
Genome-Wide Association Analysis of Over 170,000 Individuals from the UK Biobank Identifies Seven Loci Associated with Dietary Approaches to Stop Hypertension (DASH) Diet

 ,
,  , and
, and
Abstract
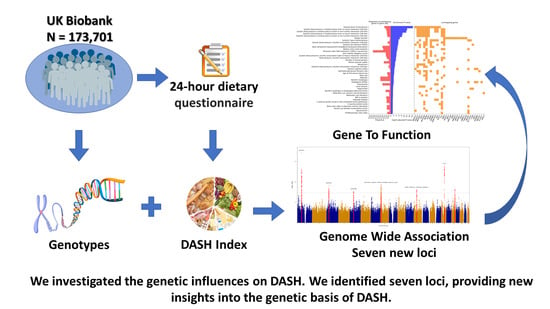
1. Introduction
2. Materials and Methods
2.1. Study Population

{kind=link}
{kind=link}
{kind=link}
| Total Participants (n = 173,701) | |
|---|---|
| Age (years) (Mean (SD)) | 56.420 (7.864) |
| Gender | |
| Female | 94,721 (54.5%) |
| Male | 78,980 (45.5%) |
| BMI (kg/m2) (Mean (SD)) | 26.462 (4.633) |
| Townsend Index (Mean (SD)) | −1.519 (2.424) |
| Smoking | |
| Yes | 98,378 (56.6%) |
| No | 75,323 (43.4%) |
| Alcohol (g/day per week) (Mean (SD)) | 13.971 (21.439) |
| Instances of 24 h FFQ answered | |
| 1 | 67,771 (39%) |
| 2 | 39,797 (22.9%) |
| 3 | 35,567 (20.5%) |
| 4 | 25,713 (14.8%) |
| 5 | 4853 (2.8%) |
| Energy intake (kcal) (Mean (SD)) | 2106.596 (595.421) |
| Fruit (Mean Servings (SD)) | 3.16 (2.52) |
| Vegetables (Mean Servings (SD)) | 3.49 (3.06) |
| Nuts and legumes (Mean Servings (SD)) | 0.9 (1.06) |
| Whole grains (Mean Servings (SD)) | 4.05 (3.12) |
| Low-fat dairy (Mean Servings (SD)) | 0.66 (0.84) |
| Sodium (mg) (Mean (SD)) | 2998.9 (3046.66) |
| Red and processed meat (Mean Servings (SD)) | 1.66 (1.75) |
| Sweetened beverages (Mean Servings (SD)) | 0.47 (0.89) |
| DASH (Mean (SD)) | 24 (4.24) |
2.2. Genome Wide Association Analysis
2.3. Mapping and Conditional Analysis
2.4. Pathway and Colocalization Analysis
2.5. Shared Genetic Architecture with Disease
2.6. Mendelian Randomization
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Kompaniyets, L.; Goodman, A.B.; Belay, B.; Freedman, D.S.; Sucosky, M.S.; Lange, S.J.; Gundlapalli, A.V.; Boehmer, T.K.; Blanck, H.M. Body Mass Index and Risk for COVID-19-Related Hospitalization, Intensive Care Unit Admission, Invasive Mechanical Ventilation, and Death—United States, March-December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 355–361. [Google Scholar] [CrossRef]
- Roberts, C.K.; Barnard, R.J. Effects of exercise and diet on chronic disease. J. Appl. Physiol. 2005, 98, 3–30. [Google Scholar] [CrossRef]
- Kivimäki, M.; Lawlor, D.A.; Singh-Manoux, A.; Batty, G.D.; Ferrie, J.E.; Shipley, M.J.; Nabi, H.; Sabia, S.; Marmot, M.G.; Jokela, M. Common mental disorder and obesity: Insight from four repeat measures over 19 years: Prospective Whitehall II cohort study. BMJ 2009, 339, b3765. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Rosenberg, I.; Uauy, R. History of modern nutrition science-implications for current research, dietary guidelines, and food policy. BMJ 2018, 361, k2392. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pinilla, F. Brain foods: The effects of nutrients on brain function. Nat. Rev. Neurosci. 2008, 9, 568–578. [Google Scholar] [CrossRef]
- Deane, K.H.O.; Jimoh, O.F.; Biswas, P.; O’Brien, A.; Hanson, S.; Abdelhamid, A.S.; Fox, C.; Hooper, L. Omega-3 and polyunsaturated fat for prevention of depression and anxiety symptoms: Systematic review and meta-analysis of randomised trials. Br. J. Psychiatry J. Ment. Sci. 2021, 218, 135–142. [Google Scholar] [CrossRef]
- Chu, A.Y.; Workalemahu, T.; Paynter, N.P.; Rose, L.M.; Giulianini, F.; Tanaka, T.; Ngwa, J.S.; Qi, Q.; Curhan, G.C.; Rimm, E.B.; et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum. Mol. Genet. 2013, 22, 1895–1902. [Google Scholar] [CrossRef]
- Meddens, S.F.W.; de Vlaming, R.; Bowers, P.; Burik, C.A.P.; Linnér, R.K.; Lee, C.; Okbay, A.; Turley, P.; Rietveld, C.A.; Fontana, M.A.; et al. Genomic analysis of diet composition finds novel loci and associations with health and lifestyle. Mol. Psychiatry 2020, 26, 2056–2069. [Google Scholar] [CrossRef]
- Merino, J.; Dashti, H.S.; Li, S.X.; Sarnowski, C.; Justice, A.E.; Graff, M.; Papoutsakis, C.; Smith, C.E.; Dedoussis, G.V.; Lemaitre, R.N.; et al. Genome-wide meta-analysis of macronutrient intake of 91,114 European ancestry participants from the cohorts for heart and aging research in genomic epidemiology consortium. Mol. Psychiatry 2019, 24, 1920–1932. [Google Scholar] [CrossRef]
- Tanaka, T.; Ngwa, J.S.; van Rooij, F.J.; Zillikens, M.C.; Wojczynski, M.K.; Frazier-Wood, A.C.; Houston, D.K.; Kanoni, S.; Lemaitre, R.N.; Luan, J.; et al. Genome-wide meta-analysis of observational studies shows common genetic variants associated with macronutrient intake. Am. J. Clin. Nutr. 2013, 97, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.B.; Florez, J.C.; Hirschhorn, J.N. Comprehensive genomic analysis of dietary habits in UK Biobank identifies hundreds of genetic associations. Nat. Commun. 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Niarchou, M.; Byrne, E.M.; Trzaskowski, M.; Sidorenko, J.; Kemper, K.E.; McGrath, J.J.; MC, O.D.; Owen, M.J.; Wray, N.R. Genome-wide association study of dietary intake in the UK biobank study and its associations with schizophrenia and other traits. Transl. Psychiatry 2020, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- May-Wilson, S.; Matoba, N.; Wade, K.H.; Hottenga, J.-J.; Concas, M.P.; Mangino, M.; Grzeszkowiak, E.J.; Menni, C.; Gasparini, P.; Timpson, N.J.; et al. Large-scale GWAS of food liking reveals genetic determinants and genetic correlations with distinct neurophysiological traits. Nat. Commun. 2022, 13, 2743. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Dietary pattern analysis: A new direction in nutritional epidemiology. Curr. Opin. Lipidol. 2002, 13, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Waijers, P.M.; Feskens, E.J.; Ocké, M.C. A critical review of predefined diet quality scores. Br. J. Nutr. 2007, 97, 219–231. [Google Scholar] [CrossRef]
- Vergnaud, A.-C.; Norat, T.; Mouw, T.; Romaguera, D.; May, A.M.; Bueno-de-Mesquita, H.B.; van der A, D.; Agudo, A.; Wareham, N.; Khaw, K.-T.; et al. Macronutrient composition of the diet and prospective weight change in participants of the EPIC-PANACEA study. PLoS ONE 2013, 8, e57300. [Google Scholar] [CrossRef]
- Mompeo, O.; Berry, S.E.; Spector, T.D.; Menni, C.; Mangino, M.; Gibson, R. Differential associations between a priori diet quality scores and markers of cardiovascular health in women: Cross-sectional analyses from TwinsUK. Br. J. Nutr. 2020, 126, 1017–1027. [Google Scholar] [CrossRef]
- Mompeo, O.; Gibson, R.; Christofidou, P.; Spector, T.D.; Menni, C.; Mangino, M. Genetic and Environmental Influences of Dietary Indices in a UK Female Twin Cohort. Twin Res. Hum. Genet. Off. J. Int. Soc. Twin Stud. 2020, 23, 330–337. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Allen, N.E.; Sudlow, C.; Peakman, T.; Collins, R. UK biobank data: Come and get it. Sci. Transl. Med. 2014, 6, 224ed224. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.T.; Chiuve, S.E.; McCullough, M.L.; Rexrode, K.M.; Logroscino, G.; Hu, F.B. Adherence to a DASH-style diet and risk of coronary heart disease and stroke in women. Arch. Intern. Med. 2008, 168, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, M.D.; St Jeor, S.T.; Hill, L.A.; Scott, B.J.; Daugherty, S.A.; Koh, Y.O. A new predictive equation for resting energy expenditure in healthy individuals. Am. J. Clin. Nutr. 1990, 51, 241–247. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef]
- Huang, J.; Howie, B.; McCarthy, S.; Memari, Y.; Walter, K.; Min, J.L.; Danecek, P.; Malerba, G.; Trabetti, E.; Zheng, H.F.; et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun. 2015, 6, 8111. [Google Scholar] [CrossRef]
- Loh, P.R.; Tucker, G.; Bulik-Sullivan, B.K.; Vilhjálmsson, B.J.; Finucane, H.K.; Salem, R.M.; Chasman, D.I.; Ridker, P.M.; Neale, B.M.; Berger, B.; et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 2015, 47, 284–290. [Google Scholar] [CrossRef]
- Townsend, P.; Phillimore, P.; Beattie, A. Health and Deprivation: Inequality and the North; Routledge: London, UK, 1988. [Google Scholar]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2018, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: An expanded tool for searching human genotype–phenotype associations. Bioinformatics 2019, 35, 4851–4853. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Mountjoy, E.; Carmona, M.; Peat, G.; Schmidt, E.M.; Hercules, A.; Fumis, L.; Miranda, A.; Carvalho-Silva, D.; Buniello, A.; et al. Open Targets Genetics: Systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2020, 49, D1311–D1320. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Macgregor, S. VEGAS2: Software for More Flexible Gene-Based Testing. Twin Res. Hum. Genet. Off. J. Int. Soc. Twin Stud. 2015, 18, 86–91. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Vosa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Kasela, S.; et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018, 447367. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zheng, J.; Erzurumluoglu, A.M.; Elsworth, B.L.; Kemp, J.P.; Howe, L.; Haycock, P.C.; Hemani, G.; Tansey, K.; Laurin, C.; Pourcain, B.S.; et al. LD Hub: A centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 2017, 33, 272–279. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Day, F.R.; Gustafsson, S.; Buchkovich, M.L.; Na, J.; Bataille, V.; Cousminer, D.L.; Dastani, Z.; Drong, A.W.; Esko, T.; et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun. 2016, 7, 10495. [Google Scholar] [CrossRef]
- de Moor, M.H.; van den Berg, S.M.; Verweij, K.J.; Krueger, R.F.; Luciano, M.; Arias Vasquez, A.; Matteson, L.K.; Derringer, J.; Esko, T.; Amin, N.; et al. Meta-analysis of Genome-wide Association Studies for Neuroticism, and the Polygenic Association With Major Depressive Disorder. JAMA Psychiatry 2015, 72, 642–650. [Google Scholar] [CrossRef]
- Rietveld, C.A.; Medland, S.E.; Derringer, J.; Yang, J.; Esko, T.; Martin, N.W.; Westra, H.J.; Shakhbazov, K.; Abdellaoui, A.; Agrawal, A.; et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Sicence 2013, 340, 1467–1471. [Google Scholar] [CrossRef]
- Privé, F.; Aschard, H.; Ziyatdinov, A.; Blum, M.G.B. Efficient analysis of large-scale genome-wide data with two R packages: Bigstatsr and bigsnpr. Bioinformatics 2018, 34, 2781–2787. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Roeder, K. Genomic control for association studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.M.; Adams, M.J.; Clarke, T.K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Karlsson Linnér, R.; Biroli, P.; Kong, E.; Meddens, S.F.W.; Wedow, R.; Fontana, M.A.; Lebreton, M.; Tino, S.P.; Abdellaoui, A.; Hammerschlag, A.R.; et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 2019, 51, 245–257. [Google Scholar] [CrossRef]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; de Leeuw, C.A.; Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: Implications for schizophrenia, autism and cancer. Mol. Psychiatry 2009, 14, 563–589. [Google Scholar] [CrossRef]
- Feitosa, M.F.; Kraja, A.T.; Chasman, D.I.; Sung, Y.J.; Winkler, T.W.; Ntalla, I.; Guo, X.; Franceschini, N.; Cheng, C.Y.; Sim, X.; et al. Novel genetic associations for blood pressure identified via gene-alcohol interaction in up to 570K individuals across multiple ancestries. PLoS ONE 2018, 13, e0198166. [Google Scholar] [CrossRef]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Cheng, I.; Pendergrass, S.A.; Kucharska-Newton, A.M.; Lim, U.; Ambite, J.L.; Caberto, C.P.; Monroe, K.R.; Schumacher, F.; Hindorff, L.A.; et al. Association of the FTO obesity risk variant rs8050136 with percentage of energy intake from fat in multiple racial/ethnic populations: The PAGE study. Am. J. Epidemiol. 2013, 178, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Boender, A.J.; van Gestel, M.A.; Garner, K.M.; Luijendijk, M.C.; Adan, R.A. The obesity-associated gene Negr1 regulates aspects of energy balance in rat hypothalamic areas. Physiol. Rep. 2014, 2, e12083. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Monnig, J.; Kemp, B.E.; Mattson, M.P. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J. Mol. Neurosci. MN 2001, 17, 45–58. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.P.; Kalra, P.S. NPY and cohorts in regulating appetite, obesity and metabolic syndrome: Beneficial effects of gene therapy. Neuropeptides 2004, 38, 201–211. [Google Scholar] [CrossRef]
- Anderson, K.A.; Means, R.L.; Huang, Q.H.; Kemp, B.E.; Goldstein, E.G.; Selbert, M.A.; Edelman, A.M.; Fremeau, R.T.; Means, A.R. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J. Biol. Chem. 1998, 273, 31880–31889. [Google Scholar] [CrossRef]
- Qi, T.; Wu, Y.; Zeng, J.; Zhang, F.; Xue, A.; Jiang, L.; Zhu, Z.; Kemper, K.; Yengo, L.; Zheng, Z.; et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 2018, 9, 2282. [Google Scholar] [CrossRef]
- Huffman, J.E. Examining the current standards for genetic discovery and replication in the era of mega-biobanks. Nat. Commun. 2018, 9, 5054. [Google Scholar] [CrossRef]
- Ramírez, J.; Duijvenboden, S.V.; Ntalla, I.; Mifsud, B.; Warren, H.R.; Tzanis, E.; Orini, M.; Tinker, A.; Lambiase, P.D.; Munroe, P.B. Thirty loci identified for heart rate response to exercise and recovery implicate autonomic nervous system. Nat. Commun. 2018, 9, 1947. [Google Scholar] [CrossRef]
- Verweij, N.; van de Vegte, Y.J.; van der Harst, P. Genetic study links components of the autonomous nervous system to heart-rate profile during exercise. Nat. Commun. 2018, 9, 898. [Google Scholar] [CrossRef] [PubMed]


| Locus | Chromosome | Locus Starts | Locus End | Top SNP | Top SNP Position | Effect Allele | Non-Effect Allele | Effect Allele Frequency | Beta | Standard Error | p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 72,511,514 | 72,956,535 | rs66495454 | 72,748,567 | GTCCT | G | 0.38 | 0.126 | 0.01 | 7.60 × 10−18 |
| 2 | 3 | 35,778,773 | 35,913,342 | rs56331918 | 35,801,168 | G | C | 0.28 | −0.1 | 0.02 | 6.90 × 10−10 |
| 3 | 5 | 60,613,826 | 60,844,213 | rs544711163 | 60,775,743 | CT | C | 0.38 | −0.082 | 0.01 | 1.90 × 10−8 |
| 4 | 8 | 8,088,230 | 11,463,015 | rs73195303 | 10,200,253 | T | C | 0.23 | −0.105 | 0.02 | 5.30 × 10−10 |
| 5 | 12 | 49,385,679 | 49,479,968 | rs1054442 | 49,389,320 | C | A | 0.37 | 0.085 | 0.01 | 7.70 × 10−9 |
| 6 | 16 | 53,797,908 | 53,845,487 | rs56094641 | 53,806,453 | G | A | 0.40 | 0.111 | 0.01 | 1.30 × 10−14 |
| 7 | 18 | 57,732,418 | 57,912,226 | rs35614134 | 57,832,856 | AC | A | 0.24 | 0.097 | 0.02 | 6.30 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mompeo, O.; Freidin, M.B.; Gibson, R.; Hysi, P.G.; Christofidou, P.; Segal, E.; Valdes, A.M.; Spector, T.D.; Menni, C.; Mangino, M. Genome-Wide Association Analysis of Over 170,000 Individuals from the UK Biobank Identifies Seven Loci Associated with Dietary Approaches to Stop Hypertension (DASH) Diet. Nutrients 2022, 14, 4431. https://doi.org/10.3390/nu14204431
Mompeo O, Freidin MB, Gibson R, Hysi PG, Christofidou P, Segal E, Valdes AM, Spector TD, Menni C, Mangino M. Genome-Wide Association Analysis of Over 170,000 Individuals from the UK Biobank Identifies Seven Loci Associated with Dietary Approaches to Stop Hypertension (DASH) Diet. Nutrients. 2022; 14(20):4431. https://doi.org/10.3390/nu14204431
Chicago/Turabian StyleMompeo, Olatz, Maxim B. Freidin, Rachel Gibson, Pirro G. Hysi, Paraskevi Christofidou, Eran Segal, Ana M. Valdes, Tim D. Spector, Cristina Menni, and Massimo Mangino. 2022. "Genome-Wide Association Analysis of Over 170,000 Individuals from the UK Biobank Identifies Seven Loci Associated with Dietary Approaches to Stop Hypertension (DASH) Diet" Nutrients 14, no. 20: 4431. https://doi.org/10.3390/nu14204431
APA StyleMompeo, O., Freidin, M. B., Gibson, R., Hysi, P. G., Christofidou, P., Segal, E., Valdes, A. M., Spector, T. D., Menni, C., & Mangino, M. (2022). Genome-Wide Association Analysis of Over 170,000 Individuals from the UK Biobank Identifies Seven Loci Associated with Dietary Approaches to Stop Hypertension (DASH) Diet. Nutrients, 14(20), 4431. https://doi.org/10.3390/nu14204431