S.R.; Kundu, C.N. 1,3-Bis(2-chloroethyl)-1-nitrosourea enhances the inhibitory effect of resveratrol on 5-fluorouracil sensitive/resistant colon cancer cells. World J. Gastroenterol. 2013, 19, 7374–7388. [Google Scholar] [CrossRef]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar]
- Zhang, W.; Jiang, H.; Chen, Y.; Ren, F. Resveratrol chemosensitizes adriamycin-resistant breast cancer cells by modulating miR-122-5p. J. Cell Biochem. 2019, 120, 16283–16292. [Google Scholar] [CrossRef]
- Hernandez-Valencia, J.; Garcia-Villa, E.; Arenas-Hernandez, A.; Garcia-Mena, J.; Diaz-Chavez, J.; Gariglio, P. Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin. Nutrients 2018, 10, 1148. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Joe, A.K.; Liu, H.; Suzui, M.; Vural, M.E.; Xiao, D.; Weinstein, I.B. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin. Cancer Res. 2002, 8, 893–903. [Google Scholar]
- Singh, S.K.; Banerjee, S.; Acosta, E.P.; Lillard, J.W.; Singh, R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget 2017, 8, 17216–17228. [Google Scholar] [CrossRef] [Green Version]
- De Amicis, F.; Giordano, F.; Vivacqua, A.; Pellegrino, M.; Panno, M.L.; Tramontano, D.; Fuqua, S.A.; Ando, S. Resveratrol, through NF-Y/p53/Sin3/HDAC1 complex phosphorylation, inhibits estrogen receptor alpha gene expression via p38MAPK/CK2 signaling in human breast cancer cells. FASEB J. 2011, 25, 3695–3707. [Google Scholar] [CrossRef] [Green Version]
- Duraj, J.; Bodo, J.; Sulikova, M.; Rauko, P.; Sedlak, J. Diverse resveratrol sensitization to apoptosis induced by anticancer drugs in sensitive and resistant leukemia cells. Neoplasma 2006, 53, 384–392. [Google Scholar] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, V.V.; Chubarev, V.N.; Ashraf, G.M.; Dostdar, S.A.; Sokolov, A.V.; Melnikova, T.I.; Sologova, S.S.; Grigorevskich, E.M.; Makhmutova, C.A.; Kinzirsky, A.S.; et al. How Cancer Cells Resist Chemotherapy: Design and Development of Drugs Targeting Protein-Protein Interactions. Curr. Top. Med. Chem. 2019, 19, 394–412. [Google Scholar] [CrossRef]
- Shi, X.P.; Miao, S.; Wu, Y.; Zhang, W.; Zhang, X.F.; Ma, H.Z.; Xin, H.L.; Feng, J.; Wen, A.D.; Li, Y. Resveratrol sensitizes tamoxifen in antiestrogen-resistant breast cancer cells with epithelial-mesenchymal transition features. Int. J. Mol. Sci. 2013, 14, 15655–15668. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Shayan, P.; Kraehe, P.; Popper, B.; Goel, A.; Shakibaei, M. Resveratrol induces chemosensitization to 5-fluorouracil through up-regulation of intercellular junctions, Epithelial-to-mesenchymal transition and apoptosis in colorectal cancer. Biochem. Pharmacol. 2015, 98, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Bai, J.Y.; Yang, K.X. Effect of resveratrol on doxorubicin resistance in breast neoplasm cells by modulating PI3K/Akt signaling pathway. IUBMB Life 2018, 70, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Sprouse, A.A.; Herbert, B.S. Resveratrol augments paclitaxel treatment in MDA-MB-231 and paclitaxel-resistant MDA-MB-231 breast cancer cells. Anticancer Res. 2014, 34, 5363–5374. [Google Scholar]
- Uzawa, K.; Amelio, A.L.; Kasamatsu, A.; Saito, T.; Kita, A.; Fukamachi, M.; Sawai, Y.; Toeda, Y.; Eizuka, K.; Hayashi, F.; et al. Resveratrol Targets Urokinase-Type Plasminogen Activator Receptor Expression to Overcome Cetuximab-Resistance in Oral Squamous Cell Carcinoma. Sci. Rep. 2019, 9, 12179. [Google Scholar] [CrossRef]
- Luo, H.; Umebayashi, M.; Doi, K.; Morisaki, T.; Shirasawa, S.; Tsunoda, T. Resveratrol Overcomes Cellular Resistance to Vemurafenib Through Dephosphorylation of AKT in BRAF-mutated Melanoma Cells. Anticancer Res. 2016, 36, 3585–3589. [Google Scholar]
- Bharti, A.C.; Aggarwal, B.B. Chemopreventive agents induce suppression of nuclear factor-kappaB leading to chemosensitization. Ann. N. Y. Acad. Sci. 2002, 973, 392–395. [Google Scholar] [CrossRef]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; Chen, X.; Zhong, B.; Gilvary, D.L.; Zhou, J.; Wei, S.; Djeu, J.Y. Clusterin mediates TRAIL resistance in prostate tumor cells. Mol. Cancer Ther. 2007, 6, 2938–2947. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Baribeau, S.; Chaudhry, P.; Parent, S.; Asselin, E. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS ONE 2014, 9, e86987. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petpiroon, N.; Sritularak, B.; Chanvorachote, P. Phoyunnanin E inhibits migration of non-small cell lung cancer cells via suppression of epithelial-to-mesenchymal transition and integrin alphav and integrin beta3. BMC Complement. Altern. Med. 2017, 17, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xie, J.; Guo, J.; Manning, H.C.; Gore, J.C.; Guo, N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 2012, 28, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Wei, Y.; Liu, Y.; Lu, X.; Ding, F.; Wang, J.; Yang, S. Resveratrol promotes sensitization to Doxorubicin by inhibiting epithelial-mesenchymal transition and modulating SIRT1/beta-catenin signaling pathway in breast cancer. Cancer Med. 2019, 8, 1246–1257. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.S.; Tsai, C.W.; Yang, J.S.; Hsu, Y.M.; Shih, L.C.; Chiu, H.Y.; Bau, D.T.; Tsai, F.J. Resveratrol inhibited the metastatic behaviors of cisplatin-resistant human oral cancer cells via phosphorylation of ERK/p-38 and suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef]
| Target | Regulatory Molecules | Cellular Effect | ||
|---|---|---|---|---|
| ↑ Upregulation | ↓ Downregulation | ↑ Upregulation | ↓ Downregulation | |
| Drug transporters and drug-metabolizing enzymes | AMPK | ABCG2, GST, LRP1, MDR1, MRP1, Nrf2, p-AKT, p-CREB, p-NF-κB, PI3K | Cellular accumulation | ABC transporters ATPase activity Detoxification |
| DNA damage, repair, and replication | APC, Topo-II, γ-H2AX | DDB2, FEN-1, POLH, POL-β, Rad51 | DNA damage | DNA repair DNA replication |
| Cell cycle regulation | miR-122-5p, p21, p53, PTEN | CDC2, CDK2, CDK4, CDK6, Cyclin D1, ERα, IRS1 | Cell cycle arrest | - |
| Pro-apoptotic and anti-apoptotic action | AIF, AMPK, Apaf-1, Bad, Bax, Caspae-3, Caspase-7, Caspase-8, Caspase-9, CHK2, CK1, Endo G, miR-122-5p, p53, p-p53(S20), PTEN, PUMA, TSC1, TSC2 | Bcl-2, Bcl-xL, Clusterin, Integrin β1, p-AKT, p-Bad(s136), p-EGFR, p-ERK1/2, p-FAK, PI3K, p-IkBα, p-Jak, p-mTOR, p-NF-κB, p-p53(S15, S46), p-Src, p-Stat1, Survivin, uPAR | Apoptosis Cell death Senescence Sub-G1 arrest | Cell proliferation Tumor volume |
| Autophagy regulation | Atg3, Atg5, Atg7, Atg14, Atg12, Atg16L1, Beclin-1, LC3-Ⅱ, p62, p-AMPKα, p-JNK | p-AKT, p-mTOR, Rubicon | Autophagy | - |
| Migration, invasion, metastasis, EMT, and CSC | E-cadherin, SIRT1, γ-catenin | ALDH1, CD133, CD44, CXCR4, Fibronectin, MMP-2, MMP-9, N-Cadherin, p-ERK, p-NF-κB, p-p38, p-Smad2, p-Smad3, Slug, Snail, TGF-β, Vimentin, β-Catenin | Intracellular junction | Cell migration, invasion, and metastasis Colony formation CSC EMT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, C.Y.; Lim, S.-C.; Lee, T.-B.; Han, S.I. Molecular Basis of Resveratrol-Induced Resensitization of Acquired Drug-Resistant Cancer Cells. Nutrients 2022, 14, 699. https://doi.org/10.3390/nu14030699
Choi CY, Lim S-C, Lee T-B, Han SI. Molecular Basis of Resveratrol-Induced Resensitization of Acquired Drug-Resistant Cancer Cells. Nutrients. 2022; 14(3):699. https://doi.org/10.3390/nu14030699
Chicago/Turabian StyleChoi, Chul Yung, Sung-Chul Lim, Tae-Bum Lee, and Song Iy Han. 2022. "Molecular Basis of Resveratrol-Induced Resensitization of Acquired Drug-Resistant Cancer Cells" Nutrients 14, no. 3: 699. https://doi.org/10.3390/nu14030699
APA StyleChoi, C. Y., Lim, S.-C., Lee, T.-B., & Han, S. I. (2022). Molecular Basis of Resveratrol-Induced Resensitization of Acquired Drug-Resistant Cancer Cells. Nutrients, 14(3), 699. https://doi.org/10.3390/nu14030699