
Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Isolation of Bacterial Extracellular Vesicles (BEVs)
2.2. Generation and Stimulation of Human Monocyte Derived-DCs (mo-DCs)
2.3. DC/Naïve CD4+ T-Cell Co-Cultures
2.4. Cytokine Quantification
2.5. Isolati, on of DC-Derived Extracellular Vesicles (EVs)
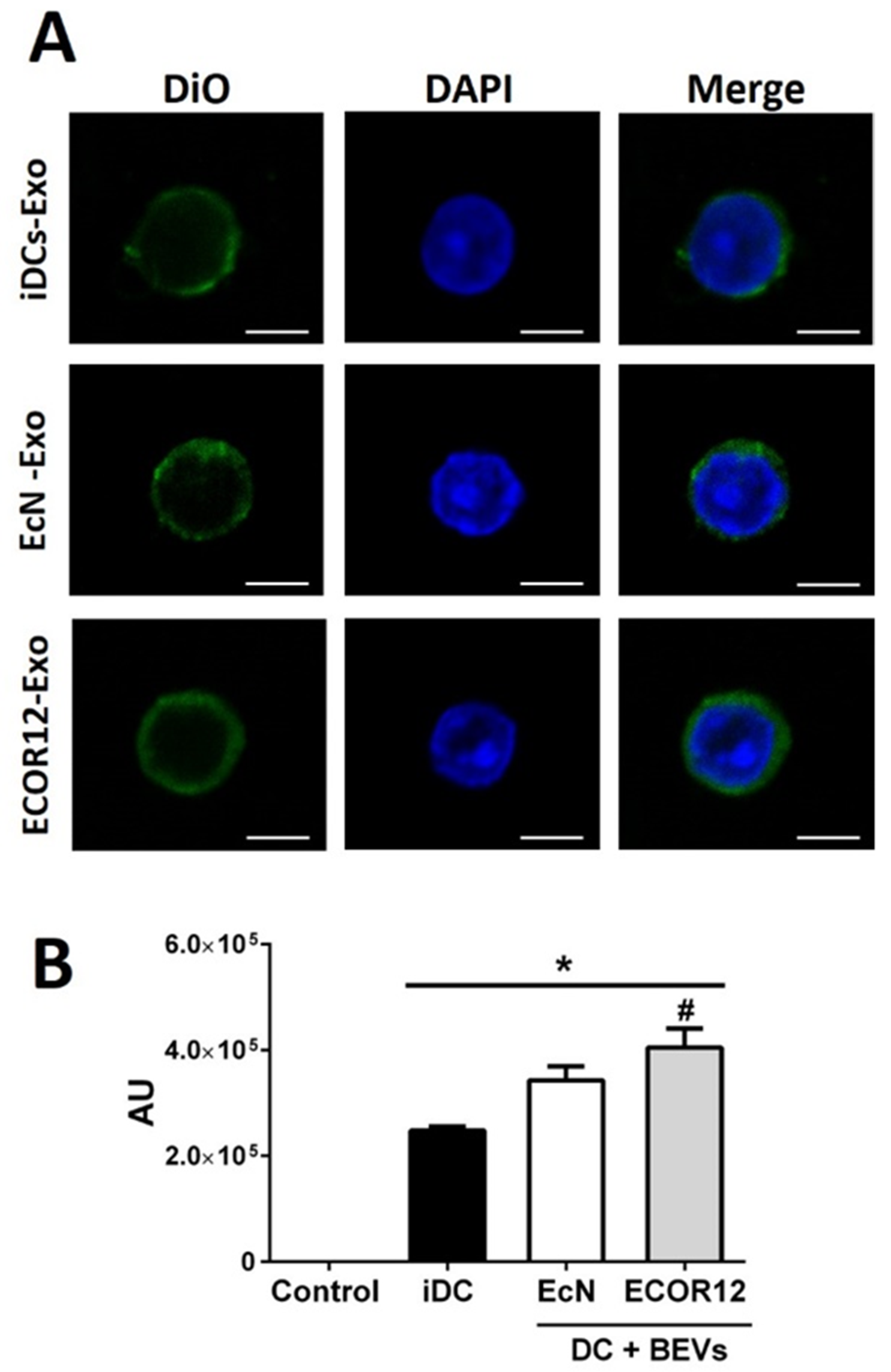
2.6. Uptake of DiO-Labeled DC-EVs by T-Cells
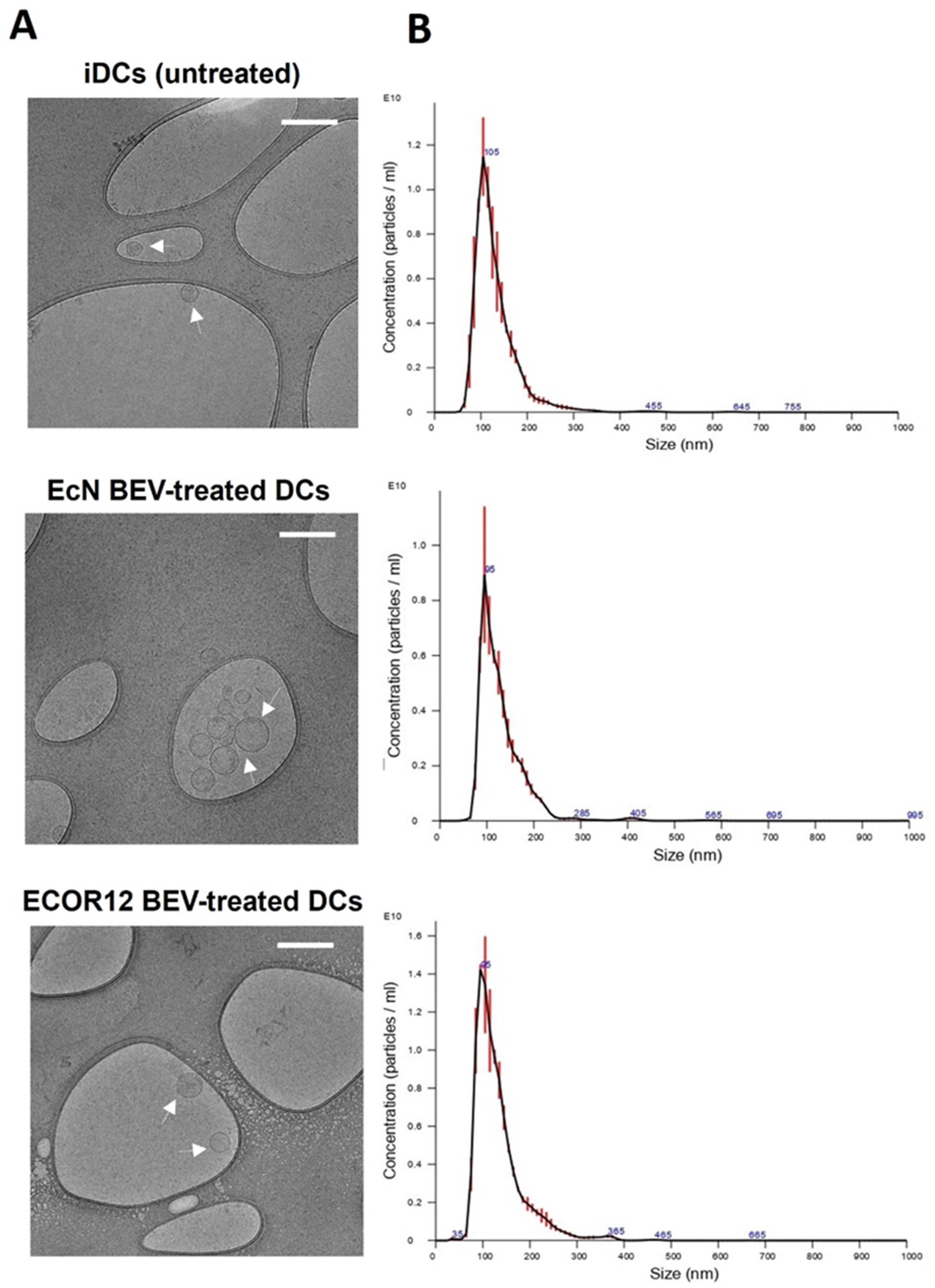
2.7. Cryo-Transmission Electron Microscopy (Cryo-TEM) of DC-Derived EVs
2.8. Nanoparticle Tracking Analysis (NTA)
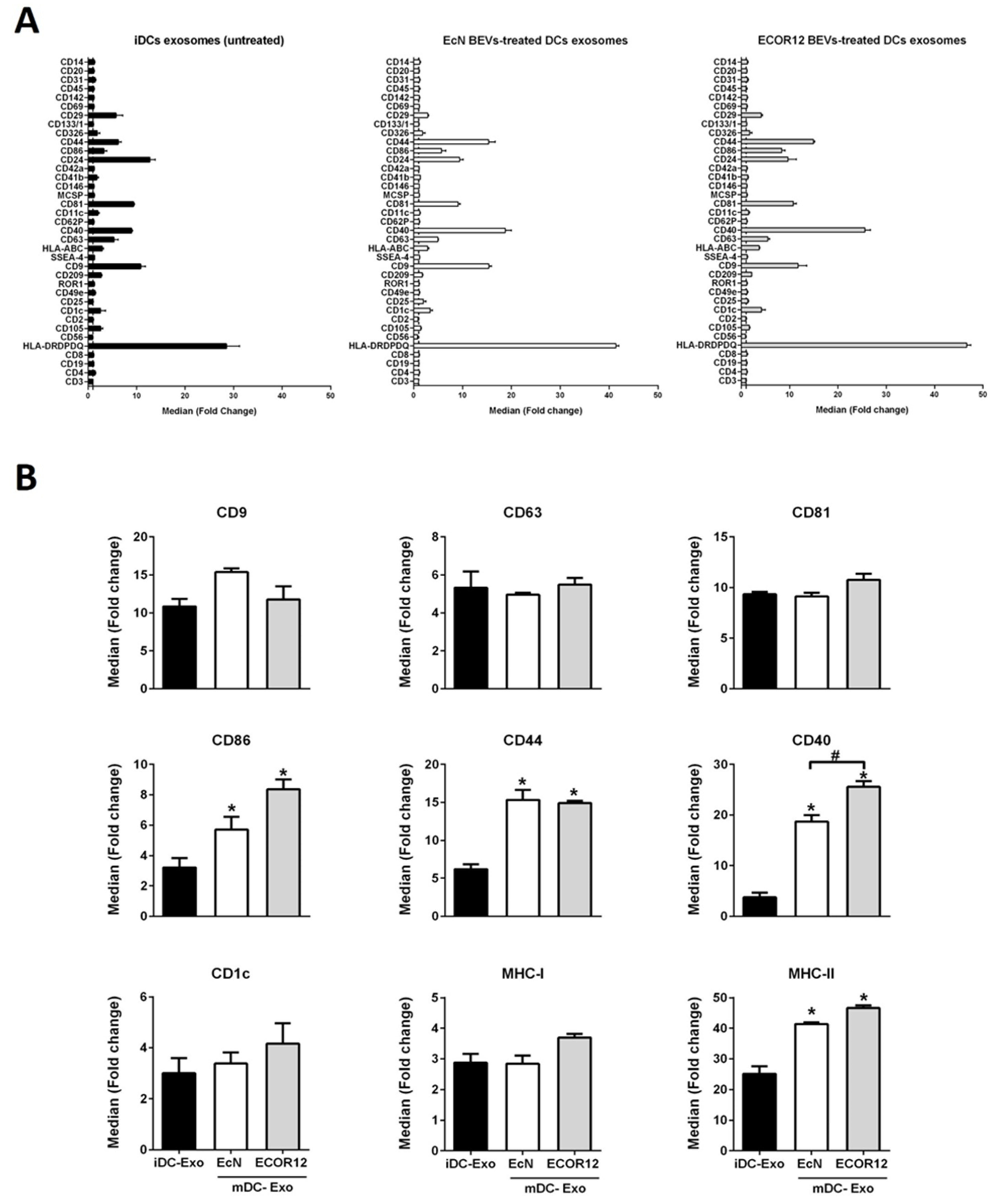
2.9. Multiplex Surface Marker Analysis
2.10. RNA Isolation
2.11. miRNA Expression Analysis by RT-qPCR
2.12. Ethics
2.13. Statistical Analysis
3. Results
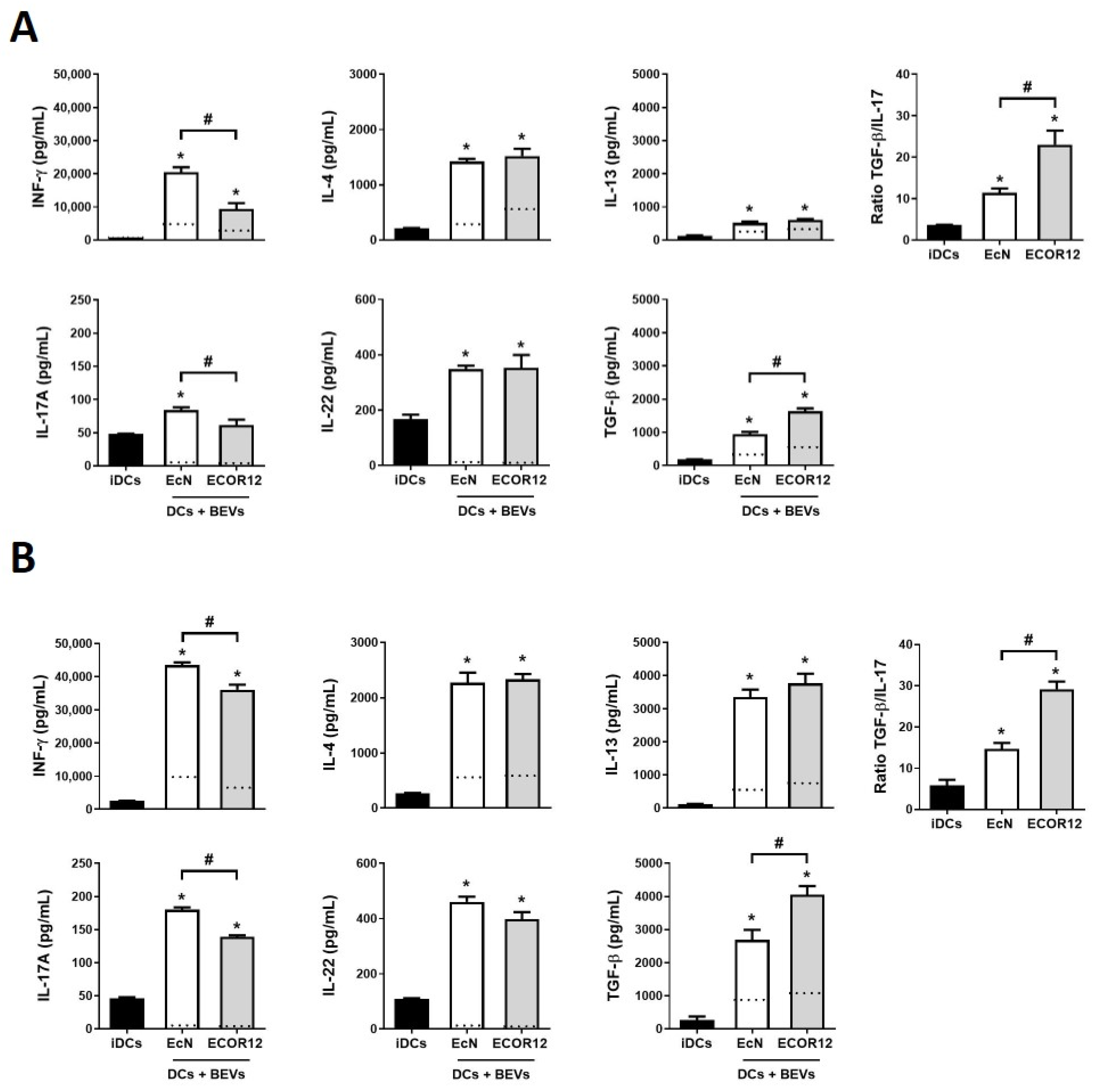
3.1. BEVs from Gut Microbiota Elicit a Distinctive Cytokine Profile in Human mo-DCs
3.2. Indirect and Direct Co-Cultures of BEV-Activated DCs/Naïve CD4+ T-Cells Generate Th-Specific Responses
3.3. Characterization of EV Secreted by mo-DCs in Response to Microbiota BEVs
3.4. EVs Secreted by BEV-Activated DCs Express a Different Profile of Surface Antigens and Costimulatory Molecules
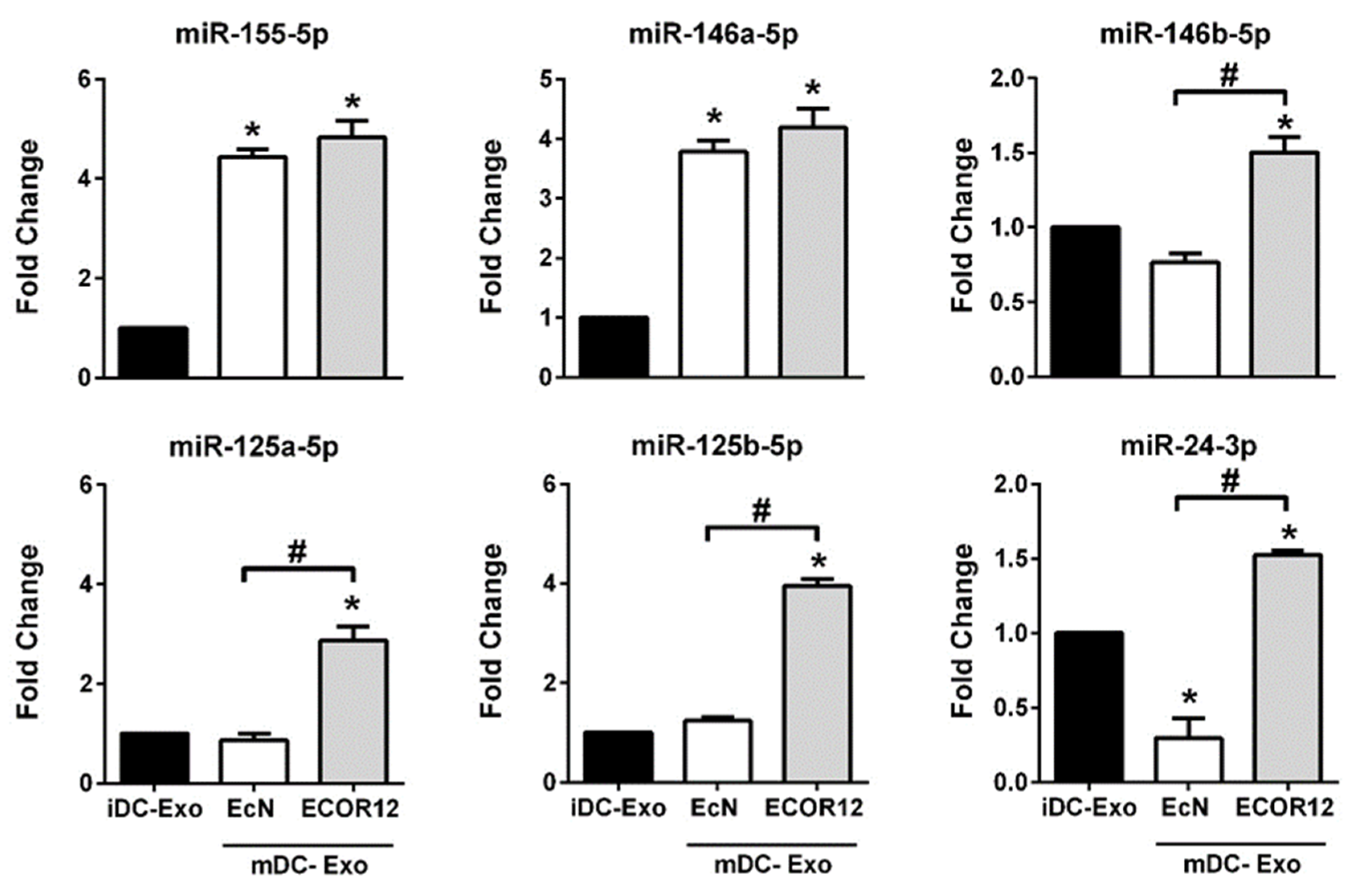
3.5. DC-Derived EVs Differentially Carry miRNAs Involved in Immune Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, S.; Pamer, E.G. Microbiota-mediated inflammation and antimicrobial defense in the intestine. Annu. Rev. Immunol. 2015, 33, 227–256. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Stagg, A.J. Intestinal dendritic cells in health and gut inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Manicassamy, S.; Pulendran, B. Dendritic cell control of tolerogenic responses. Immunol. Rev. 2011, 241, 206–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J. T helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, L.; Li, C.; Yu, Y.; Yi, Y.; Wang, J.; Chen, D. Exosome-induced regulation in inflammatory bowel disease. Front. Immunol. 2019, 10, 1464. [Google Scholar] [CrossRef]
- Thery, C.; Duban, L.; Segura, E.; Veron, P.; Lantz, O.; Amigorena, S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Nolte-‘t Hoen, E.N.M.; Buschow, S.I.; Anderton, S.M.; Stoorvogel, W.; Wauben, M.H.M. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood 2009, 113, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.M.; Round, J.L.; O’Connell, R.M. No small matter: Emerging roles for exosomal miRNAs in the immune system. FEBS J. 2021. [Google Scholar] [CrossRef]
- Diaz-Garrido, N.; Cordero, C.; Olivo-Martinez, Y.; Badia, J.; Baldomà, L. Cell-to-cell communication by host-released extracellular vesicles in the gut: Implications in health and disease. Int. J. Mol. Sci. 2021, 22, 2213. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, B.; Urdaci, M.C.; Margolles, A. Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa-bacteria interactions. Microbiology 2010, 156, 3232–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Huang, S.; Wang, Y.; Cai, S.; Yu, H.; Liu, H.; Zeng, X.; Zhang, G.; Qiao, S. Bridging intestinal immunity and gut microbiota by metabolites. Cell. Mol. Life. Sci. 2019, 76, 3917–3937. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ren, Y.; Fu, X. Inter-kingdom signaling between gut microbiota and their host. Cell. Mol. Life Sci. 2019, 76, 2383–2389. [Google Scholar] [CrossRef]
- Haurat, M.F.; Elhenawy, W.; Feldman, M.F. Prokaryotic membrane vesicles: New insights on biogenesis and biological roles. Biol. Chem. 2015, 396, 95–109. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef]
- Nissle, A. Ueber die Grundlagen einer neuen ursaechlichen Bekaempfung der pathologischen Darmflora (On the fundamentals for new causal control of pathological intestinal microflora). Dt Med. Wschr 1916, 42, 1181–1184. [Google Scholar]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañas, M.A.; Gimenez, R.; Fabrega, M.J.; Toloza, L.; Baldoma, L.; Badia, J. Outer membrane vesicles from the probiotic Escherichia coli Nissle 1917 and the commensal ECOR12 enter intestinal epithelial cells via clathrin-dependent endocytosis and elicit differential effects on DNA damage. PLoS ONE 2016, 11, e0160374. [Google Scholar] [CrossRef]
- Cañas, M.A.; Fabrega, M.J.; Gimenez, R.; Badia, J.; Baldoma, L. Outer membrane vesicles from probiotic and commensal Escherichia coli activate NOD1-mediated immune responses in intestinal epithelial cells. Front. Microbiol. 2018, 9, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, C.S.; Badia, J.; Bosch, M.; Gimenez, R.; Baldoma, L. Outer membrane vesicles and soluble factors released by probiotic Escherichia coli Nissle 1917 and commensal ECOR63 enhance barrier function by regulating expression of tight junction proteins in intestinal epithelial cells. Front. Microbiol. 2016, 7, 1981. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.-S.; Giménez, R.; Cañas, M.-A.; Vera, R.; Díaz-Garrido, N.; Badia, J.; Baldomà, L. Extracellular vesicles and soluble factors secreted by Escherichia coli Nissle 1917 and ECOR63 protect against enteropathogenic E. coli-induced intestinal epithelial barrier dysfunction. BMC Microbiol. 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrega, M.J.; Aguilera, L.; Gimenez, R.; Varela, E.; Alexandra Canas, M.; Antolin, M.; Badia, J.; Baldoma, L. Activation of immune and defense responses in the intestinal mucosa by outer membrane vesicles of commensal and probiotic Escherichia coli strains. Front. Microbiol. 2016, 7, 705. [Google Scholar] [CrossRef] [Green Version]
- Fabrega, M.J.; Rodriguez-Nogales, A.; Garrido-Mesa, J.; Algieri, F.; Badia, J.; Gimenez, R.; Galvez, J.; Baldoma, L. Intestinal anti-inflammatory effects of outer membrane vesicles from Escherichia coli Nissle 1917 in DSS-experimental colitis in mice. Front. Microbiol. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garrido, N.; Fábrega, M.J.; Vera, R.; Giménez, R.; Badia, J.; Baldomà, L. Membrane vesicles from the probiotic Nissle 1917 and gut resident Escherichia coli strains distinctly modulate human dendritic cells and subsequent T cell responses. J. Funct. Foods 2019, 61, 103495. [Google Scholar] [CrossRef]
- Díaz-Garrido, N.; Bonnin, S.; Riera, M.; Gíménez, R.; Badia, J.; Baldomà, L. Transcriptomic microRNA profiling of dendritic cells in response to gut microbiota-secreted vesicles. Cells 2020, 9, 1534. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, L.; Toloza, L.; Gimenez, R.; Odena, A.; Oliveira, E.; Aguilar, J.; Badia, J.; Baldoma, L. Proteomic analysis of outer membrane vesicles from the probiotic strain Escherichia coli Nissle 1917. Proteomics 2014, 14, 222–229. [Google Scholar] [CrossRef]
- Tkach, M.; Kowal, J.; Zucchetti, A.E.; Enserink, L.; Jouve, M.; Lankar, D.; Saitakis, M.; Martin-Jaular, L.; Thery, C. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. 2017, 36, 3012–3028. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Koerhuis, D.G.J.; Borg, E.G.F.; van ’t Veld, E.M.; Driedonks, T.A.P.; Wubbolts, R.; Stoorvogel, W.; Boes, M. Bystander T-cells support clonal T-cell activation by controlling the release of dendritic cell-derived immune-stimulatory extracellular vesicles. Front. Immunol. 2019, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Kabat, A.M.; Srinivasan, N.; Maloy, K.J. Modulation of immune development and function by intestinal microbiota. Trends Immunol. 2014, 35, 507–517. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Obregon, C.; Rothen-Rutishauser, B.; Gitahi, S.K.; Gehr, P.; Nicod, L.P. Exovesicles from human activated dendritic cells fuse with resting dendritic cells, allowing them to present alloantigens. Am. J. Pathol. 2006, 169, 2127–2136. [Google Scholar] [CrossRef] [Green Version]
- Segura, E.; Nicco, C.; Lombard, B.; Véron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Théry, C. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.-C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Haase, C.; Michelsen, B.K.; Jørgensen, T.N. CD40 is necessary for activation of naïve T cells by a dendritic cell line in vivo but not in vitro. Scand. J. Immunol. 2004, 59, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front. Immunol. 2019, 10, 1325. [Google Scholar] [CrossRef]
- Termeer, C.; Averbeck, M.; Hara, H.; Eibel, H.; Herrlich, P.; Sleeman, J.; Simon, J.C. Targeting dendritic cells with CD44 monoclonal antibodies selectively inhibits the proliferation of naive CD4+ T-helper cells by induction of FAS-independent T-cell apoptosis. Immunology 2003, 109, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur. J. Immunol. 2006, 36, 1772–1781. [Google Scholar] [CrossRef]
- Amado, T.; Schmolka, N.; Metwally, H.; Silva-Santos, B.; Gomes, A.Q. Cross-regulation between cytokine and microRNA pathways in T cells. Eur. J. Immunol. 2015, 45, 1584–1595. [Google Scholar] [CrossRef] [Green Version]
- Jeker, L.T.; Bluestone, J.A. MicroRNA regulation of T-cell differentiation and function. Immunol. Rev. 2013, 253, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejad, C.; Stunden, H.J.; Gantier, M.P. A guide to miRNAs in inflammation and innate immune responses. FEBS J. 2018, 285, 3695–3716. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J. Biol. Chem. 2015, 290, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gao, D.; Shao, Z.; Zheng, Q.; Yu, Q. miR-155 indicates the fate of CD4(+) T cells. Immunol. Lett. 2020, 224, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Garavelli, S.; De Rosa, V.; de Candia, P. The multifaceted interface between cytokines and microRNAs: An ancient mechanism to regulate the good and the bad of inflammation. Front. Immunol. 2018, 9, 3012. [Google Scholar] [CrossRef] [Green Version]
- Huffaker, T.B.; Hu, R.; Runtsch, M.C.; Bake, E.; Chen, X.; Zhao, J.; Round, J.L.; Baltimore, D.; O’Connell, R.M. Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell Rep. 2012, 2, 1697–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, R.; Ma, Y.-L.; Liang, W.; Li, H.-H.; Ma, Z.-J.; Yu, X.; Liao, Y.-H. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS ONE 2012, 7, e46082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Galán, A.; Fernández-Messina, L.; Sánchez-Madrid, F. Control of immunoregulatory molecules by miRNAs in T cell activation. Front. Immunol. 2018, 9, 2148. [Google Scholar] [CrossRef]
- Lu, L.-F.; Boldin, M.P.; Chaudhry, A.; Lin, L.-L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.R.; Fordham, J.B.; Ganesh, B.; Nares, S. miR-24, miR-30b and miR-142-3p interfere with antigen processing and presentation by primary macrophages and dendritic cells. Sci. Rep. 2016, 6, 32925. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.-B.; Zhang, H.; Cai, T.-T.; Liu, Y.-N.; Ni, J.-J.; He, J.; Peng, J.-Y.; Chen, Q.-Y.; Mo, H.-Y.; Jun-Cui; et al. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef]
- Baradaran Ghavami, S.; Asadzadeh Aghdaei, H.; Sorrentino, D.; Shahrokh, S.; Farmani, M.; Ashrafian, F.; Dore, M.P.; Keshavarz Azizi Raftar, S.; Mobin Khoramjoo, S.; Zali, M.R. Probiotic-induced tolerogenic dendritic cells: A novel therapy for inflammatory bowel disease? Int. J. Mol. Sci. 2021, 22, 8274. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Garrido, N.; Badia, J.; Baldomà, L. Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo. Nutrients 2022, 14, 344. https://doi.org/10.3390/nu14020344
Diaz-Garrido N, Badia J, Baldomà L. Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo. Nutrients. 2022; 14(2):344. https://doi.org/10.3390/nu14020344
Chicago/Turabian StyleDiaz-Garrido, Natalia, Josefa Badia, and Laura Baldomà. 2022. "Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo" Nutrients 14, no. 2: 344. https://doi.org/10.3390/nu14020344
APA StyleDiaz-Garrido, N., Badia, J., & Baldomà, L. (2022). Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo. Nutrients, 14(2), 344. https://doi.org/10.3390/nu14020344