
Gut Microbiome and Metabolome Variations in Self-Identified Muscle Builders Who Report Using Protein Supplements

 , ,
, ,
Abstract
:1. Introduction
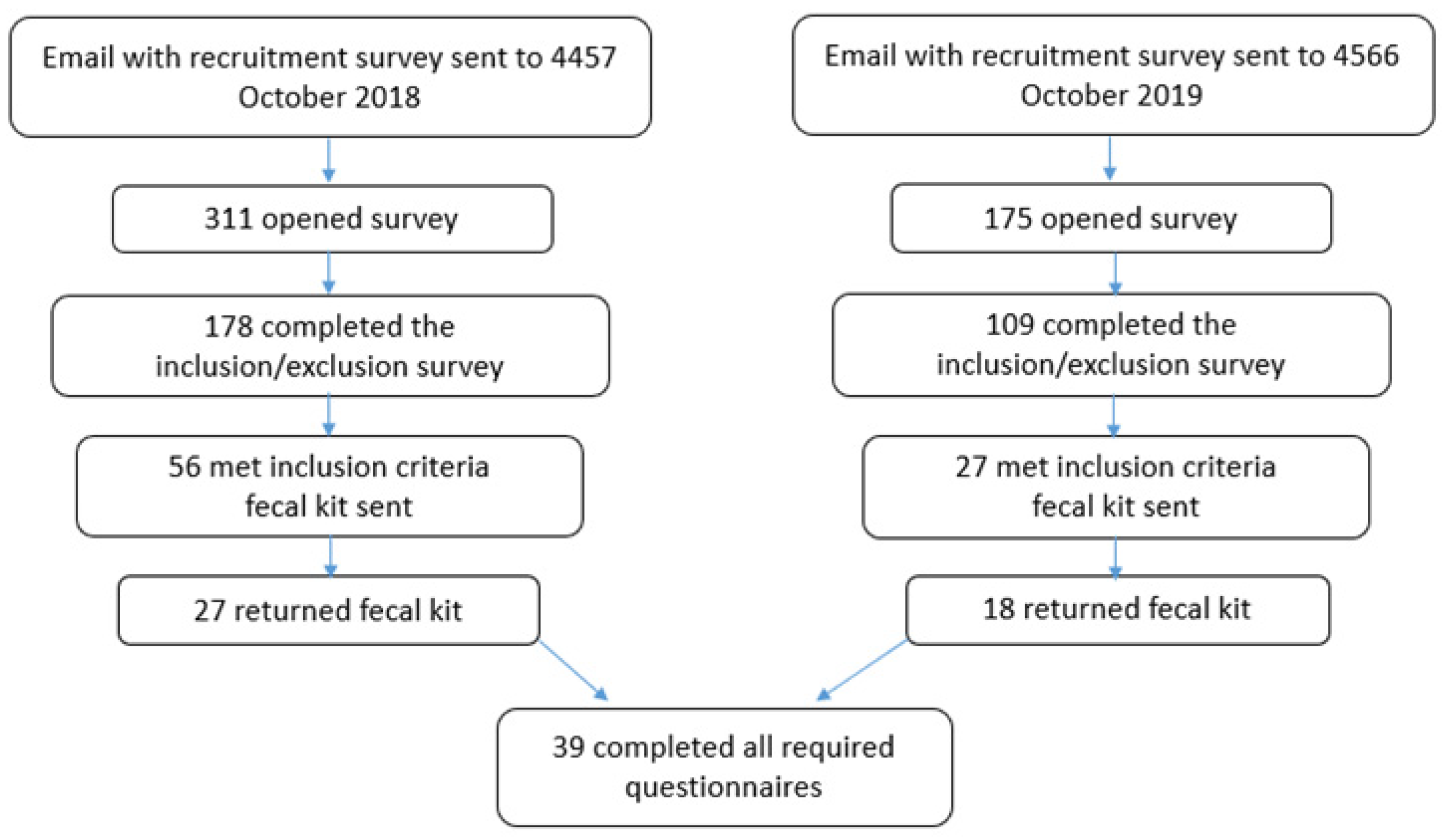
2. Materials and Methods
2.1. Experimental Approach
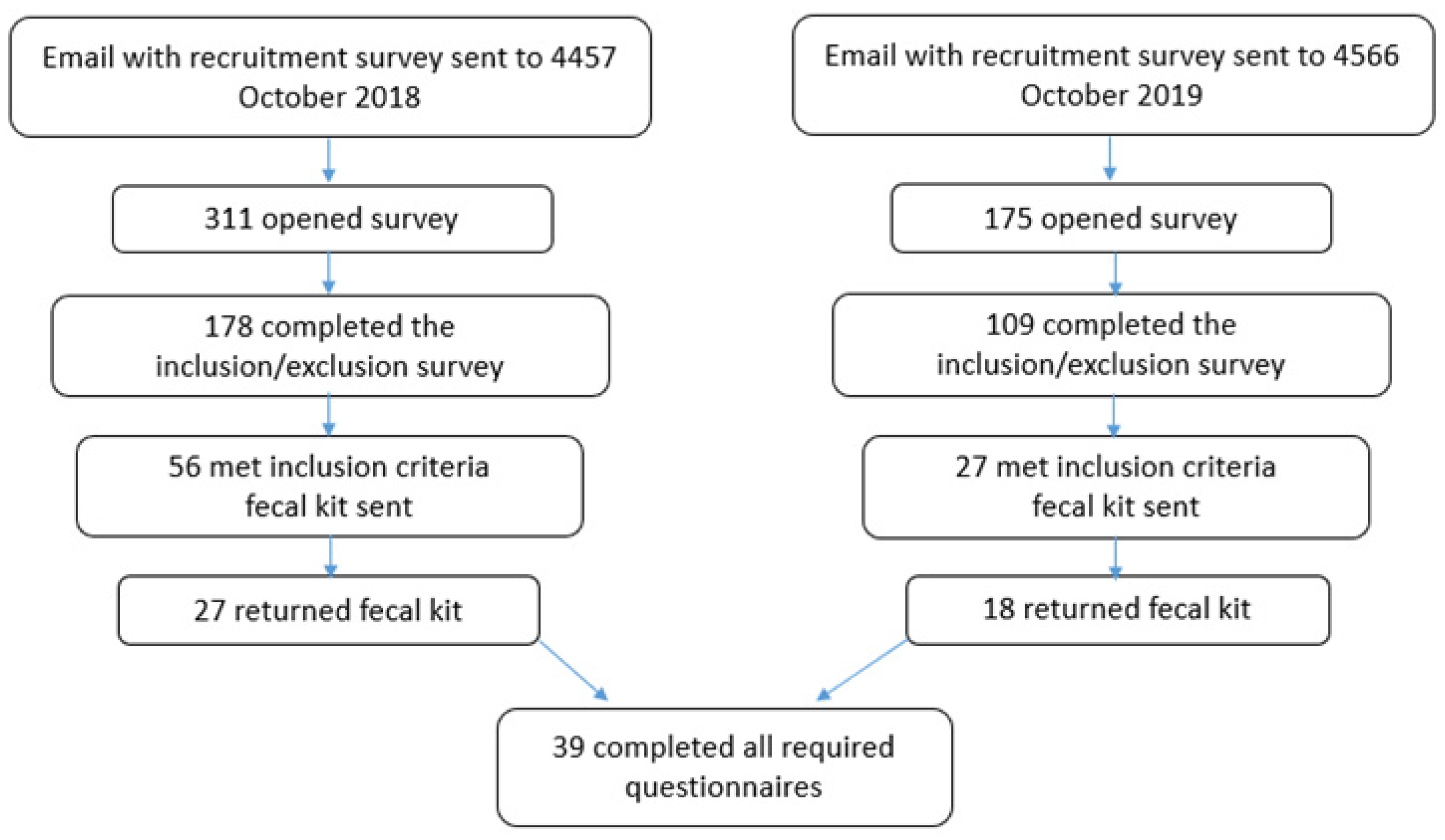
2.2. Sample Collection
2.3. Dietary Assessment
2.4. Physical Activity Assessment
2.5. Microbial Community Analysis
2.6. Prediction of Metabolic Profile
2.7. Metabolomics
2.8. Statistical Analysis
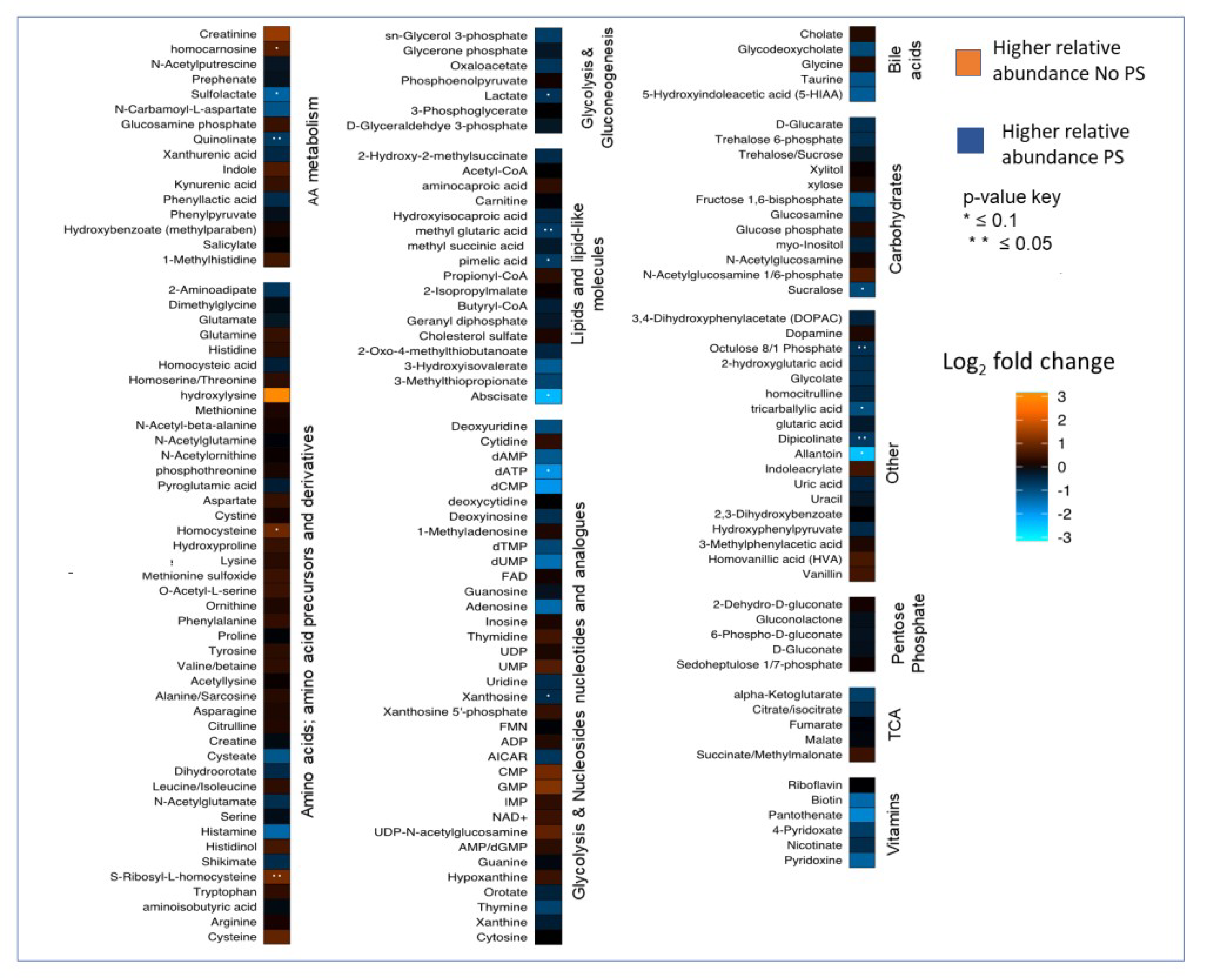
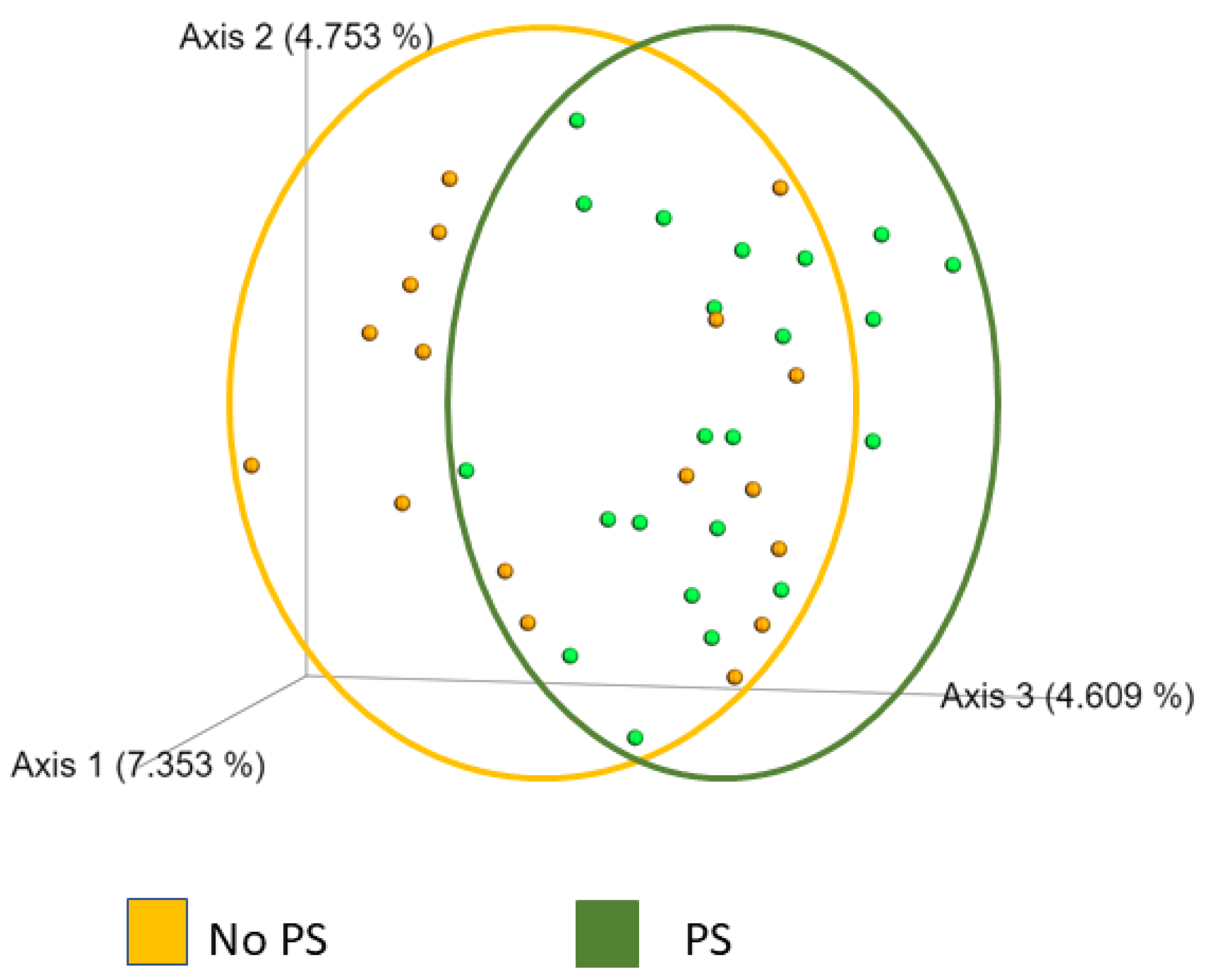
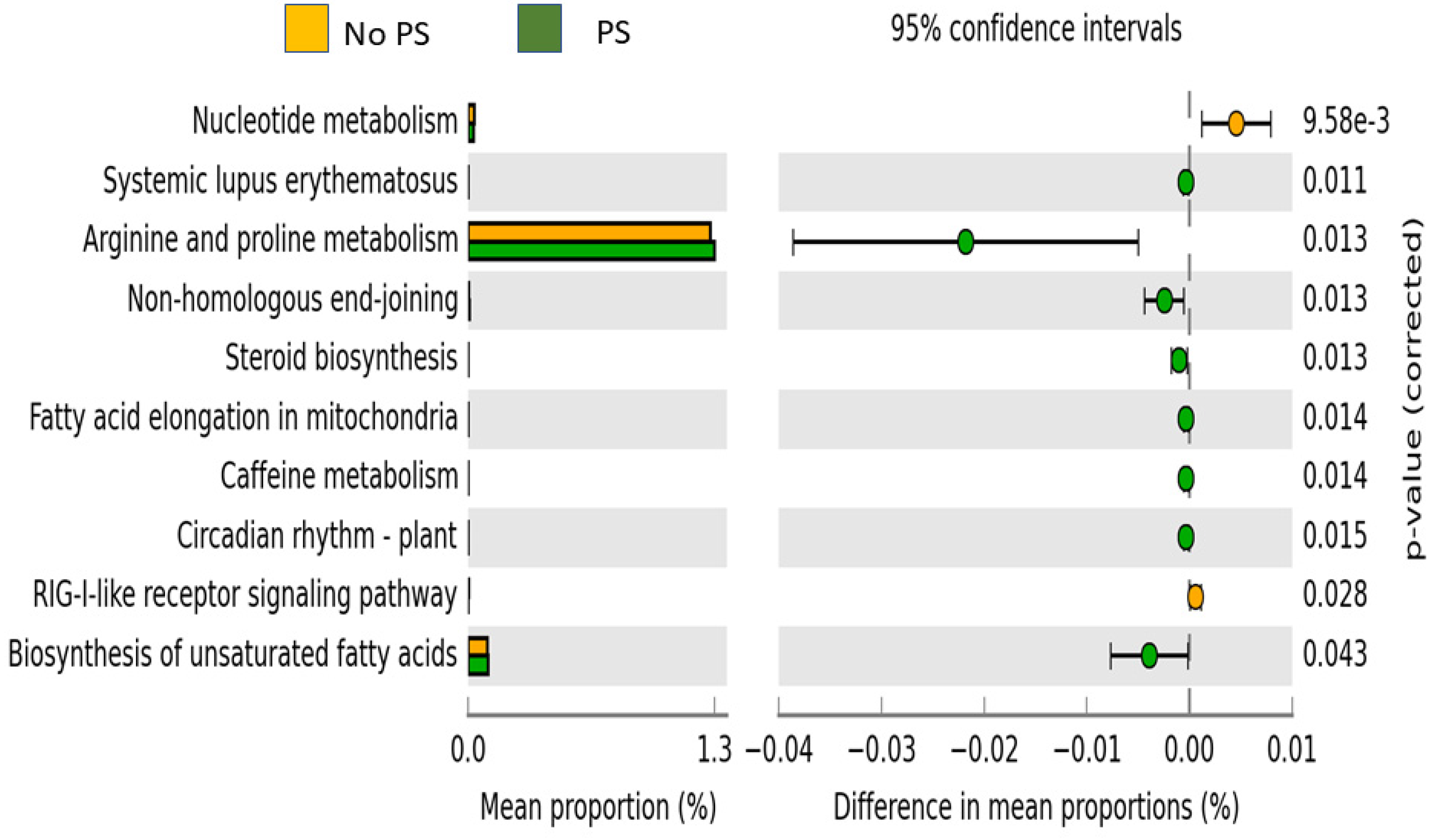
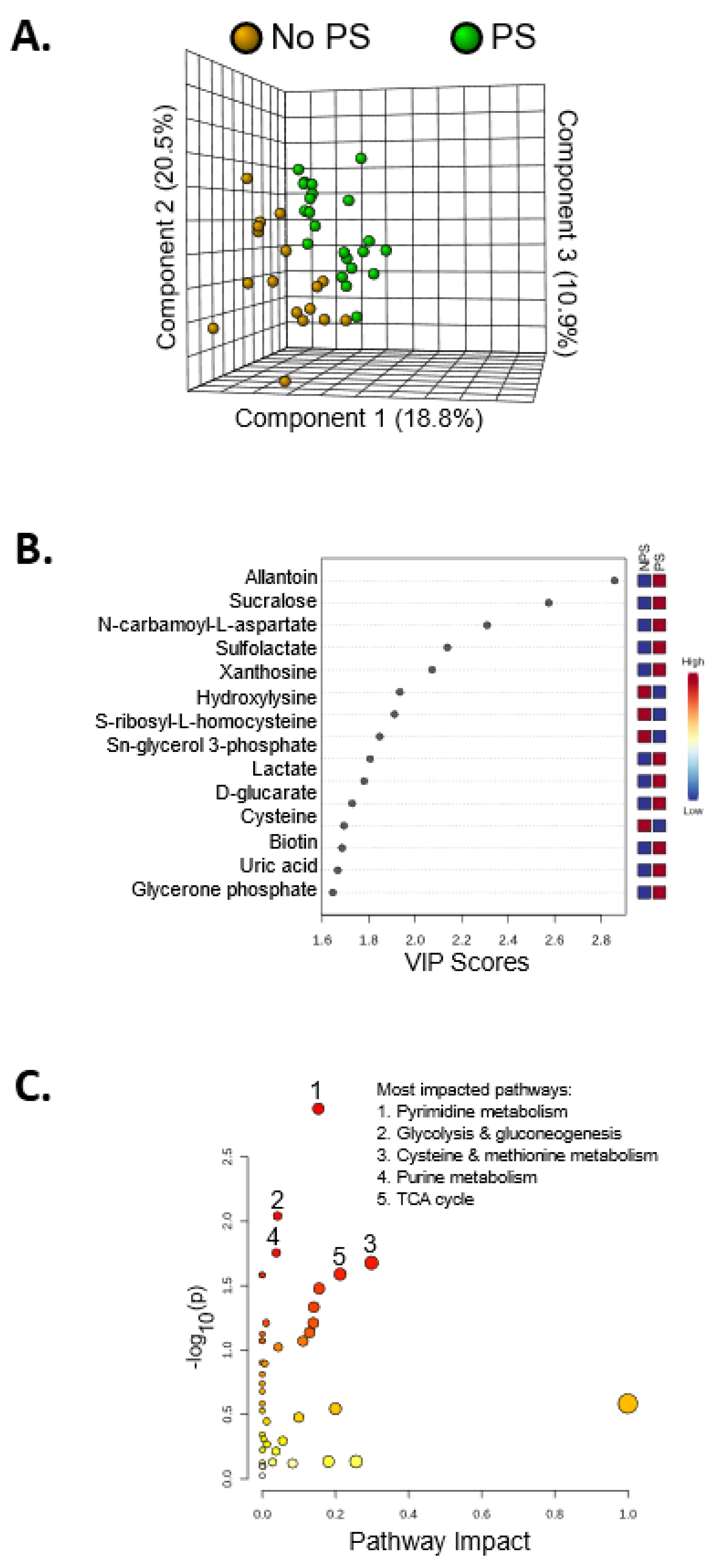
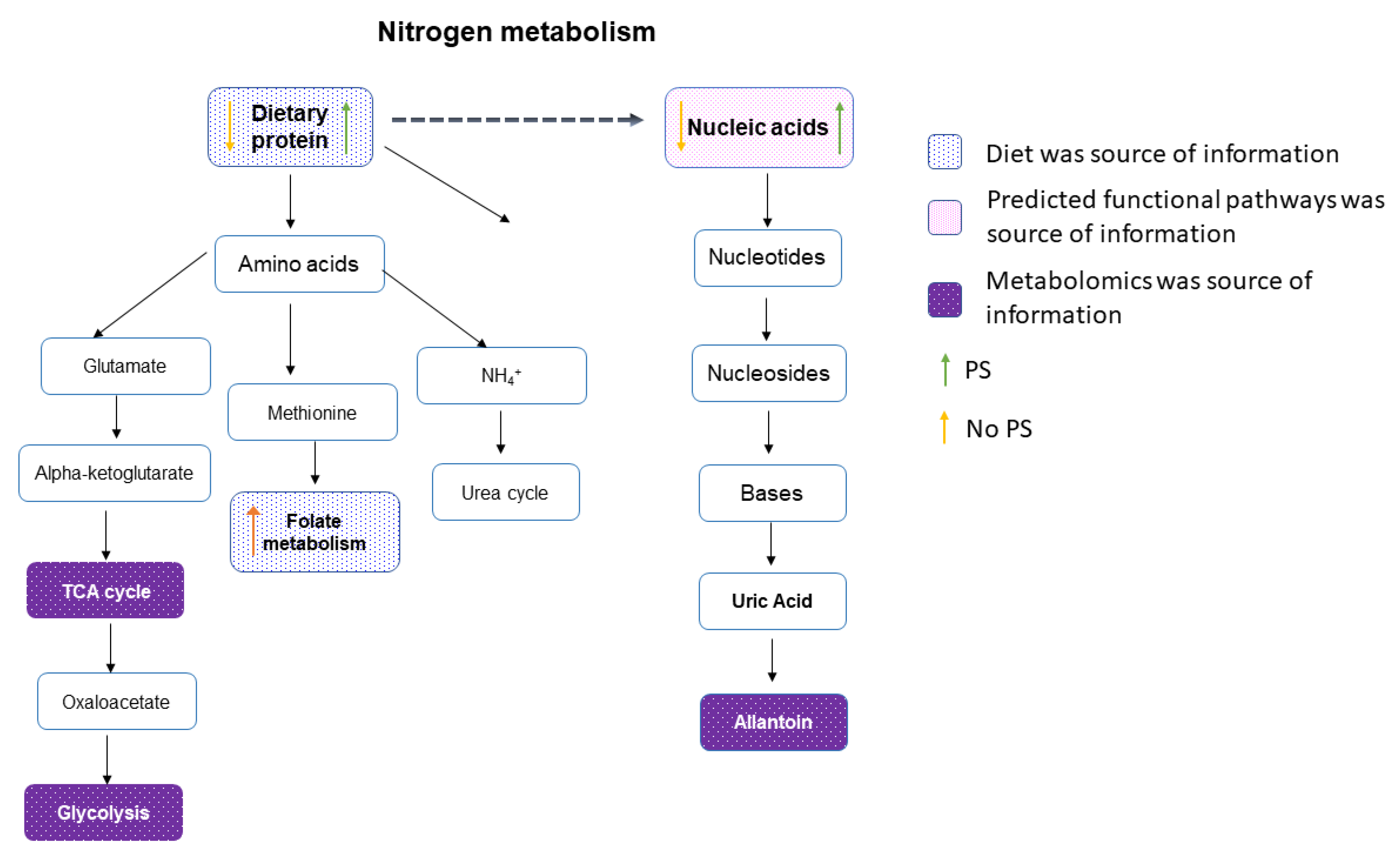
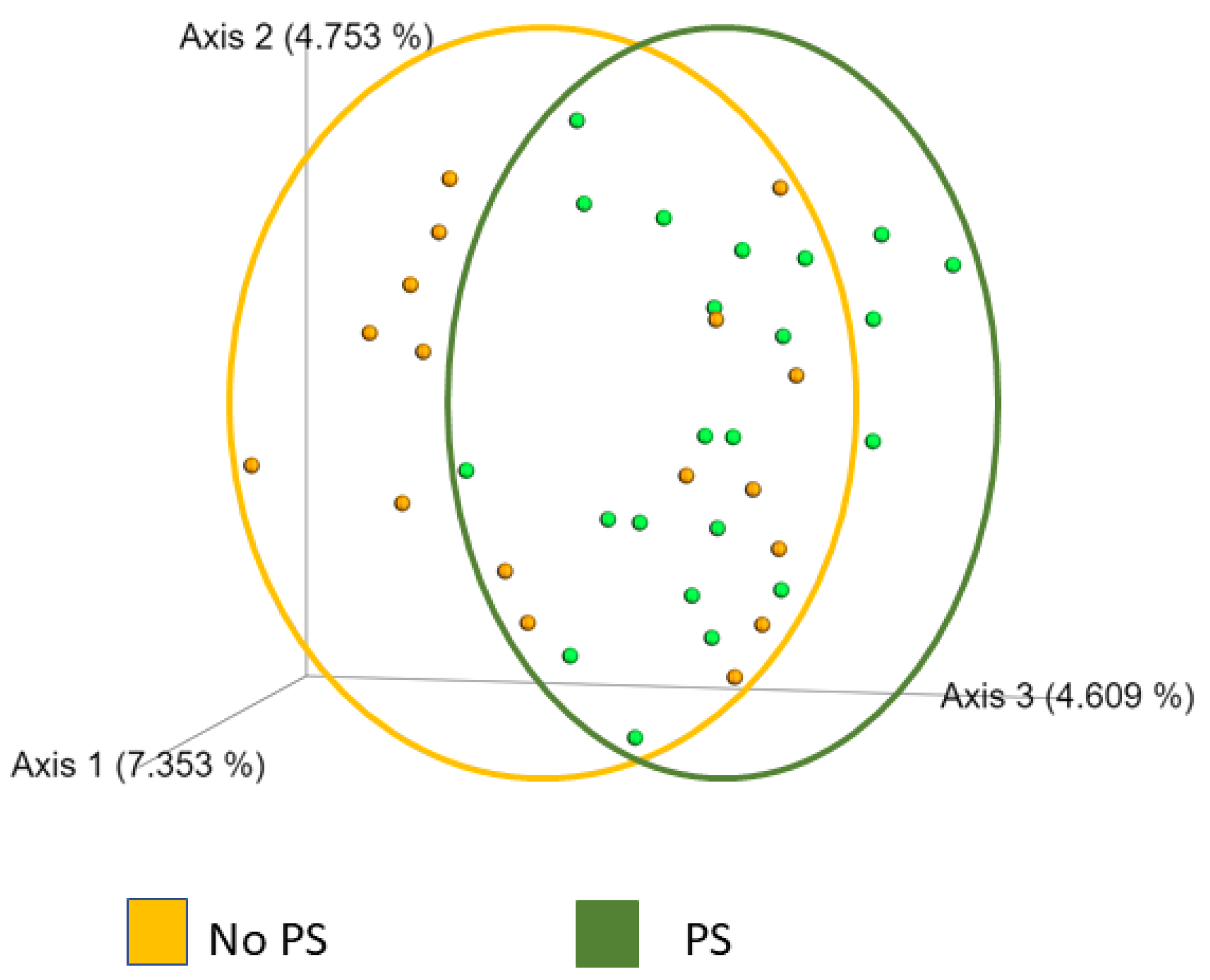
3. Results
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Garthe, I.; Maughan, R.J. Athletes and Supplements: Prevalence and Perspectives. Int. J. Sport Nutr. Exerc. Metab. 2018, 28, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, M.; Lynch, H.; Dickinson, J.M.; Reed, K.E. No Difference Between the Effects of Supplementing With Soy Protein Versus Animal Protein on Gains in Muscle Mass and Strength in Response to Resistance Exercise. Int. J. Sport Nutr. Exerc. Metab. 2018, 28, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Sears, C.L. A dynamic partnership: Celebrating our gut flora. Anaerobe 2005, 11, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Estaki, M.; Pither, J.; Baumeister, P.; Little, J.P.; Gill, S.K.; Ghosh, S.; Ahmadi-Vand, Z.; Marsden, K.R.; Gibson, D.L. Cardiorespiratory fitness as a predictor of intestinal microbial diversity and distinct metagenomic functions. Microbiome 2016, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Bycura, D.; Santos, A.C.; Shiffer, A.; Kyman, S.; Winfree, K.; Sutliffe, J.; Pearson, T.; Sonderegger, D.; Cope, E.; Caporaso, J.G. Impact of Different Exercise Modalities on the Human Gut Microbiome. Sports 2021, 9, 14. [Google Scholar] [CrossRef]
- Jang, L.G.; Choi, G.; Kim, S.W.; Kim, B.Y.; Lee, S.; Park, H. The combination of sport and sport-specific diet is associated with characteristics of gut microbiota: An observational study. J. Int. Soc. Sports Nutr. 2019, 16, 21. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef]
- Groennebaek, T.; Vissing, K. Impact of Resistance Training on Skeletal Muscle Mitochondrial Biogenesis, Content, and Function. Front. Physiol. 2017, 8, 713. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Mach, N. The Crosstalk between the Gut Microbiota and Mitochondria during Exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gao, S.; Jun, G.; Zhao, R.; Yang, X. Supplementing the maternal diet of rats with butyrate enhances mitochondrial biogenesis in the skeletal muscles of weaned offspring. Br. J. Nutr. 2017, 117, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.M.; Wei, L.; Chiu, Y.S.; Hsu, Y.J.; Tsai, T.Y.; Wang, M.F.; Huang, C.C. Lactobacillus plantarum TWK10 Supplementation Improves Exercise Performance and Increases Muscle Mass in Mice. Nutrients 2016, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Liong, M.T.; Tsai, Y.C. New perspectives of Lactobacillus plantarum as a probiotic: The gut-heart-brain axis. J. Microbiol. 2018, 56, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.W.; Murphy, K.T.; McKellar, S.R.; Schoenfeld, B.J.; Henselmans, M.; Helms, E.; Aragon, A.A.; Devries, M.C.; Banfield, L.; Krieger, J.W.; et al. A systematic review, meta-analysis and meta-regression of the effect of protein supplementation on resistance training-induced gains in muscle mass and strength in healthy adults. Br. J. Sports Med. 2018, 52, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Cronin, O.; Barton, W.; Skuse, P.; Penney, N.C.; Garcia-Perez, I.; Murphy, E.F.; Woods, T.; Nugent, H.; Fanning, A.; Melgar, S.O.; et al. A Prospective Metagenomic and Metabolomic Analysis of the Impact of Exercise and/or Whey Protein Supplementation on the Gut Microbiome of Sedentary Adults. mSystems 2018, 3, e00014-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subar, A.F.; Kirkpatrick, S.I.; Mittl, B.; Zimmerman, T.P.; Thompson, F.E.; Bingley, C.; Willis, G.; Islam, N.G.; Baranowski, T.; McNutt, S.; et al. The Automated Self-Administered 24-hour dietary recall (ASA24): A resource for researchers, clinicians, and educators from the National Cancer Institute. J. Acad. Nutr. Diet 2012, 112, 1134–1137. [Google Scholar] [CrossRef] [Green Version]
- Haff, G. Quantifying Workloads in Resistance Training: A Brief Review. Prof. Strength Cond. 2010, 10, 31–40. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshua, D.; Rabinowitz, E.K. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007, 79, 6167–6173. [Google Scholar]
- Lu, W.; Clasquin, M.F.; Melamud, E.; Amador-Noguez, D.; Caudy, A.A.; Rabinowitz, J.D. Metabolomic Analysis via Reversed-Phase Ion-Pairing Liquid Chromatography Coupled to a Stand Alone Orbitrap Mass Spectrometer. Anal. Chem. 2010, 82, 3212–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stough, J.M.A.; Dearth, S.P.; Denny, J.E.; LeCleir, G.R.; Schmidt, N.W.; Campagna, S.R.; Wilhelm, S.W. Functional Characteristics of the Gut Microbiome in C57BL/6 Mice Differentially Susceptible to Plasmodium yoelii. Front. Microbiol. 2016, 7, 1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Römpp, A.; Neumann, S.; Pizarro, A.D.; et al. mzML—a community standard for mass spectrometry data. Mol. Cell. Proteom. MCP 2011, 10, R110.000133. [Google Scholar] [CrossRef] [Green Version]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 37, 14. [Google Scholar]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic Analysis and Visualization Engine for LC−MS Data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Medicine, I.O. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids; The National Academies Press: Washington, DC, USA, 2005; p. 1358. [Google Scholar]
- Thomas, D.T.; Erdman, K.A.; Burke, L.M. Position of the Academy of Nutrition and Dietetics, Dietitians of Canada, and the American College of Sports Medicine: Nutrition and Athletic Performance. J. Acad. Nutr. Diet 2016, 116, 501–528. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.A.; Sladen, G.E.; Dawson, A.M. Protein absorption and ammonia production: The effects of dietary protein and removal of the colon. Br. J. Nutr. 1976, 35, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laitinen, K.; Mokkala, K. Overall Dietary Quality Relates to Gut Microbiota Diversity and Abundance. Int. J. Mol. Sci. 2019, 20, 1835. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Ley, R. The human gut microbiome: Ecology and recent evolutionary changes. Annu. Rev. Microbiol. 2011, 65, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Ghosh, T.S.; Kedia, S.; Rampal, R.; Saxena, S.; Bag, S.; Mitra, R.; Dayal, M.; Mehta, O.; Surendranath, A.; et al. Analysis of the Gut Microbiome of Rural and Urban Healthy Indians Living in Sea Level and High Altitude Areas. Sci. Rep. 2018, 8, 10104. [Google Scholar] [CrossRef]
- Yamauchi, T.; Oi, A.; Kosakamoto, H.; Akuzawa-Tokita, Y.; Murakami, T.; Mori, H.; Miura, M.; Obata, F. Gut Bacterial Species Distinctively Impact Host Purine Metabolites during Aging in Drosophila. iScience 2020, 23, 101477. [Google Scholar] [CrossRef]
- Hsieh, M.-W.; Chen, H.-Y.; Tsai, C.-C. Screening and Evaluation of Purine-Nucleoside-Degrading Lactic Acid Bacteria Isolated from Winemaking Byproducts In Vitro and Their Uric Acid-Lowering Effects In Vivo. Fermentation 2021, 7, 74. [Google Scholar] [CrossRef]
- Chen, Y.-R.; Zheng, H.-M.; Zhang, G.-X.; Chen, F.-L.; Chen, L.-D.; Yang, Z.-C. High Oscillospira abundance indicates constipation and low BMI in the Guangdong Gut Microbiome Project. Sci. Rep. 2020, 10, 9364. [Google Scholar] [CrossRef]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria-From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Guo, X.; Zhang, J.; Zhang, M.; Ou, Z.; Peng, Y. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp. Ther. Med. 2017, 14, 3122–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Liu, Q.; Li, H.; Wen, C.; He, Z. Alterations of the Gut Microbiome Associated With the Treatment of Hyperuricaemia in Male Rats. Front. Microbiol. 2018, 9, 2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villegas, R.; Xiang, Y.B.; Elasy, T.; Xu, W.H.; Cai, H.; Cai, Q.; Linton, M.F.; Fazio, S.; Zheng, W.; Shu, X.O. Purine-rich foods, protein intake, and the prevalence of hyperuricemia: The Shanghai Men’s Health Study. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 409–416. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
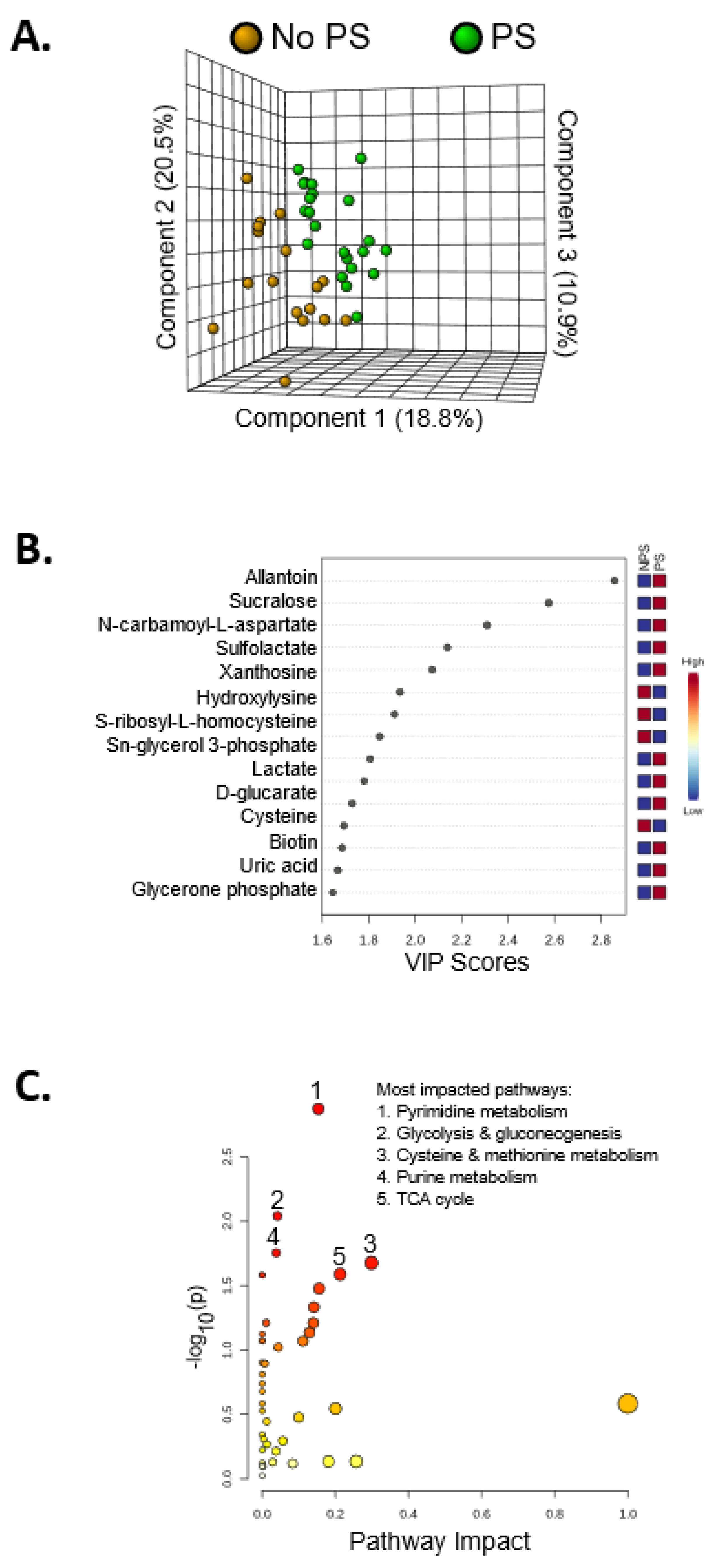{kind=link}
{kind=link}
{kind=link}
| No PS (No Protein Supplement) (n = 17) | PS (Protein Supplement) (n = 22) | p-Value | |
|---|---|---|---|
| Age (years) | 33 ± 2 1 | 32 ± 1 | 0.84 |
| Weight (lbs) | 176 ± 8 | 173 ± 7 | 0.79 |
| Height (inches) | 67 ± 1 | 67 ± 1 | 0.72 |
| BMI (kg/m2) | 27 ± 1 | 27 ± 1 | 0.99 |
| Males | 12 | 14 | |
| Females | 5 | 8 | |
| Total Physical Activity (MET-minutes/week) | 7040 ± 1282 | 12,081 ± 1870 | 0.03 |
| Total Vigorous Activity (MET-minutes/week) | 2535 ± 830 | 6331 ± 1284 | 0.02 |
| Upper Body Resistance Exercise Volume (kg/week) | 15,743 ± 13,103 | 31,067 ± 49,323 | 0.15 |
| Lower Body Resistance Exercise Volume (kg/week) | 16,694 ± 19,430 | 56,464 ± 127,594 | 0.16 |
| Bristol Scale (arbitrary units) | 3.8 ± 0.4 | 3.3 ± 0.2 | 0.25 |
| Number of Supplements (count) | 0.9 ± 1.5 | 1.4 ± 2.4 | 0.45 |
| No PS (n = 17) | PS (n = 22) | p-Value | |
|---|---|---|---|
| Number of Foods | 19 ± 2 1 | 21 ± 2 | 0.26 |
| Energy (kcal) | 2551 ± 429 | 2452 ± 199 | 0.84 |
| Protein (g) | 117.6 ± 11.8 | 169.3 ± 17.6 | 0.02 |
| Protein (g/kg body weight) | 1.49 ± 0.14 | 2.15 ± 0.19 | 0.009 |
| Calories from protein (%) | 20.1 ± 1.5 | 27.5 ± 1.7 | 0.003 |
| Carbohydrate (g) | 228.3 ± 37.9 | 239.3 ± 25.9 | 0.81 |
| Calories from carbohydrates (%) | 36.5 ± 2.9 | 39.3 ± 2.6 | 0.48 |
| Fiber (g) | 18.9 ± 2.1 | 27.3 ± 3.1 | 0.03 |
| Total Sugar (g) | 93.3 ± 20.7 | 88.1 ± 13.4 | 0.84 |
| Kcal from sugar (%) | 13.7 ± 1.7 | 14.1 ± 1.50 | 0.84 |
| Ratio of protein to carbohydrate (g:g) | 0.67 ± 0.11 | 0.89 ± 0.16 | 0.26 |
| Fat (g) | 109.0 ± 13.7 | 92.9 ± 9.0 | 0.33 |
| Total saturated fatty acids (g) | 34.1 ± 6.0 | 29.3 ± 3.2 | 0.49 |
| Total polyunsaturated fatty acids (g) | 24.8 ± 2.7 | 19.6 ± 2.1 | 0.14 |
| Total monounsaturated fatty acids (g) | 40.6 ± 5.1 | 35.6 ± 4.2 | 0.46 |
| Calories from fat (%) | 40.8 ± 2.7 | 33.9 ± 1.9 | 0.05 |
| Iron (mg) | 14.7 ± 1.5 | 19.5 ± 1.8 | 0.05 |
| Magnesium (mg) | 369 ± 30 | 533 ± 77 | 0.03 |
| Potassium (mg) | 3064 ± 303 | 3996 ± 395 | 0.07 |
| Vitamin C (mg) | 120 ± 22 | 217 ± 49 | 0.08 |
| Folate, food (mcg) | 263 ± 30 | 375 ± 56 | 0.09 |
| Intact fruits (whole or cut) of citrus, melons, and berries (cup eq.) | 0.065 ± 0.04 | 0.443 ± 0.15 | 0.02 |
| Beans and Peas (legumes) computed as protein foods (oz.eq.) | 0.132 ± 0.12 | 0.97 ± 0.4 | 0.045 |
| Beans and Peas (legumes) computed as vegetables (cup eq.) | 0.032 ± 0.03 | 0.24 ± 0.10 | 0.045 |
| Healthy Eating Index (HEI) | 54.0 ± 13.3 | 61.8 ± 15.1 | 0.088 |
| Water (g) | 3914 ± 304 | 4266 ± 441 | 0.51 |
| Alcohol (g) (14 g = 1 standard drink) | 31.4 ± 25.8 | 1.92 ± 1.1 | 0.27 |
| Caffeine (mg) | 180.3 ± 42.3 | 156.1 ± 28.5 | 0.64 |
| Bacteria | No PS | PS | Association with Dietary Protein | Association with Dietary Fat | |||
|---|---|---|---|---|---|---|---|
| Relative Abundance | p-Value | Correlation Coefficient | p-value | Correlation Coefficient | p-Value | ||
| p__Actinobacteria | |||||||
| c__Coriobacteriia | 94.9 ± 33.6 1 | 565.5 ± 158.0 | 0.008 | 0.332 | 0.039 | 0.513 | 0.0008 |
| c__Coriobacteriia;o__Coriobacteriales | 94.9 ± 33.6 | 565.5 ± 158.0 | 0.008 | 0.332 | 0.039 | 0.513 | 0.0008 |
| c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae | 94.9 ± 33.6 | 565.5 ± 158.0 | 0.008 | 0.332 | 0.039 | 0.513 | 0.0008 |
| c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Adlercreutzia | 4.3 ± 2.1 | 54.3 ± 14.1 | 0.002 | ||||
| p__Bacteroidetes | |||||||
| c__Bacteroidia | 8923.1 ± 1941.1 | 16,425.5 ± 1362.1 | 0.004 | 0.333 | 0.038 | 0.393 | 0.013 |
| c__Bacteroidia;o__Bacteroidales | 8923.1 ± 1941.1 | 16,425.5 ± 1362.1 | 0.004 | 0.333 | 0.038 | 0.393 | 0.013 |
| c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides;__ | 181.6 ± 45.4 | 438.4 ± 81.4 | 0.0096 | ||||
| c__Bacteroidia;o__Bacteroidales;f__Rikenellaceae | 857.8 ± 269.3 | 2083.7 ± 265.7 | 0.003 | ||||
| c__Bacteroidia;o__Bacteroidales;f__Rikenellaceae;g__ | 850.2 ± 267.7 | 2068.4 ± 263.4 | 0.0026 | ||||
| p__Firmicutes | |||||||
| c__Bacilli | 30.5 ± 6.8 | 195.3 ± 42.3 | 0.0009 | ||||
| c__Bacilli;o__Lactobacillales | 22.6 ± 4.9 | 136.5 ± 38.1 | 0.007 | ||||
| c__Bacilli;o__Turicibacterales | 7.1 ± 4.5 | 58.5 ± 17.6 | 0.009 | ||||
| c__Bacilli;o__Turicibacterales;f__Turicibacteraceae | 7.1 ± 4.5 | 58.5 ± 17.6 | 0.009 | ||||
| c__Bacilli;o__Turicibacterales;f__Turicibacteraceae;g__Turicibacter | 7.1 ± 4.5 | 58.5 ± 17.6 | 0.0094 | ||||
| c__Clostridia | 6433.2 ± 1303.2 | 16,415.6 ± 1526.5 | 0.00002 | 0.413 | 0.009 | ||
| c__Clostridia;o__Clostridiales | 6431.3 ± 1303.0 | 16,403.7 ± 1522.5 | 0.00002 | 0.414 | 0.009 | ||
| c__Clostridia;o__Clostridiales;f__ | 242.7 ± 74.3 | 739.8 ± 153.0 | 0.007 | 0.322 | 0.046 | 0.462 | 0.003 |
| c__Clostridia;o__Clostridiales;f__;g__ | 242.7 ± 74.3 | 739.8 ± 153.0 | 0.0066 | 0.322 | 0.046 | 0.462 | 0.003 |
| c__Clostridia;o__Clostridiales;f__Clostridiaceae | 97.8 ± 23.3 | 250.4 ± 47.2 | 0.007 | ||||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae | 2960.9 ± 639.1 | 7179.0 ± 686.7 | 0.0001 | 0.382 | 0.016 | ||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__ | 665.6 ± 147.1 | 1401.9 ± 118.5 | 0.0004 | 0.344 | 0.032 | ||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__[Ruminococcus] | 190.7 ± 42.5 | 539.7 ± 80.0 | 0.0005 | 0.380 | 0.017 | ||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__[Ruminococcus];s__torques | 13.7 ± 6.4 | 110 ± 32.7 | 0.008 | ||||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Anaerostipes | 17.2 ± 4.9 | 56.4 ± 12.4 | 0.0066 | ||||
| c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Coprococcus | 371 ± 123 | 1238.5 ± 191.7 | 0.0006 | ||||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae | 2328.5 ± 565.8 | 6663.8 ± 732.5 | 0.00004 | 0.373 | 0.019 | ||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae;__ | 55.9 ± 10.8 | 373.2 ± 75.4 | 0.0004 | ||||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__ | 259.7 ± 74.4 | 798.3 ± 178.9 | 0.0097 | 0.443 | 0.005 | ||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__Faecalibacterium | 1372.5 ± 450.4 | 3488.4 ± 257.8 | 0.0004 | ||||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__Oscillospira | 318.6 ± 73.6 | 822.5 ± 111.8 | 0.0006 | 0.445 | 0.005 | ||
| c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__Ruminococcus | 314 ± 84 | 1167.9 ± 275.1 | 0.0065 | ||||
| c__Clostridia;o__Clostridiales;f__Veillonellaceae | 569.2 ± 122.6 | 1127.6 ± 147.1 | 0.00600 | 0.345 | 0.031 | 0.362 | 0.024 |
| c__Clostridia;o__Clostridiales;f__Veillonellaceae;g__Phascolarctobacterium | 171.1 ± 78.3 | 746.2 ± 151.8 | 0.0021 | 0.518 | 0.0007 | ||
| c__Clostridia;o__Clostridiales;f__Veillonellaceae;g__Phascolarctobacterium;s__ | 205.5 ± 79.7 | 719.7 ± 155.8 | 0.0062 | 0.518 | 0.0007 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byerley, L.O.; Gallivan, K.M.; Christopher, C.J.; Taylor, C.M.; Luo, M.; Dowd, S.E.; Davis, G.M.; Castro, H.F.; Campagna, S.R.; Ondrak, K.S. Gut Microbiome and Metabolome Variations in Self-Identified Muscle Builders Who Report Using Protein Supplements. Nutrients 2022, 14, 533. https://doi.org/10.3390/nu14030533
Byerley LO, Gallivan KM, Christopher CJ, Taylor CM, Luo M, Dowd SE, Davis GM, Castro HF, Campagna SR, Ondrak KS. Gut Microbiome and Metabolome Variations in Self-Identified Muscle Builders Who Report Using Protein Supplements. Nutrients. 2022; 14(3):533. https://doi.org/10.3390/nu14030533
Chicago/Turabian StyleByerley, Lauri O., Karyn M. Gallivan, Courtney J. Christopher, Christopher M. Taylor, Meng Luo, Scot E. Dowd, Gregory M. Davis, Hector F. Castro, Shawn R. Campagna, and Kristin S. Ondrak. 2022. "Gut Microbiome and Metabolome Variations in Self-Identified Muscle Builders Who Report Using Protein Supplements" Nutrients 14, no. 3: 533. https://doi.org/10.3390/nu14030533
APA StyleByerley, L. O., Gallivan, K. M., Christopher, C. J., Taylor, C. M., Luo, M., Dowd, S. E., Davis, G. M., Castro, H. F., Campagna, S. R., & Ondrak, K. S. (2022). Gut Microbiome and Metabolome Variations in Self-Identified Muscle Builders Who Report Using Protein Supplements. Nutrients, 14(3), 533. https://doi.org/10.3390/nu14030533