
Resistant Starch as a Dietary Intervention to Limit the Progression of Diabetic Kidney Disease



Abstract
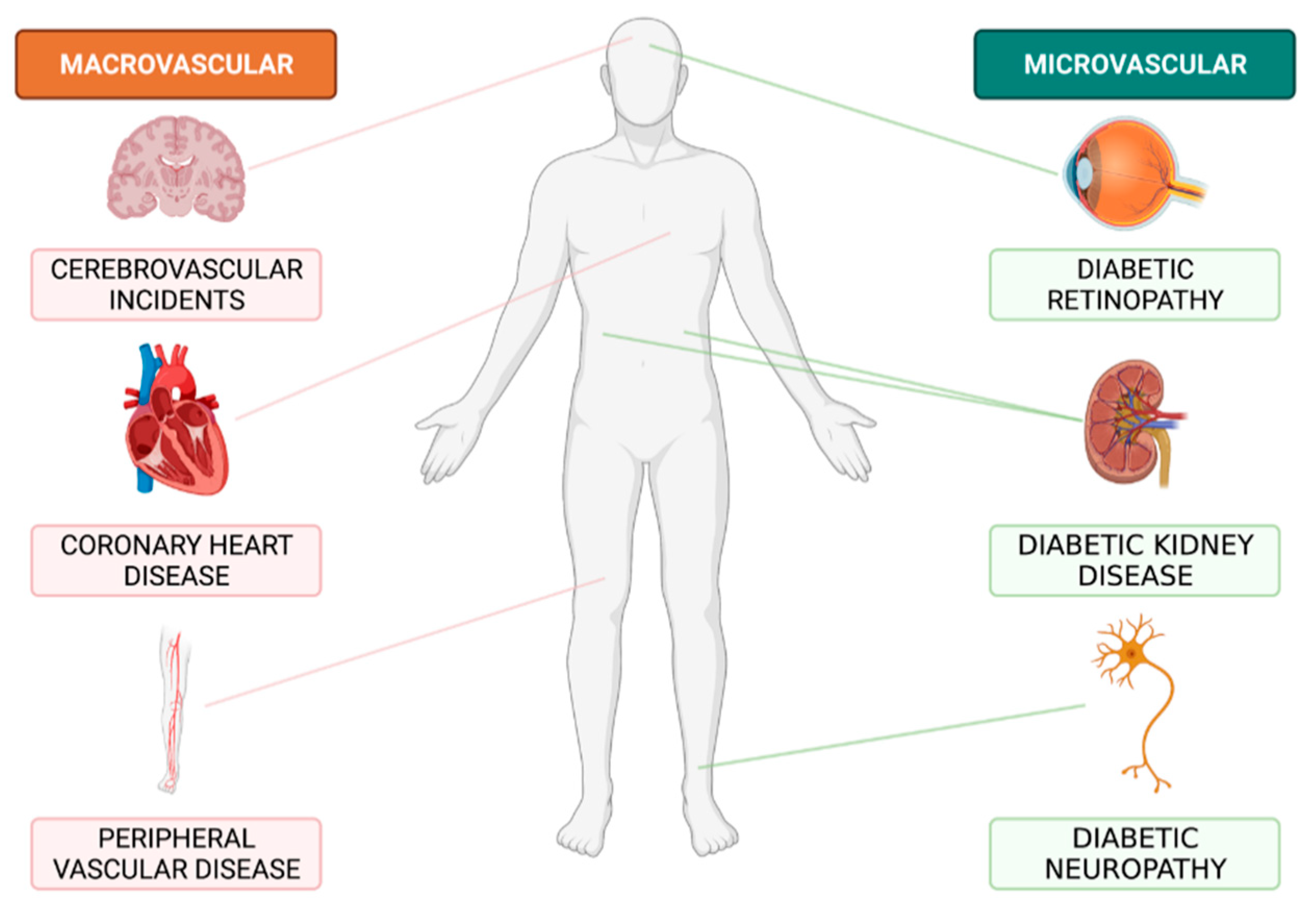
1. Introduction
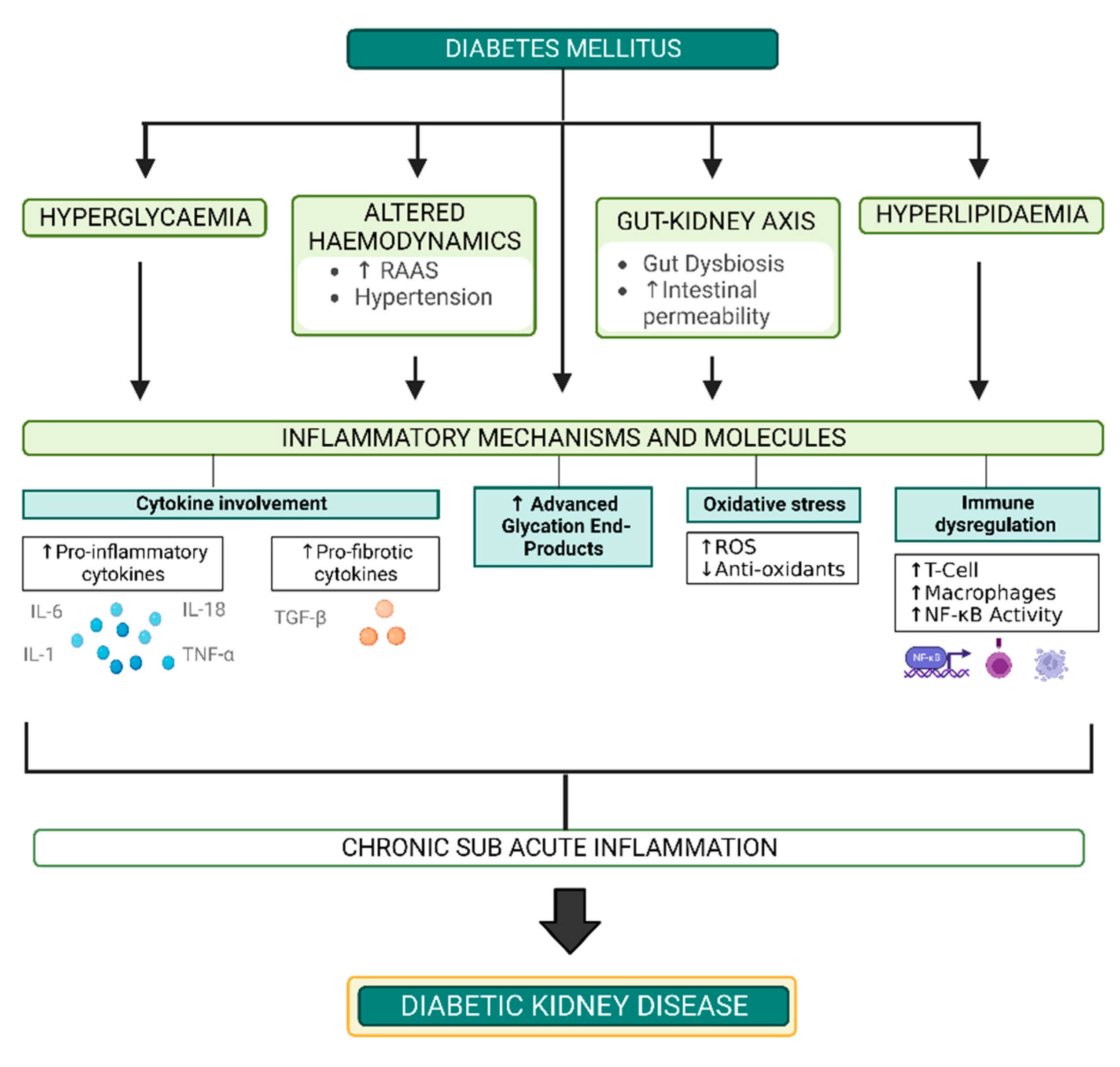
2. Pathogenesis of DKD
2.1. Hyperglycaemia
2.2. Haemodynamic Factors
2.3. Inflammatory and Immune Mechanisms
2.4. Oxidative Stress
2.5. Advanced Glycation Endproducts
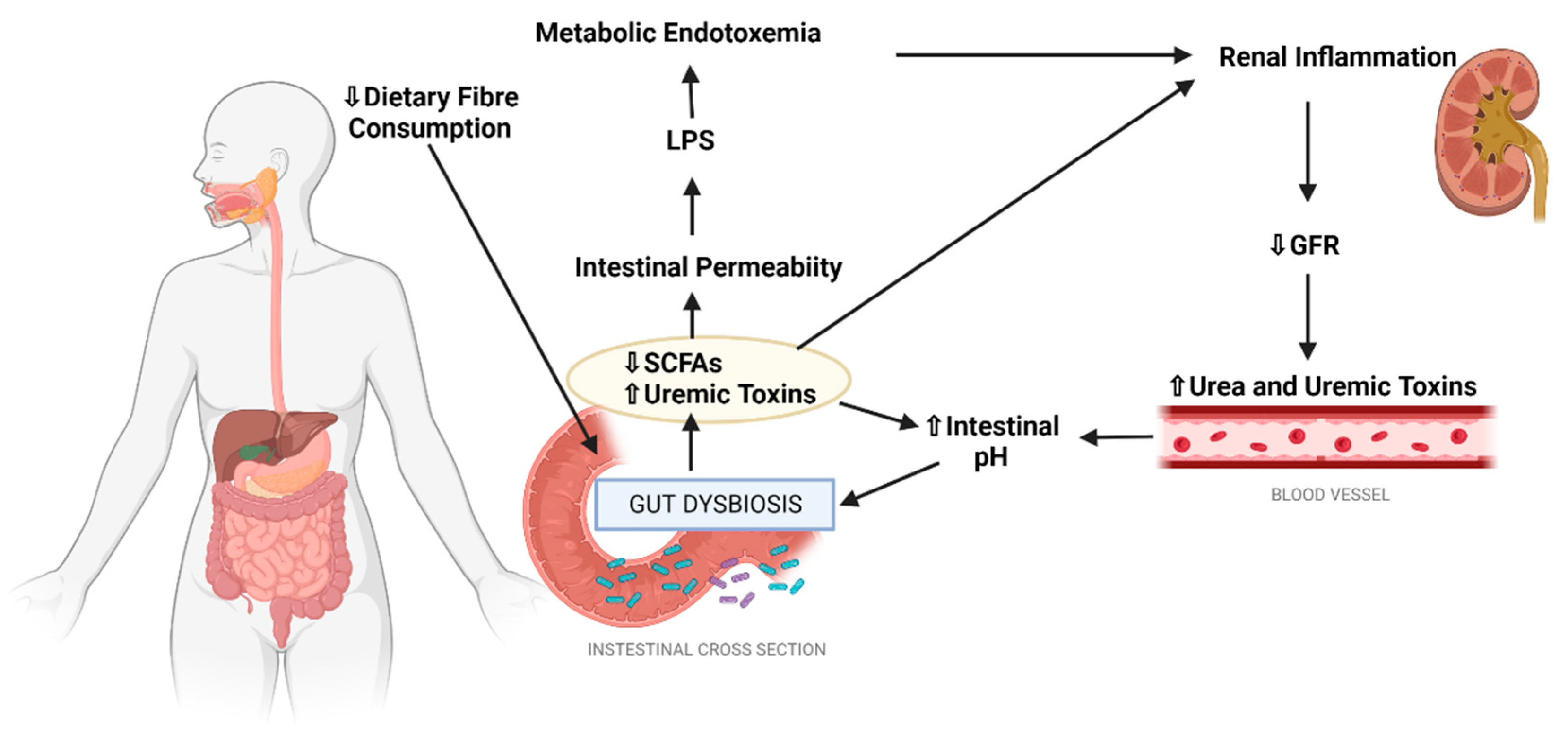
3. The Gut-Kidney Axis
3.1. The Gut Microbiome and DKD
3.2. Intestinal Barrier Disruption and DKD
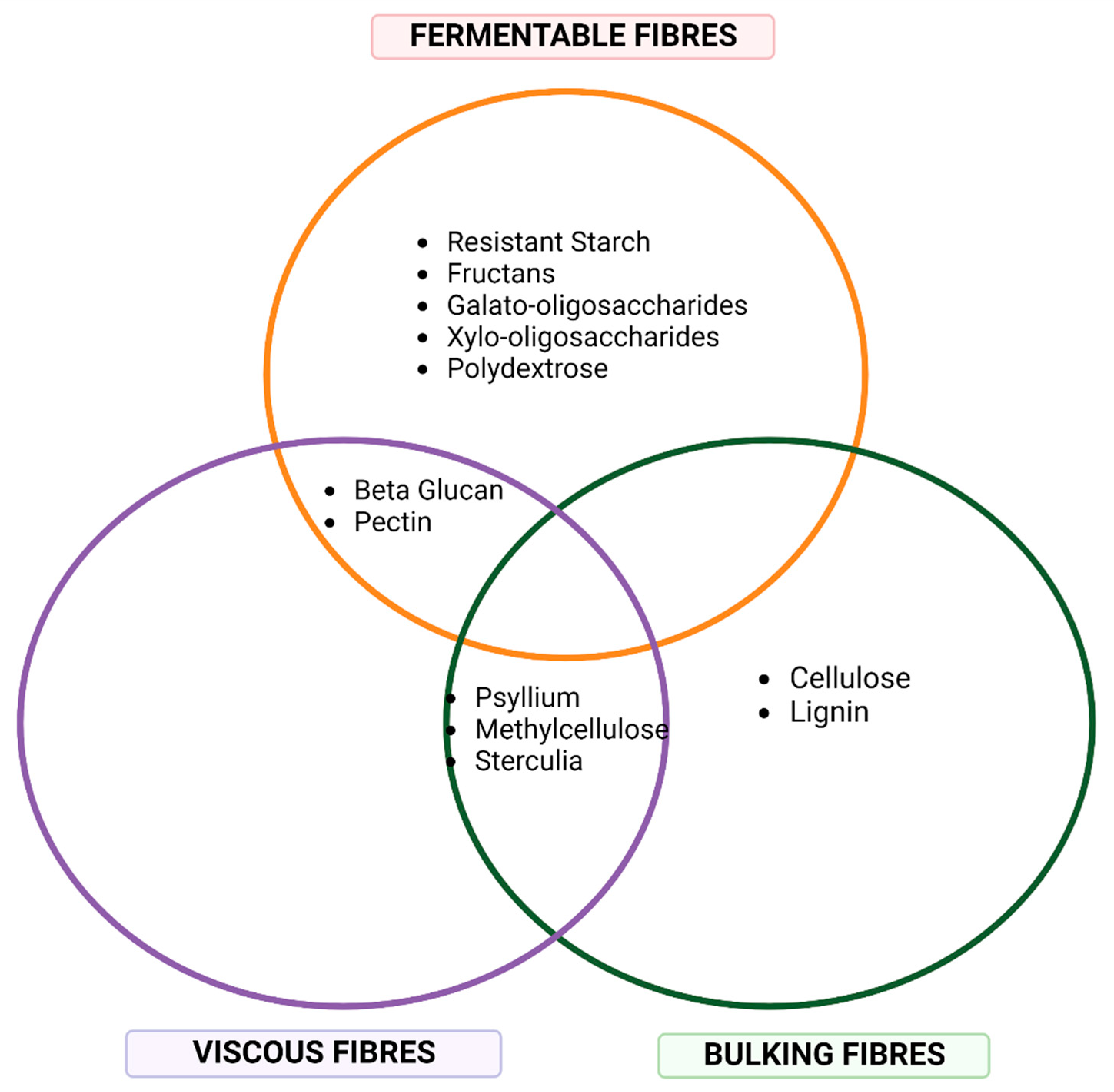
4. Dietary Fibre
4.1. Resistant Starch
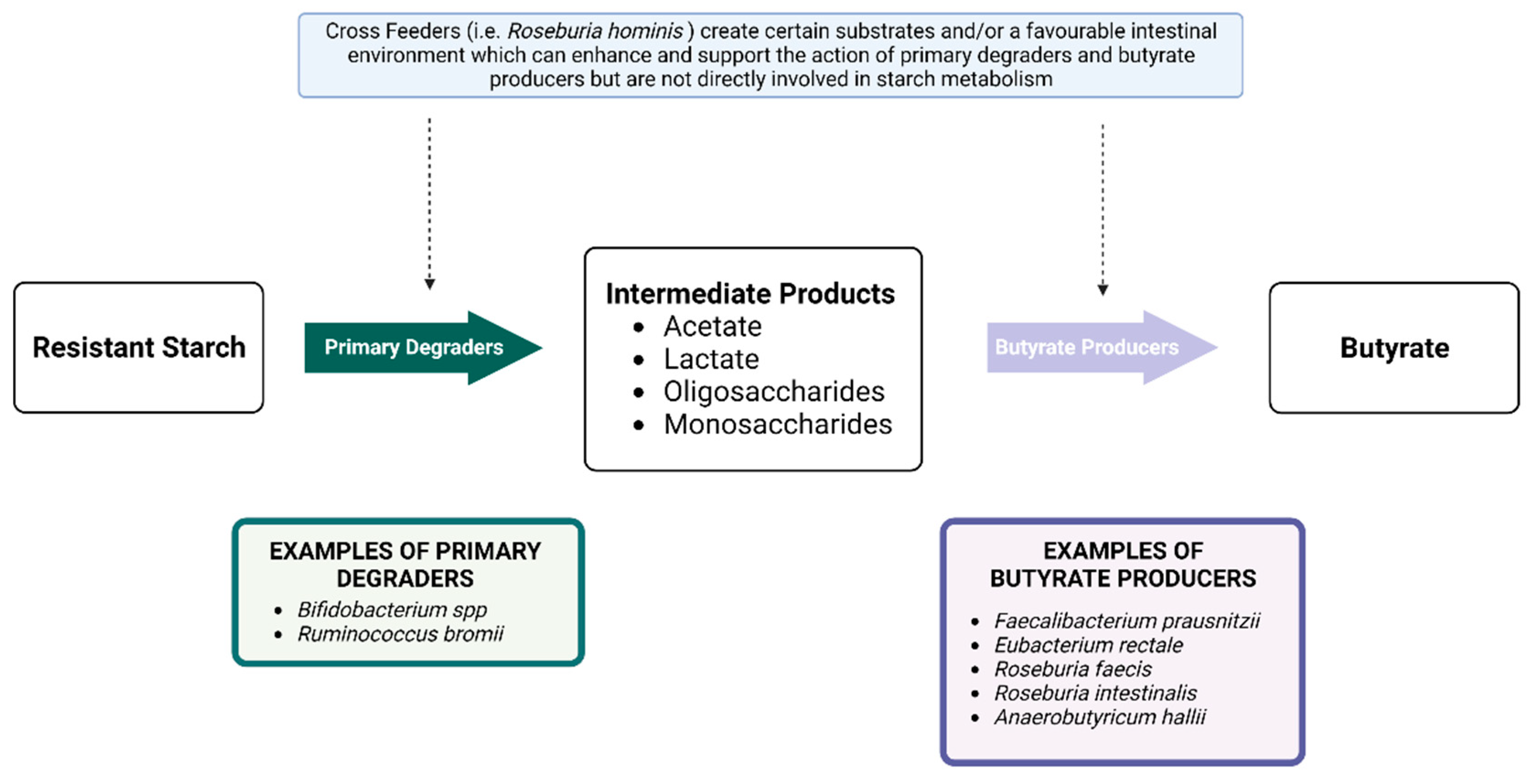
4.1.1. Resistant Starch and the Gut Microbiota
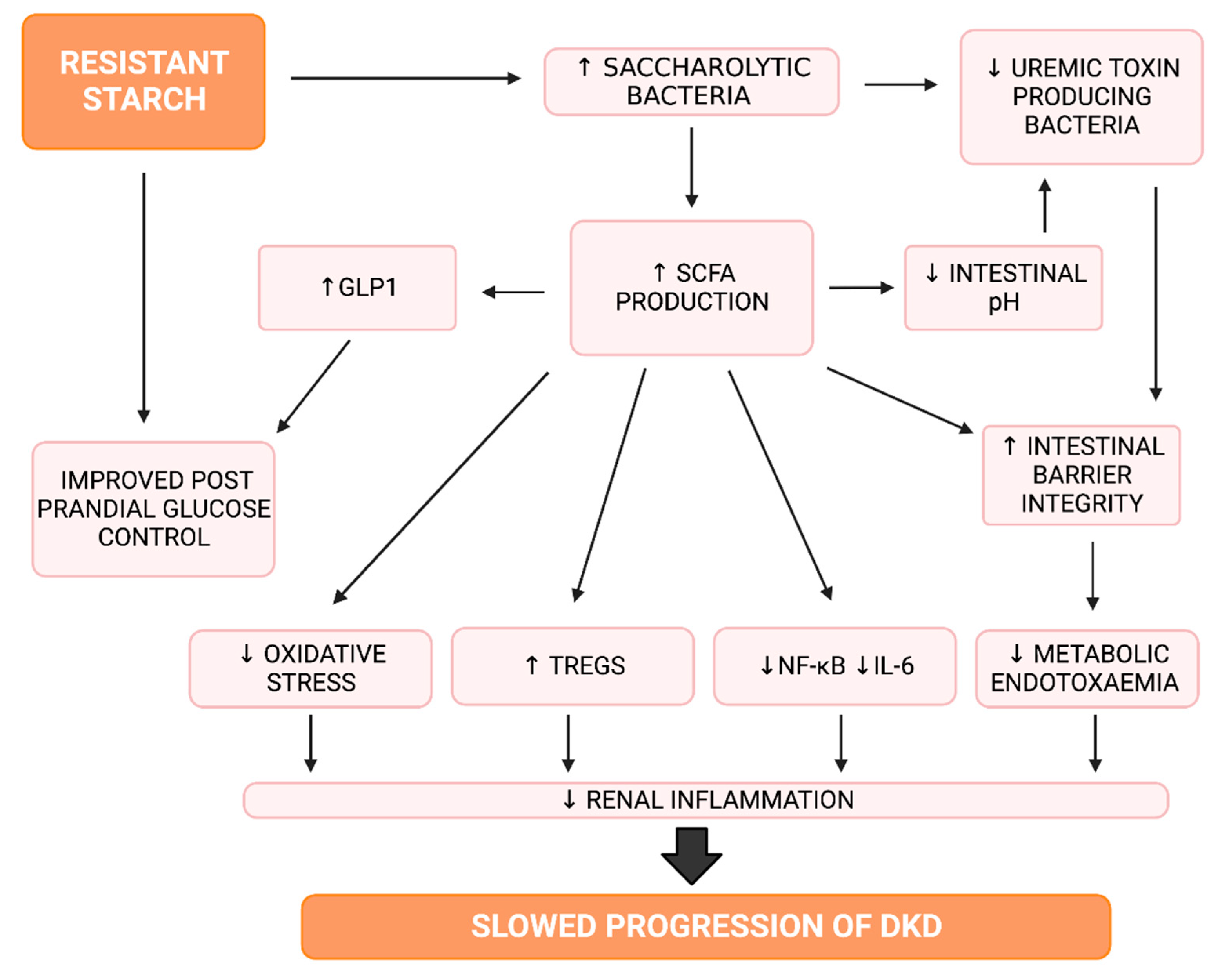
4.1.2. Resistant Starch and Short Chain Fatty Acids
4.1.3. Resistant Starch, Inflammation and Oxidative Stress
4.1.4. Resistant Starch and Glucose Control
4.1.5. Factors Influencing the Effects of Resistant Starch
4.2. Resistant Starch and Diabetic Kidney Disease—Animal Models
4.2.1. Resistant Starch in T2DM Models
4.2.2. Resistant Starch in T1DM Models
4.2.3. Resistant Starch in CKD Models
4.3. Resistant Starch and Diabetic Kidney Disease—Clinical Trials
4.3.1. Resistant Starch in Early Stage DKD
4.3.2. Resistant Starch in End Stage Kidney Disease
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Magliano, D.J.; Boyko, E.J.; Balkau, B.; Barengo, N.; Barr, E.; Basit, A.; Bhata, D.; Bommer, C.; Booth, G.; Cariou, B.; et al. IDF Diabetes Atlas, 10th ed. 2022. Available online: https://diabetesatlas.org/atlas/tenth-edition (accessed on 15 March 2022).
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Prim. 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Thornton Snider, J.; Sullivan, J.; van Eijndhoven, E.; Hansen, M.K.; Bellosillo, N.; Neslusan, C.; O’Brien, E.; Riley, R.; Seabury, S.; Kasiske, B.L. Lifetime benefits of early detection and treatment of diabetic kidney disease. PLoS ONE 2019, 14, e0217487. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- White, S.; Chadban, S. Diabetic kidney disease in Australia: Current burden and future projections. Nephrology 2014, 19, 450–458. [Google Scholar] [CrossRef]
- Meng, Y.; Bai, H.; Yu, Q.; Yan, J.; Zhao, L.; Wang, S.; Li, Z.; Wang, Q.; Chen, L. High-Resistant Starch, Low-Protein Flour Intervention on Patients With Early Type 2 Diabetic Nephropathy: A Randomized Trial. J. Ren. Nutr. 2019, 29, 386–393. [Google Scholar] [CrossRef]
- De Boer, I.H.; Rue, T.C.; Hall, Y.N.; Heagerty, P.J.; Weiss, N.S.; Himmelfarb, J. Temporal Trends in the Prevalence of Diabetic Kidney Disease in the United States. JAMA 2011, 305, 2532–2539. [Google Scholar] [CrossRef]
- AIHW. Chronic Kidney Disease. Available online: https://www.aihw.gov.au/reports/chronic-kidney-disease/chronic-kidney-disease-compendium/contents/how-many-australians-have-chronic-kidney-disease (accessed on 17 March 2022).
- Lim, H.; Johnson, D.W.; Hawley, C.; Lok, C. Type 2 diabetes in patients with end-stage kidney disease: Influence on cardiovascular disease-related mortality risk. Med. J. Aust. 2018, 209, 440–446. [Google Scholar] [CrossRef]
- Stewart, J.H.; McCredie, M.R.; McDonald, S.P. The incidence of treated end-stage renal disease in New Zealand Maori and Pacific Island people and in Indigenous Australians. Nephrol. Dial. Transpl. 2004, 19, 678–685. [Google Scholar] [CrossRef]
- AIHW. Kidney Disease. Available online: https://www.indigenoushpf.gov.au/measures/1-10-kidney-disease (accessed on 17 March 2022).
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Zac-Varghese, S.; Winocour, P. Managing diabetic kidney disease. Br. Med. Bull. 2018, 125, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lee, K.; Ni, Z.; He, J.C. Diabetic Kidney Disease: Challenges, Advances, and Opportunities. Kidney Dis. 2020, 6, 215–225. [Google Scholar] [CrossRef]
- Fernandes, R.; Viana, S.D.; Nunes, S.; Reis, F. Diabetic gut microbiota dysbiosis as an inflammaging and immunosenescence condition that fosters progression of retinopathy and nephropathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1876–1897. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Iatcu, C.O.; Steen, A.; Covasa, M. Gut Microbiota and Complications of Type-2 Diabetes. Nutrients 2021, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, R.J.; Jerums, G.; Ekinci, E.I. Effects of glycaemic management on diabetic kidney disease. World J. Diabetes 2017, 8, 172–186. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Toth-Manikowski, S.; Atta, M. Diabetic Kidney Disease: Pathophysiology and Therapeutic Target. J. Diabetes Res. 2015, 2015, 697010. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Farrington, K. Oxidative stress in early diabetic nephropathy: Fueling the fire. Nat. Rev. Endocrinol. 2011, 7, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.D.; Weigert, C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Usui, H.K.; Sharma, K. Regulation of transforming growth factor beta in diabetic nephropathy: Implications for treatment. Semin. Nephrol. 2007, 27, 153–160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, W.; Cao, R.; He, Y.C.; Guan, Y.F.; Ruan, X.Z. Crosstalk of Hyperglycemia and Dyslipidemia in Diabetic Kidney Disease. Kidney Dis. 2017, 3, 171–180. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Cheng, H.; Harris, R.C. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 22–33. [Google Scholar] [CrossRef]
- Lozano-Maneiro, L.; Puente-García, A. Renin-Angiotensin-Aldosterone System Blockade in Diabetic Nephropathy. Present Evidences. J. Clin. Med. 2015, 4, 1908–1937. [Google Scholar] [CrossRef]
- Sachse, A.; Wolf, G. Angiotensin II-induced reactive oxygen species and the kidney. J. Am. Soc. Nephrol. 2007, 18, 2439–2446. [Google Scholar] [CrossRef]
- Więcek, A.; Chudek, J.; Kokot, F. Role of angiotensin II in the progression of diabetic nephropathy—Therapeutic implications. Nephrol. Dial. Transplant. 2003, 18, v16–v20. [Google Scholar] [CrossRef]
- Singh, R.; Alavi, N.; Singh, A.K.; Leehey, D.J. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes 1999, 48, 2066–2073. [Google Scholar] [CrossRef]
- Rüster, C.; Wolf, G. Renin-angiotensin-aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.K.; Thaiss, F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors11See Editorial by Luft, p. 2272. Kidney Int. 2002, 61, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Garcia-Perez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Snelson, M.; Østergaard, J.A.; Coughlan, M.T. The Complement Pathway: New Insights into Immunometabolic Signaling in Diabetic Kidney Disease. Antioxid. Redox Signal. 2022, 37, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Ziemann, M.; Thallas-Bonke, V.; Snelson, M.; Kumar, V.; Laskowski, A.; Nguyen, T.-V.; Huynh, K.; Clarke, M.V.; Libianto, R.; et al. Complement C5a Induces Renal Injury in Diabetic Kidney Disease by Disrupting Mitochondrial Metabolic Agility. Diabetes 2019, 69, 83–98. [Google Scholar] [CrossRef]
- Sanghavi, S.F.; Roark, T.; Zelnick, L.R.; Najafian, B.; Andeen, N.K.; Alpers, C.E.; Pichler, R.; Ayers, E.; de Boer, I.H. Histopathologic and Clinical Features in Patients with Diabetes and Kidney Disease. Kidney360 2020, 1, 1217–1225. [Google Scholar] [CrossRef]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of Urease- and Uricase-Containing, Indole- and p-Cresol-Forming and Contraction of Short-Chain Fatty Acid-Producing Intestinal Microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef]
- Stavropoulou, E.; Kantartzi, K.; Tsigalou, C.; Konstantinidis, T.; Romanidou, G.; Voidarou, C.; Bezirtzoglou, E. Focus on the Gut-Kidney Axis in Health and Disease. Front. Med. 2020, 7, 620102. [Google Scholar] [CrossRef]
- Snelson, M.; de Pasquale, C.; Ekinci, E.I.; Coughlan, M.T. Gut microbiome, prebiotics, intestinal permeability and diabetes complications. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101507. [Google Scholar] [CrossRef]
- Tayebi Khosroshahi, H.; Vaziri, N.D.; Abedi, B.; Asl, B.H.; Ghojazadeh, M.; Jing, W.; Vatankhah, A.M. Effect of high amylose resistant starch (HAM-RS2) supplementation on biomarkers of inflammation and oxidative stress in hemodialysis patients: A randomized clinical trial. Hemodial. Int. 2018, 22, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kinsey, G.R. Regulatory T cells in acute and chronic kidney diseases. Am. J. Physiol. Ren. Physiol. 2018, 314, F679–F698. [Google Scholar] [CrossRef] [PubMed]
- Snelson, M.; Kellow, N.J.; Coughlan, M.T. Modulation of the Gut Microbiota by Resistant Starch as a Treatment of Chronic Kidney Diseases: Evidence of Efficacy and Mechanistic Insights. Adv. Nutr. 2019, 10, 303–320. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef]
- Wu, X.-Q.; Zhang, D.-D.; Wang, Y.-N.; Tan, Y.-Q.; Yu, X.-Y.; Zhao, Y.-Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic. Biol. Med. 2021, 171, 260–271. [Google Scholar] [CrossRef]
- Snelson, M.; Tan Sih, M.; Clarke Rachel, E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.-V.; Penfold Sally, A.; Harcourt Brooke, E.; Sourris Karly, C.; Lindblom Runa, S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Higgins, G.C.; Lindblom, R.S.J.; Coughlan, M.T. Exploring the role of the metabolite-sensing receptor GPR109a in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2020, 318, F835–F842. [Google Scholar] [CrossRef]
- Jayasudha, R.; Das, T.; Kalyana Chakravarthy, S.; Sai Prashanthi, G.; Bhargava, A.; Tyagi, M.; Rani, P.K.; Pappuru, R.R.; Shivaji, S. Gut mycobiomes are altered in people with type 2 Diabetes Mellitus and Diabetic Retinopathy. PLoS ONE 2020, 15, e0243077. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.T.B.; Wu, K.C.; Hsu, J.L.; Chang, C.S.; Chou, C.; Lin, C.Y.; Liao, Y.M.; Lin, P.C.; Yang, L.Y.; Lin, H.W. Effects of Non-insulin Anti-hyperglycemic Agents on Gut Microbiota: A Systematic Review on Human and Animal Studies. Front. Endocrinol. 2020, 11, 573891. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef]
- Schluter, J.; Foster, K.R. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012, 10, e1001424. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Mosterd, C.M.; Kanbay, M.; van den Born, B.J.H.; van Raalte, D.H.; Rampanelli, E. Intestinal microbiota and diabetic kidney diseases: The Role of microbiota and derived metabolites inmodulation of renal inflammation and disease progression. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101484. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Cosola, C.; Gesualdo, L.; Fiaccadori, E. Intestinal Microbiota in Type 2 Diabetes and Chronic Kidney Disease. Curr. Diabetes Rep. 2017, 17, 16. [Google Scholar] [CrossRef]
- Tao, S.; Li, L.; Li, L.; Liu, Y.; Ren, Q.; Shi, M.; Liu, J.; Jiang, J.; Ma, H.; Huang, Z.; et al. Understanding the gut–kidney axis among biopsy-proven diabetic nephropathy, type 2 diabetes mellitus and healthy controls: An analysis of the gut microbiota composition. Acta Diabetol. 2019, 56, 581–592. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Qin, X.; Yang, C.; Sabatino, A.; Kelly, J.T.; Avesani, C.M.; Carrero, J.J.; on behalf of the ERA European Renal Nutrition Working Group, an Official Body of the ERA. Fiber intake and health in people with chronic kidney disease. Clin. Kidney J. 2022, 15, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Biruete, A.; Shin, A.; Kistler, B.M.; Moe, S.M. Feeling gutted in chronic kidney disease (CKD): Gastrointestinal disorders and therapies to improve gastrointestinal health in individuals CKD, including those undergoing dialysis. Semin. Dial. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Kanbay, M.; Onal, E.M.; Afsar, B.; Dagel, T.; Yerlikaya, A.; Covic, A.; Vaziri, N.D. The crosstalk of gut microbiota and chronic kidney disease: Role of inflammation, proteinuria, hypertension, and diabetes mellitus. Int. Urol. Nephrol. 2018, 50, 1453–1466. [Google Scholar] [CrossRef]
- Wu, I.W.; Lin, C.-Y.; Chang, L.-C.; Lee, C.-C.; Chiu, C.-Y.; Hsu, H.-J.; Sun, C.-Y.; Chen, Y.-C.; Kuo, Y.-L.; Yang, C.-W.; et al. Gut Microbiota as Diagnostic Tools for Mirroring Disease Progression and Circulating Nephrotoxin Levels in Chronic Kidney Disease: Discovery and Validation Study. Int. J. Biol. Sci. 2020, 16, 420–434. [Google Scholar] [CrossRef]
- Linh, H.T.; Iwata, Y.; Senda, Y.; Sakai-Takemori, Y.; Nakade, Y.; Oshima, M.; Nakagawa-Yoneda, S.; Ogura, H.; Sato, K.; Minami, T.; et al. Intestinal Bacterial Translocation Contributes to Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1105–1119. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Rahimi, A.; Ni, Z.; Said, H.; Subramanian, V.S. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol. Dial. Transplant. 2012, 27, 2686–2693. [Google Scholar] [CrossRef]
- Shah, N.B.; Allegretti, A.S.; Nigwekar, S.U.; Kalim, S.; Zhao, S.; Lelouvier, B.; Servant, F.; Serena, G.; Thadhani, R.I.; Raj, D.S.; et al. Blood Microbiome Profile in CKD: A Pilot Study. Clin. J. Am. Soc. Nephrol. 2019, 14, 692–701. [Google Scholar] [CrossRef]
- Rebholz, C.M.; Crews, D.C.; Grams, M.E.; Steffen, L.M.; Levey, A.S.; Miller, E.R.; Appel, L.J.; Coresh, J. DASH (Dietary Approaches to Stop Hypertension) Diet and Risk of Subsequent Kidney Disease. Am. J. Kidney Dis. 2016, 68, 853–861. [Google Scholar] [CrossRef]
- Cione, E.; Fazio, A.; Curcio, R.; Tucci, P.; Lauria, G.; Cappello, A.R.R.; Dolce, V. Resistant Starches and Non-Communicable Disease: A Focus on Mediterranean Diet. Foods 2021, 10, 2062. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Gross, L.A.; de Azevedo, M.J.; Viana, L.V. Dietary Fiber Intake (Supplemental or Dietary Pattern Rich in Fiber) and Diabetic Kidney Disease: A Systematic Review of Clinical Trials. Nutrients 2019, 11, 347. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.Y.; Rowling, M.J. Resistant starch as a novel dietary strategy to maintain kidney health in diabetes mellitus. Nutr. Rev. 2017, 75, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.M.; Champ, M.M.J.; Cloran, S.J.; Fleith, M.; van Lieshout, L.; Mejborn, H.; Burley, V.J. Dietary fibre in Europe: Current state of knowledge on definitions, sources, recommendations, intakes and relationships to health. Nutr. Res. Rev. 2017, 30, 149–190. [Google Scholar] [CrossRef]
- World Health Organization. CODEX ALIMENTARIUS: Guidelines on Nutritional Labelling CXG 2-1985; 1985 (Revised 2021); World Health Organization: Geneva, Switzerland, 1985. [Google Scholar]
- So, D.; Gibson, P.R.; Muir, J.G.; Yao, C.K. Dietary fibres and IBS: Translating functional characteristics to clinical value in the era of personalised medicine. Gut 2021, 70, 2383–2394. [Google Scholar] [CrossRef]
- Dai, F.-J.; Chau, C.-F. Classification and regulatory perspectives of dietary fiber. J. Food Drug Anal. 2017, 25, 37–42. [Google Scholar] [CrossRef]
- Dhingra, D.; Michael, M.; Rajput, H.; Patil, R.T. Dietary fibre in foods: A review. J. Food Sci. Technol. 2012, 49, 255–266. [Google Scholar] [CrossRef]
- Włodarczyk, M.; Śliżewska, K. Efficiency of Resistant Starch and Dextrins as Prebiotics: A Review of the Existing Evidence and Clinical Trials. Nutrients 2021, 13, 3808. [Google Scholar] [CrossRef]
- Leszczyński, W. Resistant Starch–Classification, Structure, Production. Pol. J. Food Nutr. Sci. 2004, 54, 37–50. [Google Scholar]
- Snelson, M.; Coughlan, M. The Devil’s in the Detail: The Importance of Specific, Descriptive Language for Reproducibility in Nutrition Science. J. Ren. Nutr. 2020, 30, 274–275. [Google Scholar] [CrossRef]
- Bird, A.R.; Conlon, M.A.; Christophersen, C.T.; Topping, D.L. Resistant starch, large bowel fermentation and a broader perspective of prebiotics and probiotics. Benef. Microbes 2010, 1, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Kim, J.; Duffy, P.R.; Schlegel, V.L.; Walter, J. Resistant Starches Types 2 and 4 Have Differential Effects on the Composition of the Fecal Microbiota in Human Subjects. PLoS ONE 2010, 5, e15046. [Google Scholar] [CrossRef] [PubMed]
- Sobh, M.; Montroy, J.; Daham, Z.; Sibbald, S.; Lalu, M.; Stintzi, A.; Mack, D.; Fergusson, D.A. Tolerability and SCFA production after resistant starch supplementation in humans: A systematic review of randomized controlled studies. Am. J. Clin. Nutr. 2022, 115, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.F. An introductory review of resistant starch type 2 from high-amylose cereal grains and its effect on glucose and insulin homeostasis. Nutr. Rev. 2019, 77, 748–764. [Google Scholar] [CrossRef]
- Stewart, M.L.; Wilcox, M.L.; Bell, M.; Buggia, M.A.; Maki, K.C. Type-4 Resistant Starch in Substitution for Available Carbohydrate Reduces Postprandial Glycemic Response and Hunger in Acute, Randomized, Double-Blind, Controlled Study. Nutrients 2018, 10, 129. [Google Scholar] [CrossRef]
- Gutiérrez, T.J.; Tovar, J. Update of the concept of type 5 resistant starch (RS5): Self-assembled starch V-type complexes. Trends Food Sci. Technol. 2021, 109, 711–724. [Google Scholar] [CrossRef]
- Bojarczuk, A.; Skąpska, S.; Mousavi Khaneghah, A.; Marszałek, K. Health benefits of resistant starch: A review of the literature. J. Funct. Foods 2022, 93, 105094. [Google Scholar] [CrossRef]
- Miketinas, D.C.; Shankar, K.; Maiya, M.; Patterson, M.A. Usual Dietary Intake of Resistant Starch in US Adults from NHANES 2015–2016. J. Nutr. 2020, 150, 2738–2747. [Google Scholar] [CrossRef]
- CSIRO. Understanding Resistant Starch and Its Role in Gut Health. Available online: https://www.csiro.au/en/research/health-medical/nutrition/resistant-starch (accessed on 19 March 2022).
- Bede, D.; Zaixiang, L. Recent Developments in Resistant Starch as a Functional Food. Starch Stärke 2021, 73, 2000139. [Google Scholar] [CrossRef]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. The Health Benefits of Dietary Fibre. Nutrients 2020, 12, 3209. [Google Scholar] [CrossRef]
- Birt, D.F.; Boylston, T.; Hendrich, S.; Jane, J.L.; Hollis, J.; Li, L.; McClelland, J.; Moore, S.; Phillips, G.J.; Rowling, M.; et al. Resistant starch: Promise for improving human health. Adv. Nutr. 2013, 4, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Kerr, C.A.; McSweeney, C.S.; Dunne, R.A.; Shaw, J.M.; Kang, S.; Bird, A.R.; Morell, M.K.; Lockett, T.J.; Molloy, P.L.; et al. Resistant Starches Protect against Colonic DNA Damage and Alter Microbiota and Gene Expression in Rats Fed a Western Diet. J. Nutr. 2012, 142, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Willems, A.; Reading, S.; Collins, M.D. Fermentation of non-digestible oligosaccharides by human colonic bacteria. Proc. Nutr. Soc. 1996, 55, 899–912. [Google Scholar] [CrossRef]
- Shamloo, M.; Mollard, R.; Wang, H.; Kingra, K.; Tangri, N.; MacKay, D. A randomized double-blind cross-over trial to study the effects of resistant starch prebiotic in chronic kidney disease (ReSPECKD). Trials 2022, 23, 72. [Google Scholar] [CrossRef] [PubMed]
- Metzler-Zebeli, B.U.; Canibe, N.; Montagne, L.; Freire, J.; Bosi, P.; Prates, J.A.M.; Tanghe, S.; Trevisi, P. Resistant starch reduces large intestinal pH and promotes fecal lactobacilli and bifidobacteria in pigs. Animal 2019, 13, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sybille, T.; June, Z.; Michael, K.; Roy, M.; Maria, L.M. The intestinal microbiota in aged mice is modulated by dietary resistant starch and correlated with improvements in host responses. FEMS Microbiol. Ecol. 2013, 83, 299–309. [Google Scholar] [CrossRef]
- Baxter, N.T.; Schmidt, A.W.; Venkataraman, A.; Kim, K.S.; Waldron, C.; Schmidt, T.M.; Blaser, M.J.; Britton, R.; Walter, J. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 2019, 10, e02566-18. [Google Scholar] [CrossRef]
- Venkataraman, A.; Sieber, J.R.; Schmidt, A.W.; Waldron, C.; Theis, K.R.; Schmidt, T.M. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome 2016, 4, 33. [Google Scholar] [CrossRef]
- Alfa, M.J.; Strang, D.; Tappia, P.S.; Graham, M.; Van Domselaar, G.; Forbes, J.D.; Laminman, V.; Olson, N.; DeGagne, P.; Bray, D.; et al. A randomized trial to determine the impact of a digestion resistant starch composition on the gut microbiome in older and mid-age adults. Clin. Nutr. 2018, 37, 797–807. [Google Scholar] [CrossRef]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- Haenen, D.; Zhang, J.; Souza da Silva, C.; Bosch, G.; van der Meer, I.M.; van Arkel, J.; van den Borne, J.J.; Pérez Gutiérrez, O.; Smidt, H.; Kemp, B.; et al. A diet high in resistant starch modulates microbiota composition, SCFA concentrations, and gene expression in pig intestine. J. Nutr. 2013, 143, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Le Leu, R.K.; Hu, Y.; Brown, I.L.; Young, G.P. Effect of high amylose maize starches on colonic fermentation and apoptotic response to DNA-damage in the colon of rats. Nutr. Metab. 2009, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.H.; Park, J.H.; Jeon, W.M.; Han, K.S. Butyrate modulates bacterial adherence on LS174T human colorectal cells by stimulating mucin secretion and MAPK signaling pathway. Nutr. Res. Pract. 2015, 9, 343–349. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, Q.; Dorfman, R.G.; Huang, X.; Fan, T.; Zhang, H.; Zhang, J.; Yu, C. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol. 2016, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate Inhibits NF-κB Activation in Lamina Propria Macrophages of Patients with Ulcerative Colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Roshanravan, N.; Mahdavi, R.; Alizadeh, E.; Jafarabadi, M.A.; Hedayati, M.; Ghavami, A.; Alipour, S.; Alamdari, N.M.; Barati, M.; Ostadrahimi, A. Effect of Butyrate and Inulin Supplementation on Glycemic Status, Lipid Profile and Glucagon-Like Peptide 1 Level in Patients with Type 2 Diabetes: A Randomized Double-Blind, Placebo-Controlled Trial. Horm. Metab. Res. 2017, 49, 886–891. [Google Scholar] [CrossRef]
- Petersen, K.E.; Rakipovski, G.; Raun, K.; Lykkesfeldt, J. Does Glucagon-like Peptide-1 Ameliorate Oxidative Stress in Diabetes? Evidence Based on Experimental and Clinical Studies. Curr. Diabetes Rev. 2016, 12, 331–358. [Google Scholar] [CrossRef]
- Leite, K.M.; Long, A.M.; Ostroff, M.L.; Borges, L.; Braden, G. A Review of the Renoprotective Effects of Novel Antidiabetic Agents. J. Pharm. Pract. 2021, 34, 141–148. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.M.A.E.; Bast, A.; Vanhoutvin, S.A.L.W.; Fischer, M.A.J.G.; Kodde, A.; Troost, F.J.; Venema, K.; Brummer, R.-J.M. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin. Nutr. 2009, 28, 88–93. [Google Scholar] [CrossRef]
- Hadden, M.J.; Advani, A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 2630. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhuang, S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front. Physiol. 2015, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Abell, G.C.J.; Cooke, C.M.; Bennett, C.N.; Conlon, M.A.; McOrist, A.L. Phylotypes related to Ruminococcus bromii are abundant in the large bowel of humans and increase in response to a diet high in resistant starch. FEMS Microbiol. Ecol. 2008, 66, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Chiavaroli, L.; Mirrahimi, A.; Sievenpiper, J.L.; Jenkins, D.J.; Darling, P.B. Dietary fiber effects in chronic kidney disease: A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2015, 69, 761–768. [Google Scholar] [CrossRef]
- Tougaard, N.H.; Frimodt-Møller, M.; Salmenkari, H.; Stougaard, E.B.; Zawadzki, A.D.; Mattila, I.M.; Hansen, T.W.; Legido-Quigley, C.; Hörkkö, S.; Forsblom, C.; et al. Effects of Butyrate Supplementation on Inflammation and Kidney Parameters in Type 1 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Clin. Med. 2022, 11, 3573. [Google Scholar] [CrossRef]
- Vernero, M.; De Blasio, F.; Ribaldone, D.G.; Bugianesi, E.; Pellicano, R.; Saracco, G.M.; Astegiano, M.; Caviglia, G.P. The Usefulness of Microencapsulated Sodium Butyrate Add-On Therapy in Maintaining Remission in Patients with Ulcerative Colitis: A Prospective Observational Study. J. Clin. Med. 2020, 9, 3941. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, X.; Meng, Y.; Wang, Q.; Xu, H.; Chen, L. The Effects of Resistant Starch on Biomarkers of Inflammation and Oxidative Stress: A Systematic Review and Meta-Analysis. Nutr. Cancer 2022, 74, 2337–2350. [Google Scholar] [CrossRef]
- Lu, J.; Ma, B.; Qiu, X.; Sun, Z.; Xiong, K. Effects of resistant starch supplementation on oxidative stress and inflammation biomarkers: A systematic review and meta-analysis of randomized controlled trials. Asia Pac. J. Clin. Nutr. 2021, 30, 614–623. [Google Scholar] [CrossRef]
- Haghighatdoost, F.; Gholami, A.; Hariri, M. Effect of resistant starch type 2 on inflammatory mediators: A systematic review and meta-analysis of randomized controlled trials. Complement. Ther. Med. 2021, 56, 102597. [Google Scholar] [CrossRef]
- Snelson, M.; Jong, J.; Manolas, D.; Kok, S.; Louise, A.; Stern, R.; Kellow, N.J. Metabolic Effects of Resistant Starch Type 2: A Systematic Literature Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2019, 11, 1833. [Google Scholar] [CrossRef]
- Bodinham, C.L.; Smith, L.; Thomas, E.L.; Bell, J.D.; Swann, J.R.; Costabile, A.; Russell-Jones, D.; Umpleby, A.M.; Robertson, M.D. Efficacy of increased resistant starch consumption in human type 2 diabetes. Endocr. Connect. 2014, 3, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Paik, J.K.; Kim, H.I.; Kim, O.Y.; Shin, D.Y.; Kim, H.-J.; Lee, J.H.; Lee, J.H. Dietary treatment with rice containing resistant starch improves markers of endothelial function with reduction of postprandial blood glucose and oxidative stress in patients with prediabetes or newly diagnosed type 2 diabetes. Atherosclerosis 2012, 224, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Dainty, S.A.; Klingel, S.L.; Pilkey, S.E.; McDonald, E.; McKeown, B.; Emes, M.J.; Duncan, A.M. Resistant Starch Bagels Reduce Fasting and Postprandial Insulin in Adults at Risk of Type 2 Diabetes. J. Nutr. 2016, 146, 2252–2259. [Google Scholar] [CrossRef] [PubMed]
- Ble-Castillo, J.L.; Aparicio-Trápala, M.A.; Francisco-Luria, M.U.; Córdova-Uscanga, R.; Rodríguez-Hernández, A.; Méndez, J.D.; Díaz-Zagoya, J.C. Effects of Native Banana Starch Supplementation on Body Weight and Insulin Sensitivity in Obese Type 2 Diabetics. Int. J. Environ. Res. Public Health 2010, 7, 1953–1962. [Google Scholar] [CrossRef]
- Johnston, K.L.; Thomas, E.L.; Bell, J.D.; Frost, G.S.; Robertson, M.D. Resistant starch improves insulin sensitivity in metabolic syndrome. Diabet. Med. 2010, 27, 391–397. [Google Scholar] [CrossRef]
- Karimi, P.; Farhangi, M.A.; Sarmadi, B.; Gargari, B.P.; Zare Javid, A.; Pouraghaei, M.; Dehghan, P. The Therapeutic Potential of Resistant Starch in Modulation of Insulin Resistance, Endotoxemia, Oxidative Stress and Antioxidant Biomarkers in Women with Type 2 Diabetes: A Randomized Controlled Clinical Trial. Ann. Nutr. Metab. 2016, 68, 85–93. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, F.; Ren, X.; Wang, Y.; Blanchard, C. Resistant starch manipulated hyperglycemia/hyperlipidemia and related genes expression in diabetic rats. Int. J. Biol. Macromol. 2015, 75, 316–321. [Google Scholar] [CrossRef]
- Strozyk, S.; Rogowicz-Frontczak, A.; Pilacinski, S.; LeThanh-Blicharz, J.; Koperska, A.; Zozulinska-Ziolkiewicz, D. Influence of resistant starch resulting from the cooling of rice on postprandial glycemia in type 1 diabetes. Nutr. Diabetes 2022, 12, 21. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chang, D.-M.; Wu, D.-J.; Peng, H.-Y.; Chuang, L.-M. Assessment of Blood Glucose Regulation and Safety of Resistant Starch Formula-Based Diet in Healthy Normal and Subjects With Type 2 Diabetes. Medicine 2015, 94, e1332. [Google Scholar] [CrossRef]
- Bell, K.J.; Saad, S.; Tillett, B.J.; McGuire, H.M.; Bordbar, S.; Yap, Y.A.; Nguyen, L.T.; Wilkins, M.R.; Corley, S.; Brodie, S.; et al. Metabolite-based dietary supplementation in human type 1 diabetes is associated with microbiota and immune modulation. Microbiome 2022, 10, 9. [Google Scholar] [CrossRef]
- Rizkalla, S.W.; Bellisle, F.; Slama, G. Health benefits of low glycaemic index foods, such as pulses, in diabetic patients and healthy individuals. Br. J. Nutr. 2002, 88, S255–S262. [Google Scholar] [CrossRef] [PubMed]
- DeMartino, P.; Johnston, E.A.; Petersen, K.S.; Kris-Etherton, P.M.; Cockburn, D.W. Additional Resistant Starch from One Potato Side Dish per Day Alters the Gut Microbiota but Not Fecal Short-Chain Fatty Acid Concentrations. Nutrients 2022, 14, 721. [Google Scholar] [CrossRef] [PubMed]
- Penn-Marshall, M.; Holtzman, G.I.; Barbeau, W.E. African Americans May Have to Consume More Than 12 Grams a Day of Resistant Starch to Lower Their Risk for Type 2 Diabetes. J. Med. Food 2010, 13, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.Y.; Whitley, E.M.; Mancosky, K.; Loo, Y.T.; Grapentine, K.; Bowers, E.; Schalinske, K.L.; Rowling, M.J. Dietary Resistant Starch Prevents Urinary Excretion of Vitamin D Metabolites and Maintains Circulating 25-Hydroxycholecalciferol Concentrations in Zucker Diabetic Fatty Rats. J. Nutr. 2014, 144, 1667–1673. [Google Scholar] [CrossRef]
- Koh, G.Y.; Rowling, M.J.; Schalinske, K.L.; Grapentine, K.; Loo, Y.T. Consumption of Dietary Resistant Starch Partially Corrected the Growth Pattern Despite Hyperglycemia and Compromised Kidney Function in Streptozotocin-Induced Diabetic Rats. J. Agric. Food Chem. 2016, 64, 7540–7545. [Google Scholar] [CrossRef]
- Zhang, J.; Yakovlieva, L.; de Haan, B.J.; de Vos, P.; Minnaard, A.J.; Witte, M.D.; Walvoort, M.T.C. Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells. Antibiotics 2020, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Smazal, A.L.; Borcherding, N.C.; Anderegg, A.S.; Schalinske, K.L.; Whitley, E.M.; Rowling, M.J. Dietary Resistant Starch Prevents Urinary Excretion of 25-Hydroxycholecalciferol and Vitamin D-Binding Protein in Type 1 Diabetic Rats. J. Nutr. 2013, 143, 1123–1128. [Google Scholar] [CrossRef]
- Kieffer, D.A.; Piccolo, B.D.; Vaziri, N.D.; Liu, S.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Moore, M.E.; Marco, M.L.; Martin, R.J.; et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am. J. Physiol. Physiol. 2016, 310, F857–F871. [Google Scholar] [CrossRef]
- Karaduta, O.; Glazko, G.; Dvanajscak, Z.; Arthur, J.; Mackintosh, S.; Orr, L.; Rahmatallah, Y.; Yeruva, L.; Tackett, A.; Zybailov, B. Resistant starch slows the progression of CKD in the 5/6 nephrectomy mouse model. Physiol. Rep. 2020, 8, e14610. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Liu, S.-M.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Farzaneh, S.H.; Kieffer, D.A.; Adams, S.H.; Martin, R.J. High Amylose Resistant Starch Diet Ameliorates Oxidative Stress, Inflammation, and Progression of Chronic Kidney Disease. PLoS ONE 2014, 9, e114881. [Google Scholar] [CrossRef]
- Jia, L.; Dong, X.; Li, X.; Jia, R.; Zhang, H.-L. Benefits of resistant starch type 2 for patients with end-stage renal disease under maintenance hemodialysis: A systematic review and meta-analysis. Int. J. Med. Sci. 2021, 18, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Laffin, M.R.; Tayebi Khosroshahi, H.; Park, H.; Laffin, L.J.; Madsen, K.; Kafil, H.S.; Abedi, B.; Shiralizadeh, S.; Vaziri, N.D. Amylose resistant starch (HAM-RS2) supplementation increases the proportion of Faecalibacterium bacteria in end-stage renal disease patients: Microbial analysis from a randomized placebo-controlled trial. Hemodial. Int. 2019, 23, 343–347. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of Increasing Dietary Fiber on Plasma Levels of Colon-Derived Solutes in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603. [Google Scholar] [CrossRef]
- Esgalhado, M.; Kemp, J.A.; Azevedo, R.; Paiva, B.R.; Stockler-Pinto, M.B.; Dolenga, C.J.; Borges, N.A.; Nakao, L.S.; Mafra, D. Could resistant starch supplementation improve inflammatory and oxidative stress biomarkers and uremic toxins levels in hemodialysis patients? A pilot randomized controlled trial. Food Funct. 2018, 9, 6508–6516. [Google Scholar] [CrossRef]
- Khosroshahi, H.T.; Abedi, B.; Ghojazadeh, M.; Samadi, A.; Jouyban, A. Effects of fermentable high fiber diet supplementation on gut derived and conventional nitrogenous product in patients on maintenance hemodialysis: A randomized controlled trial. Nutr. Metab. 2019, 16, 18. [Google Scholar] [CrossRef]
- De Paiva, B.R.; Esgalhado, M.; Borges, N.A.; Kemp, J.A.; Alves, G.; Leite, P.E.C.; Macedo, R.; Cardozo, L.; de Brito, J.S.; Mafra, D. Resistant starch supplementation attenuates inflammation in hemodialysis patients: A pilot study. Int. Urol. Nephrol. 2020, 52, 549–555. [Google Scholar] [CrossRef]
- McFarlane, C.; Krishnasamy, R.; Stanton, T.; Savill, E.; Snelson, M.; Mihala, G.; Kelly, J.T.; Morrison, M.; Johnson, D.W.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology II (SYNERGY II): A Feasibility Randomized Controlled Trial. Nutrients 2021, 13, 4481. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Description | Food Sources |
|---|---|---|
| RS1 | RS1 refers to starch molecules encased within an intact plant cell wall. These starch molecules are physically inaccessible to the digestive enzymes of the upper alimentary tract, as humans lack the ability to digested plant cell wall components [82,86]. Therefore, the resistance of RS1 can be lost through any process which damages this protective cell wall barrier, such as milling, grinding or mastication [87]. | Legumes, Seeds, Wholegrains |
| RS2 | RS2 refers to tightly organised ungelatinized starch granules [46]. Whilst the exact mechanisms of its resistance are not fully understood [82], it is thought that its dense structure makes it difficult for digestive enzymes to effectively access and attach to these starch molecules [82,88]. | Raw potato, Unripe bananas, High Amylose Maize Starch (HAMS) [84] |
| RS3 | RS3 is created through the process of retrogradation [76]. To undergo retrogradation, starch first needs to undergo gelatinization which occurs when starch is heated and becomes more viscous as water molecules enter the starch granule [46]. As the starch cools down, retrogradation then occurs, where its structure reforms to create a more tightly packed, inaccessible crystalline structure [88]. | Heated and cooled potatoes, rice |
| RS4 | RS4 is created by the chemical modification of starch molecules. Such processes include dextrinization, substitution of functional groups and esterification [43,82]. RS4 encompasses a large range of different molecules given the various combinations of starch bases and chemical processes that are available [89]. | |
| RS5 | RS5 has traditionally referred to starch-lipid complexes, created through the combination of long side chains of amylopectin or amylose with lipids or free fatty acids. This structure limits accessibility to digestive enzymes [82] and can be both naturally or artificially derived [88]. More recently, more resistant starch complexes have been identified such as starch-protein complexes and starch-glycerol complexes [90]. |
| Study | Population | Intervention | Control | Group Size (n) | Duration (Weeks) | Alb | Ucr | CrCl | BUN | Renal Histology | Inflammatory Markers | Intestinal Markers |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Preclinical CKD Models | ||||||||||||
| [145] ^ | Male Sprague Dawley rats adenine-induced CKD | 59% HAMS | Amylopectin low fibre diet | 9 | 3 | -- | -- | ↑ | -- | ↓ tubulointerstitial injury | ↓ TGF-β, ROS, MCP-1 | Δ microbiome |
| [143] ^ | Male Sprague Dawley rats adenine-induced CKD | 59% HAMS | Amylopectin low fibre diet | 9 | 3 | -- | -- | ↑ | ↓ | -- | -- | ↓ pH Δ microbiome |
| [144] | Male C57BL6 mice 5/6 nephrectomy | 59% HAMS | Regular control diet | 4 | 4 | -- | -- | -- | ↔ | ↓tubulointerstitial injury | -- | Δ microbiome |
| Preclinical Diabetes Models | ||||||||||||
| [142] | STZ treated Male Sprague Dawley rats | 55% HAMS (20% RS) | 55% Corn Starch | 5 | 5 | ↓ | -- | -- | -- | ↓ proximal tubular injury | -- | -- |
| [139] | Male Zucker Diabetic Fatty Rats | 55% HAMS (35% RS) | Corn Starch control diet | 8 | 6 | ↓ | ↑ | -- | -- | -- | -- | -- |
| [140] | STZ-treated male Sprague Dawley rats | 13.75%, 27.5% or 55% HAMS (5%,10% or 20% RS) | Corn Starch control diet | 8 | 6 | ↔ | ↔ | -- | -- | -- | -- | -- |
| [53] | STZ-treated male Gpr109a−/− mice | 25% HAMS (12.5% RS) | 20% starch + 5% cellulose | 10–11 | 24 | ↔ | -- | -- | -- | ↔ renal hypertrophy ↔ Glomerulosclerosis Index | ↔ MCP-1 | -- |
| [17] | STZ-treated male Gpr109a−/− mice | 63.6% RS (source not outlined) | Normal Chow, Zero Fibre | 5–10 | 12 | ↓ | -- | -- | -- | ↓ Glomerular hypertrophy ↓ Podocyte injury ↓ Interstitial fibrosis | ↓ TNF-α ↓TGF-β ↓ IL6 | ↑ SCFA |
| [52] | Male db/db mice | 25% HAMS (12.5% RS) | 20% starch + 5% cellulose | 12 | 10 | ↓ | -- | ↔ | -- | -- | -- | ↓ in vivo gut permeability |
| Study | Population | Intervention | Control | Group Size (n) | Duration (Weeks) | Alb | SCr | BUN | Uremic Toxins | Inflammatory Markers | Microbiota |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chronic Kidney Disease | |||||||||||
| [149] | Stable haemodialysis | 15 g/d HAMS (60% RS) | 15 g/d waxy corn starch | 20 | 6 | -- | -- | ↔ | ↓IS ↔ PS | ↔ CRP | -- |
| [150] % | Stable haemodialysis | 26 g/d HAMS (16 g/d RS) | 20 g/d manioc flour | 15–16 | 4 | -- | ↔ | ↔ | ↓IS ↔ PS | ↓ IL-6, ↓ TBARS, ↔ hs-CRP | -- |
| [44] | Stable haemodialysis (Diabetic patients excluded) | 20 g/d 4 weeks, 25 g/d 4 weeks HAMSRS2 (60% RS) | 20 g/d 4 weeks, 25 g/d 4 weeks Wheat flour | 22 | 8 | -- | ↓ | ↓ | -- | ↓ TNF-α, ↓ IL6, ↓ MDA ↔ hs-CRP, ↔ IL-1β ↔ TAO activity | -- |
| [151] $ | Stable haemodialysis (Diabetic patients excluded) | 20 g/d 4 weeks, 25 g/d 4 weeks HAMSRS2 (60% RS) | 20 g/d 4 weeks, 25 g/d 4 weeks waxy corn starch | 21–23 | 8 | -- | ↓ | ↔ | ↓ PC ↔IS | ↔ hs-CRP | -- |
| [147] $ | Stable haemodialysis (Diabetic patients excluded) | 20 g/d 4 weeks, 25 g/d 4 weeks HAMSRS2 (RS% not stated) | 20 g/d 4 weeks, 25 g/d 4 weeks waxy corn starch | 9–11 | 8 | -- | ↔ | ↓ | ↓ IL6, ↓TNF-α ↓MDA | ↑ Faecalibacterium genus ↔ Bifidobacteria genus ↔ Ruminococcus genus ↔ Prevotella genus | |
| [152] % | Stable haemodialysis | 16 g/d RS | 16 g/day manioc flour | 8 | 4 | -- | -- | -- | -- | ↓ RANTES, ↓ PDGF-BB ↓ IP10, ↔ IL10 | -- |
| Diabetic Kidney Disease | |||||||||||
| [7] | T2DM with early stage DN aged 18–80 | 50 g/d high RS flour (17.41 g/d RS) | Control diet (not stated) | 37–38 | 12 | ↔ | ↔ | ↔ | -- | ↔ TNF-α, ↔ IL-6, ↑ SOD | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drake, A.M.; Coughlan, M.T.; Christophersen, C.T.; Snelson, M. Resistant Starch as a Dietary Intervention to Limit the Progression of Diabetic Kidney Disease. Nutrients 2022, 14, 4547. https://doi.org/10.3390/nu14214547
Drake AM, Coughlan MT, Christophersen CT, Snelson M. Resistant Starch as a Dietary Intervention to Limit the Progression of Diabetic Kidney Disease. Nutrients. 2022; 14(21):4547. https://doi.org/10.3390/nu14214547
Chicago/Turabian StyleDrake, Anna M., Melinda T. Coughlan, Claus T. Christophersen, and Matthew Snelson. 2022. "Resistant Starch as a Dietary Intervention to Limit the Progression of Diabetic Kidney Disease" Nutrients 14, no. 21: 4547. https://doi.org/10.3390/nu14214547
APA StyleDrake, A. M., Coughlan, M. T., Christophersen, C. T., & Snelson, M. (2022). Resistant Starch as a Dietary Intervention to Limit the Progression of Diabetic Kidney Disease. Nutrients, 14(21), 4547. https://doi.org/10.3390/nu14214547