
Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animal Experiment
2.2. Depletion of Gut Microbiota
2.3. Traumatic Brain Injury Model
2.4. Fecal Microbiota Transplant
2.5. Histology
2.6. Immunofluorescence Examination
2.7. RNA Sequencing
2.8. Untargeted Metabolomics Profiling
2.9. 16s rRNA Gene Sequence
2.10. Statistical Analysis
3. Results
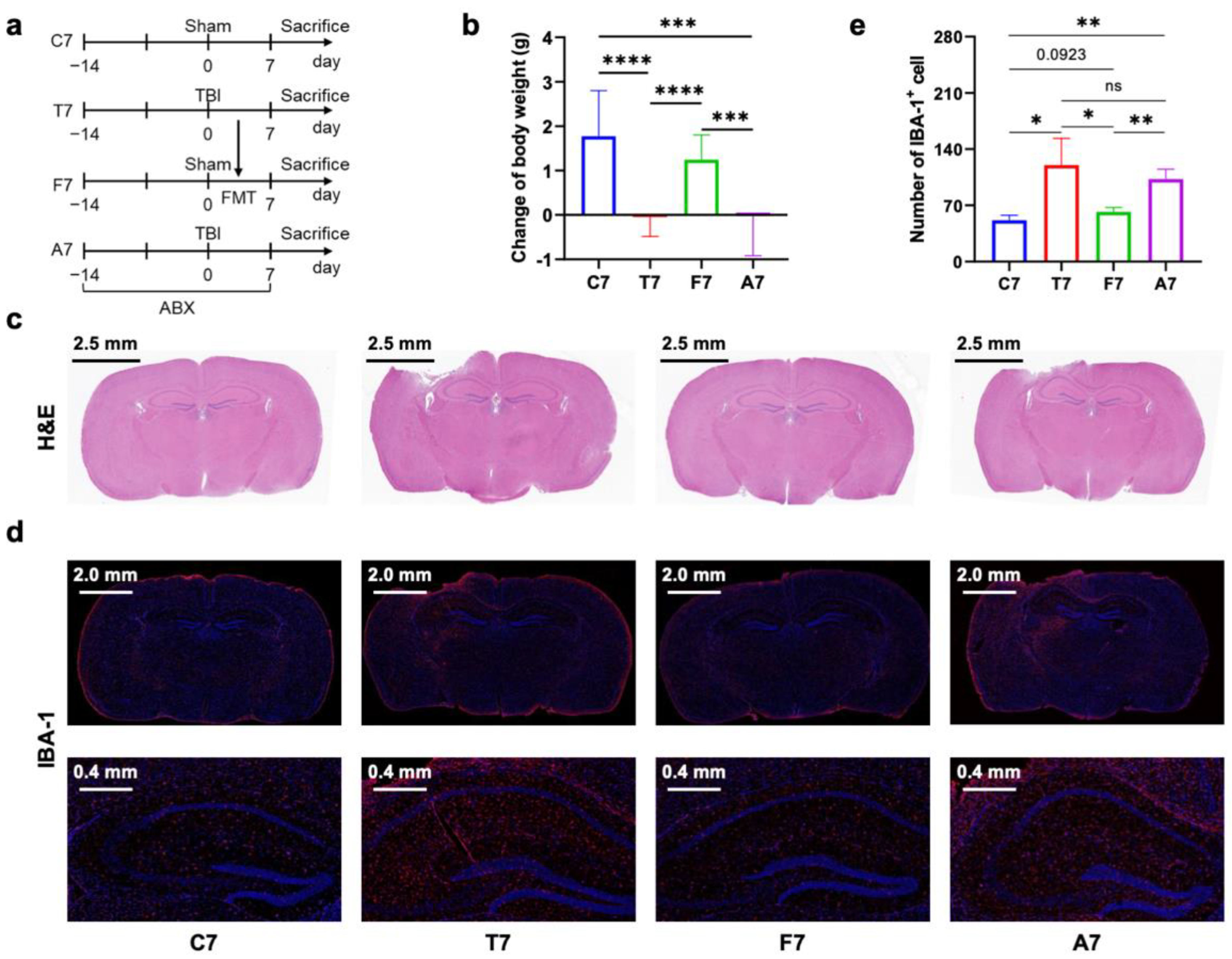
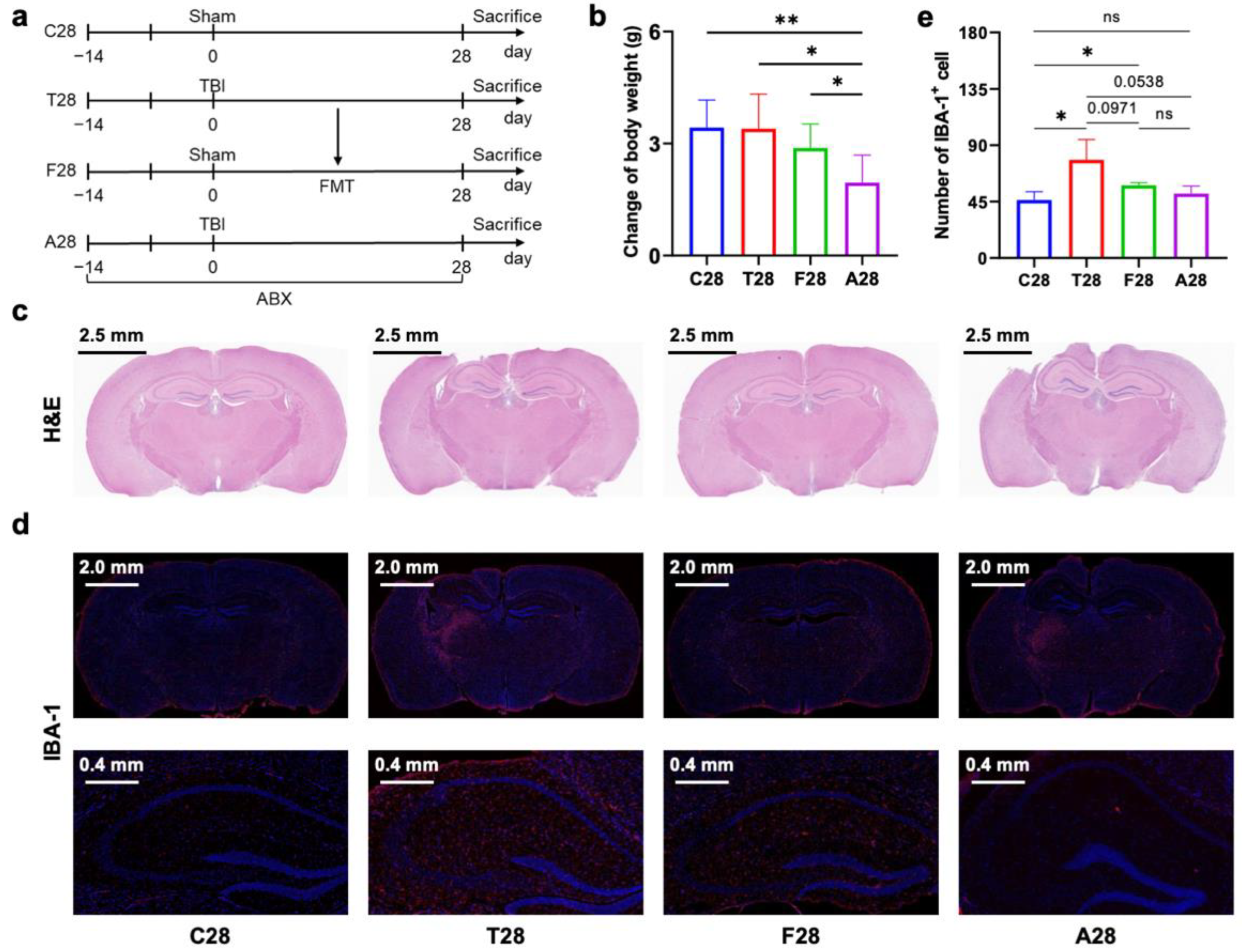
3.1. Gut Microbiota Dysbiosis Mediated Prolonged Microglial Activation in the Hippocampus of Mice with TBI
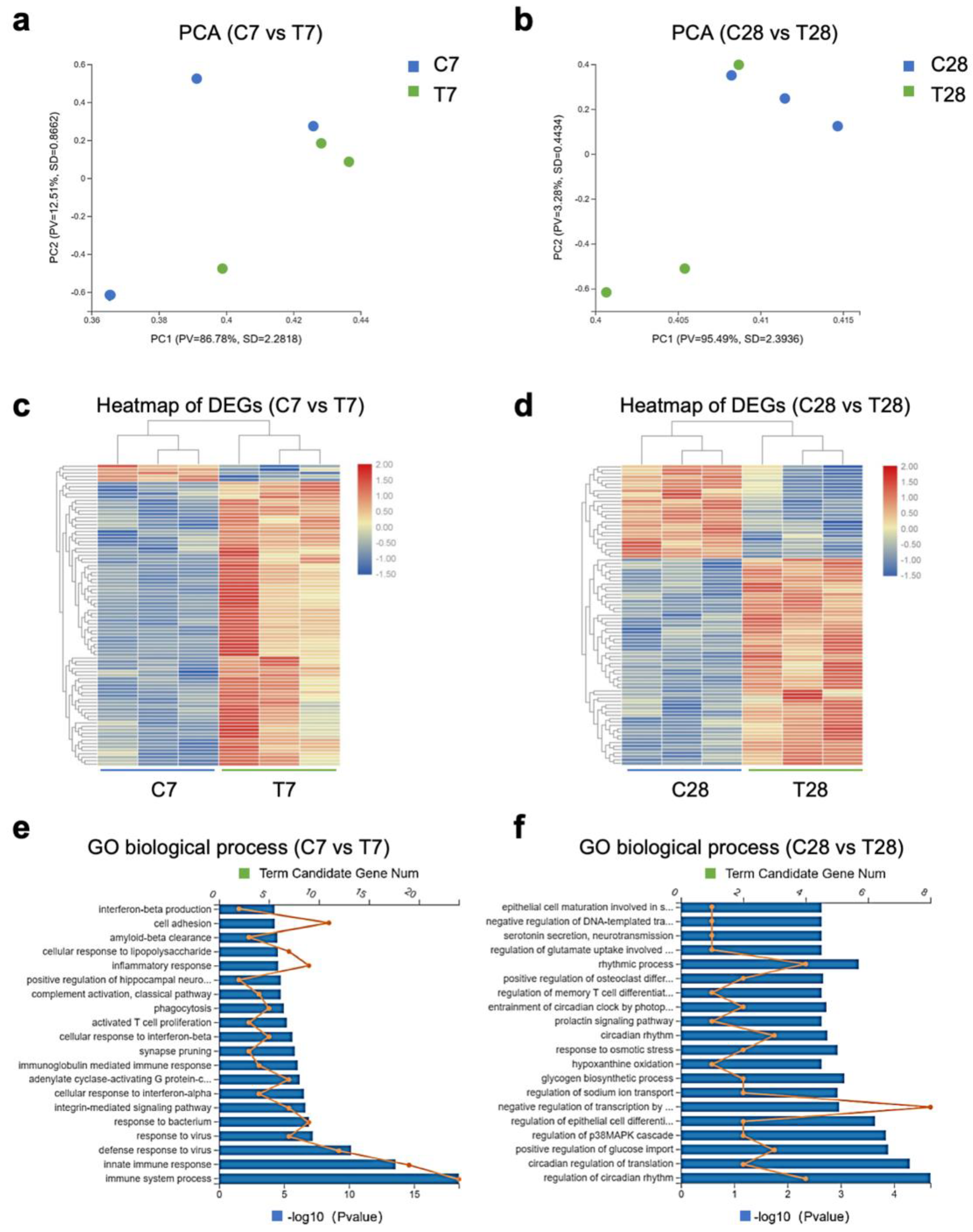
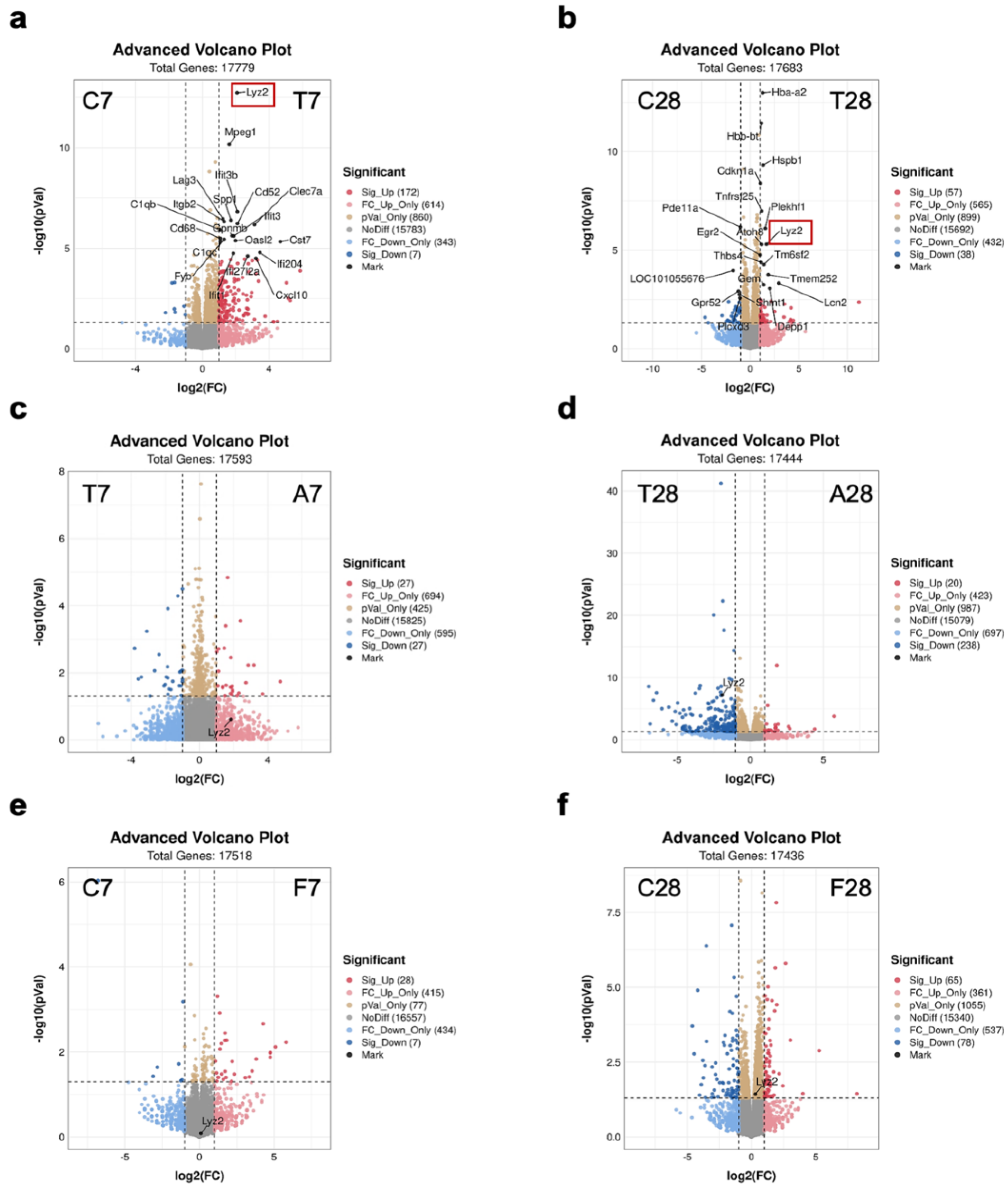
3.2. Gut Microbiota-Dependent Persistent Upregulation of Lyz2 Post-TBI Was Parallel with Prolonged Microglial Activation
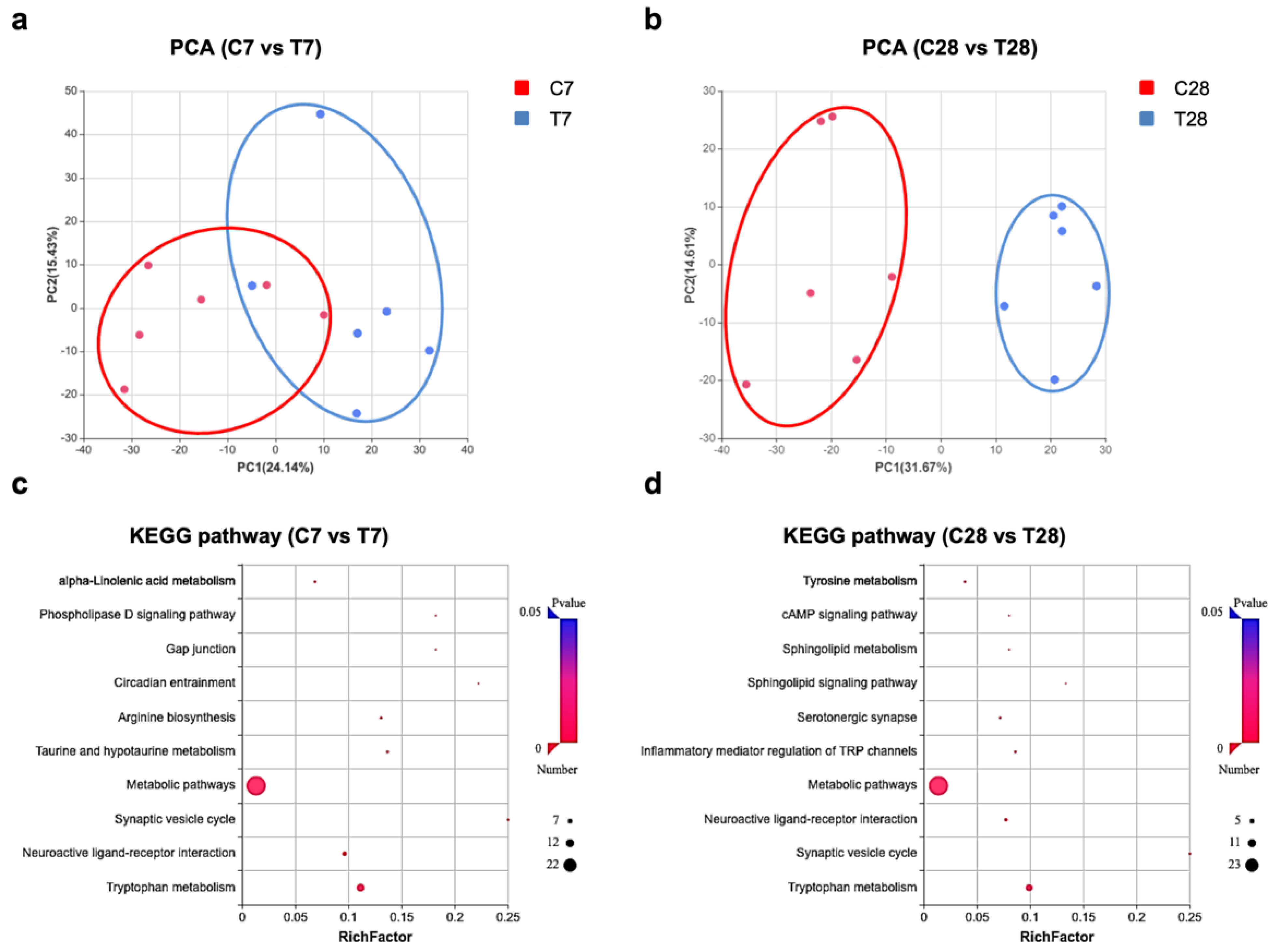
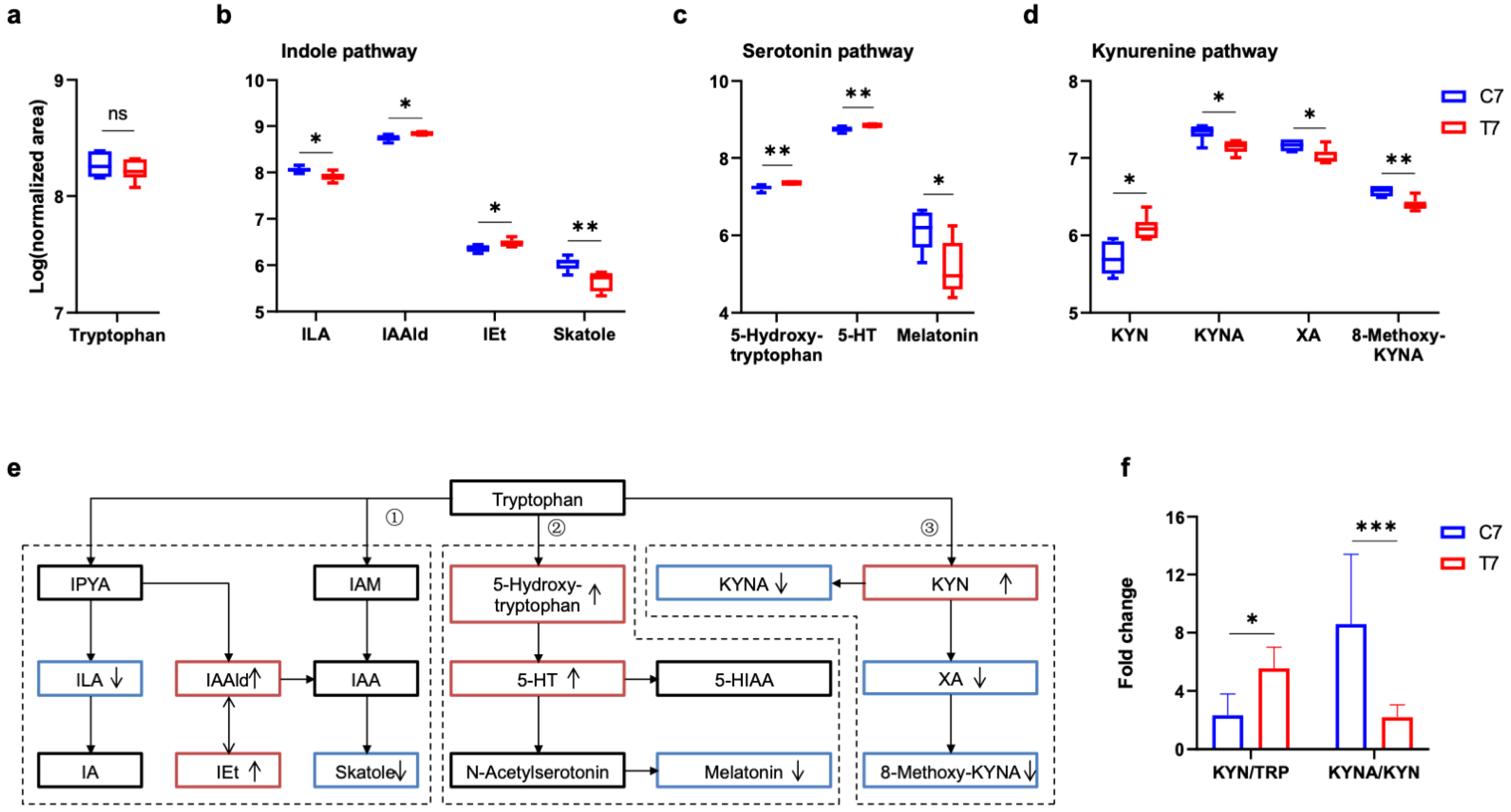
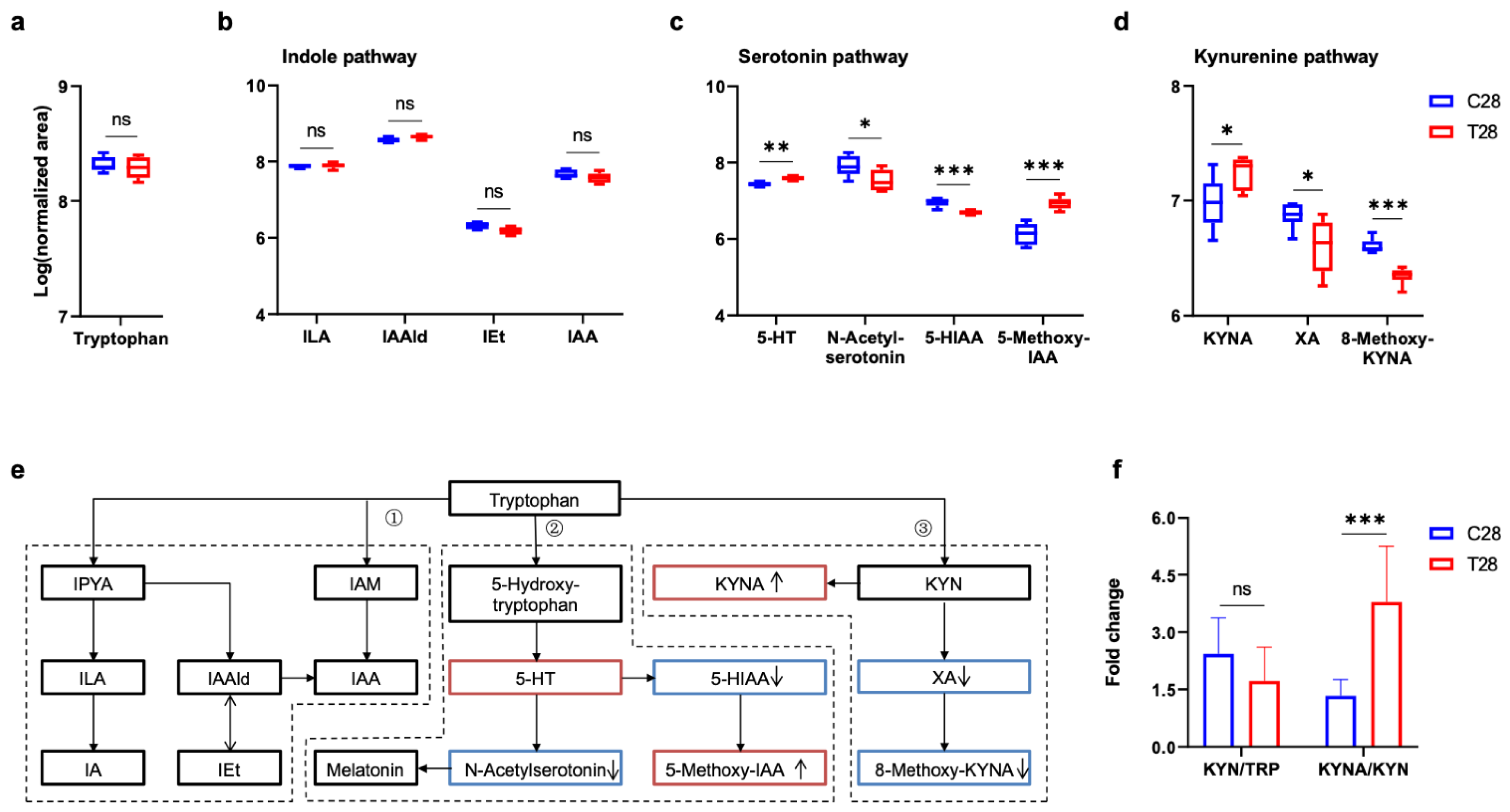
3.3. Tryptophan Metabolic Phenotype Was Differently Reshaped at 7 Days and 28 Days Post-TBI
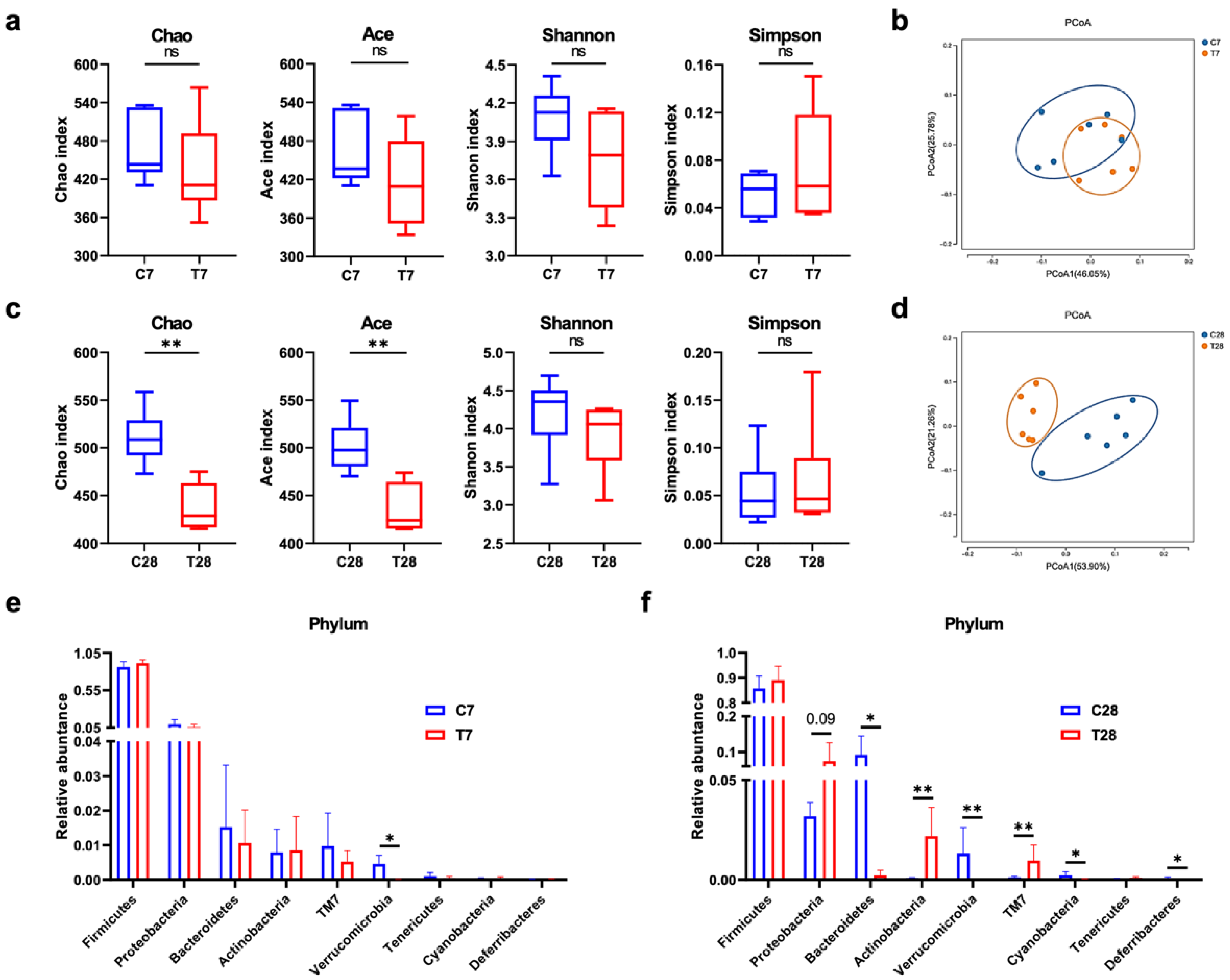
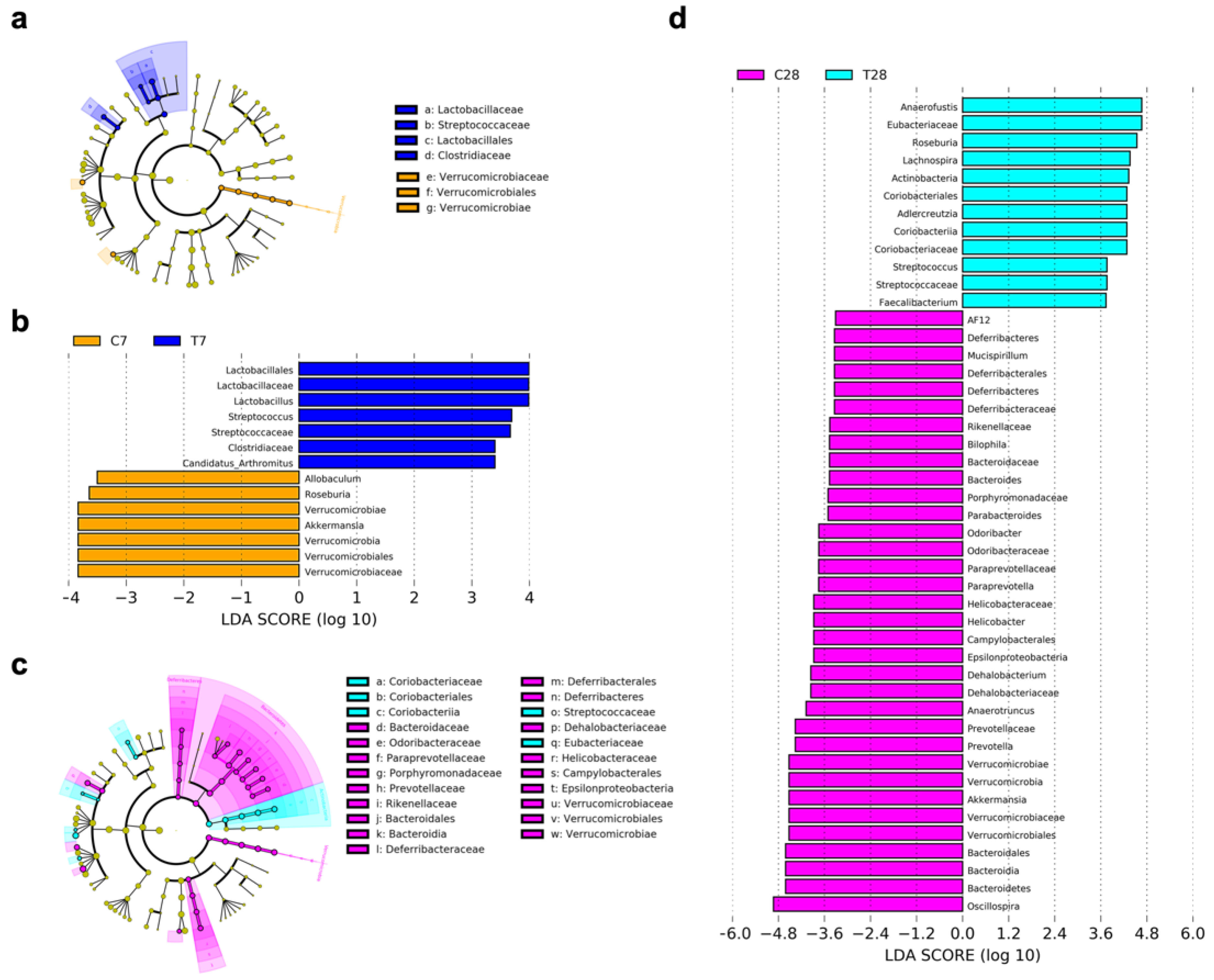
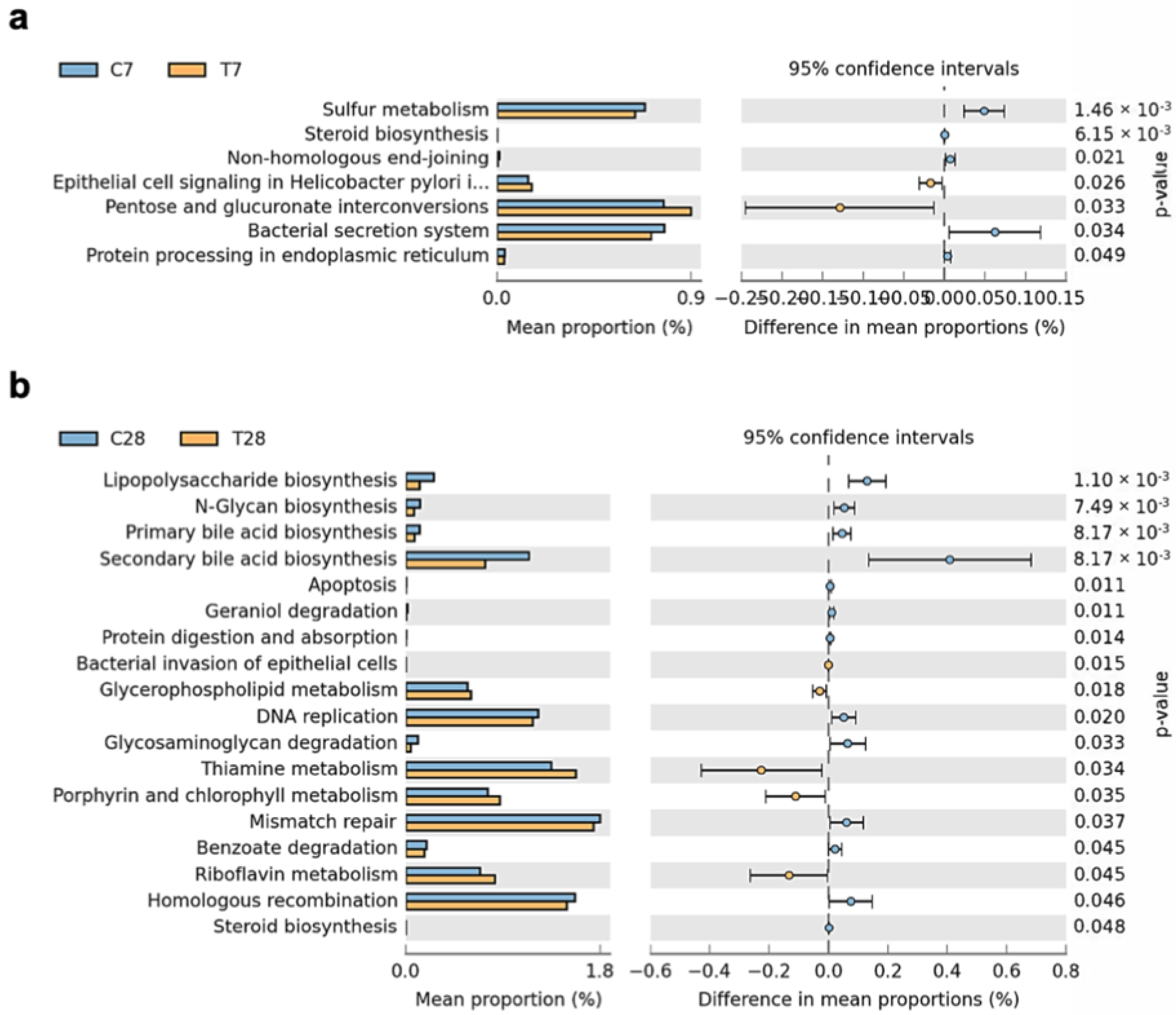
3.4. Composition and Function of Gut Microbiota Progressively Shifted to Dysbiosis Post-TBI
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, J.-Y.; Gao, G.-Y.; Feng, J.-F.; Mao, Q.; Chen, L.-G.; Yang, X.-F.; Liu, J.-F.; Wang, Y.-H.; Qiu, B.-H.; Huang, X.-J. Traumatic brain injury in China. Lancet Neurol. 2019, 18, 286–295. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2021, 91, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossmann, M.C.; Semple, B.D.; Hellewell, S.C.; Bye, N.; Ziebell, J.M. The complexity of neuroinflammation consequent to traumatic brain injury: From research evidence to potential treatments. Acta Neuropathol. 2019, 137, 731–755. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Bolte, A.C.; Lukens, J.R. Neuroimmune cleanup crews in brain injury. Trends Immunol. 2021, 42, 480–494. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Sundman, M.H.; Chen, N.-K.; Subbian, V.; Chou, Y.-H. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav. Immun. 2017, 66, 31–44. [Google Scholar] [CrossRef]
- Erny, D.; Prinz, M. How microbiota shape microglial phenotypes and epigenetics. Glia 2020, 68, 1655–1672. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.B.; Savitz, J. The Kynurenine Pathway in Traumatic Brain Injury: Implications for Psychiatric Outcomes. Biol. Psychiatry 2021, 91, 449–458. [Google Scholar] [CrossRef]
- Wu, L.; Han, Y.; Zheng, Z.; Zhu, S.; Chen, J.; Yao, Y.; Yue, S.; Teufel, A.; Weng, H.; Li, L.; et al. Obeticholic Acid Inhibits Anxiety via Alleviating Gut Microbiota-Mediated Microglia Accumulation in the Brain of High-Fat High-Sugar Diet Mice. Nutrients 2021, 13, 940. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, J.; Xu, S.; Fang, Y.; Wu, Y.; Zeng, J.; Shao, A.; Shi, L.; Lu, J.; Mei, S.; et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neu-roinflammation following traumatic brain injury. J. Neuroinflammation 2021, 18, 2. [Google Scholar] [CrossRef]
- Gong, S.; Lan, T.; Zeng, L.; Luo, H.; Yang, X.; Li, N.; Chen, X.; Liu, Z.; Li, R.; Win, S.; et al. Gut microbiota mediates diurnal variation of acetaminophen induced acute liver injury in mice. J. Hepatol. 2018, 69, 51–59. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Keane, L.; Antignano, I.; Riechers, S.P.; Zollinger, R.; Dumas, A.A.; Offermann, N.; Bernis, M.E.; Russ, J.; Graelmann, F.; McCormick, P.N.; et al. mTOR-dependent translation amplifies microglia priming in aging mice. J. Clin. Investig. 2021, 131, e132727. [Google Scholar] [CrossRef]
- Cadiz, M.P.; Jensen, T.D.; Sens, J.P.; Zhu, K.; Song, W.-M.; Bin Zhang, B.; Ebbert, M.; Chang, R.; Fryer, J.D. Culture shock: Microglial heterogeneity, activation, and disrupted single-cell microglial networks in vitro. Mol. Neurodegener. 2022, 17, 1–20. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf97994. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Depommier, C.; Derrien, M.; Everard, A.; de Vos, W.M. Akkermansia muciniphila: Paradigm for next-generation beneficial microorganisms. Nat. Rev. Gastroenterol. Hepatol. 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hanscom, M.; Loane, D.J.; Shea-Donohue, T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J. Clin. Investig. 2021, 131, e143777. [Google Scholar] [CrossRef]
- Yuan, B.; Lu, X.-J.; Wu, Q. Gut Microbiota and Acute Central Nervous System Injury: A New Target for Therapeutic Intervention. Front. Immunol. 2021, 12, 800796. [Google Scholar] [CrossRef]
- Mahajan, C.; Khurana, S.; Kapoor, I.; Sokhal, S.; Kumar, S.; Prabhakar, H.; Mathur, P.; Mani, K. Characteristics of Gut Microbiome After Traumatic Brain Injury. J. Neurosurg. Anesthesiol. 2021; online ahead of print. [Google Scholar] [CrossRef]
- Hou, Y.; Xu, L.; Song, S.; Fan, W.; Wu, Q.; Tong, X.; Yan, H. Oral Administration of Brain Protein Combined With Probiotics Induces Immune Tolerance Through the Tryptophan Pathway. Front. Mol. Neurosci. 2021, 14, 634631. [Google Scholar] [CrossRef]
- Urban, R.J.; Pyles, R.B.; Stewart, C.; Ajami, N.; Randolph, M.K.; Durham, W.J.; Danesi, C.P.; Dillon, E.L.; Summons, M.J.R.; Singh, C.K.; et al. Altered Fecal Microbiome Years after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 1037–1051. [Google Scholar] [CrossRef]
- Treangen, T.J.; Wagner, J.; Burns, M.P.; Villapol, S. Traumatic Brain Injury in Mice Induces Acute Bacterial Dysbiosis Within the Fecal Microbiome. Front. Immunol. 2018, 9, 2757. [Google Scholar] [CrossRef]
- Nicholson, S.E.; Watts, L.T.; Burmeister, D.M.; Merrill, D.; Scroggins, S.; Zou, Y.; Lai, Z.; Grandhi, R.; Lewis, A.M.; Newton, L.M.; et al. Moderate Traumatic Brain Injury Alters the Gastrointestinal Microbiome in a Time-Dependent Manner. Shock 2019, 52, 240–248. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Du, J.; Wang, F.; Fang, R.; Yu, C.; Xiong, J.; Chen, W.; Lu, Z.; Liu, J. Clostridium butyricum exerts a neuroprotective effect in a mouse model of traumatic brain injury via the gut-brain axis. Neurogastroenterol. Motil. 2017, 30, e13260. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, T.; Fu, J.; Fu, S.; Hu, C.; Sun, B.; Fan, X.; Zhu, J. Lactobacillus acidophilus Exerts Neuroprotective Effects in Mice with Traumatic Brain Injury. J. Nutr. 2019, 149, 1543–1552. [Google Scholar] [CrossRef]
- Celorrio, M.; Abellanas, M.A.; Rhodes, J.; Goodwin, V.; Moritz, J.; Vadivelu, S.; Wang, L.; Rodgers, R.; Xiao, S.; Anabayan, I.; et al. Gut microbial dysbiosis after traumatic brain injury modulates the immune response and impairs neurogenesis. Acta Neuropathol. Commun. 2021, 9, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Van Treuren, W.; Dodd, D. Microbial Contribution to the Human Metabolome: Implications for Health and Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 345–369. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, Z.; You, W.; Wang, Y.; Tu, M.; Zheng, P.; Wen, L.; Yang, X. A Retrospective Clinical Analysis of the Serum Bile Acid Alteration Caused by Traumatic Brain Injury. Front. Neurol. 2021, 12, 624378. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Zhu, Y.; Wei, A.; Du, J.; Wang, Y.; Zheng, P.; Tu, M.; Wang, H.; Wen, L.; Yang, X. Traumatic Brain Injury Induces Gastrointestinal Dysfunction and Dysbiosis of Gut Microbiota Accompanied by Alterations of Bile Acid Profile. J. Neurotrauma 2022, 39, 227–237. [Google Scholar] [CrossRef]
- Opeyemi, M.O.M.; Rogers, M.B.; Firek, B.A.; Janesko-Feldman, M.K.; Vagni, V.; Mullett, S.J.; Wendell, S.G.; Nelson, B.P.; New, L.A.; Mariño, E.; et al. Sustained Dysbiosis and Decreased Fecal Short-Chain Fatty Acids after Traumatic Brain Injury and Impact on Neurologic Outcome. J. Neurotrauma 2021, 38, 2610–2621. [Google Scholar] [CrossRef]
- Xie, Y.; Zou, X.; Han, J.; Zhang, Z.; Feng, Z.; Ouyang, Q.; Hua, S.; Liu, Z.; Li, C.; Cai, Y.; et al. Indole-3-propionic acid alleviates ischemic brain injury in a mouse middle cerebral artery occlusion model. Exp. Neurol. 2022, 353, 114081. [Google Scholar] [CrossRef]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Kong, Y.; Ye, T.; Yu, Q.; Satyanarayanan, S.K.; Su, K.-P.; Liu, J. Microbiota-derived metabolite Indoles induced aryl hydrocarbon receptor activation and inhibited neuroinflammation in APP/PS1 mice. Brain Behav. Immun. 2022, 106, 76–88. [Google Scholar] [CrossRef]
- Huang, Y.-L.; Lin, C.-H.; Tsai, T.-H.; Huang, C.-H.; Li, J.-L.; Chen, L.-K.; Li, C.-H.; Tsai, T.-F.; Wang, P.-N. Discovery of a Metabolic Signature Predisposing High Risk Patients with Mild Cognitive Impairment to Converting to Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lang, Z.; Yang, Q.; Nie, Y.; Wang, Z.; Gao, M.; Zhang, N.; Xu, X. UPLC-Q-TOF/MS-based plasma metabolome to identify biomarkers and time of injury in traumatic brain injured rats. NeuroReport 2021, 32, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Chen, C.C.; Liao, H.Y.; Wu, Y.W.; Liou, J.M.; Wu, M.S.; Kou, C.H.; Lin, C.H. Alteration of Gut Microbial Metabolites in the Systemic Cir-culation of Patients with Parkinson’s Disease. J. Parkinsons Dis. 2022, 12, 1219–1230. [Google Scholar] [CrossRef]
- Serger, E.; Luengo-Gutierrez, L.; Chadwick, J.S.; Kong, G.; Zhou, L.; Crawford, G.; Danzi, M.C.; Myridakis, A.; Brandis, A.; Bello, A.T.; et al. The gut metabolite indole-3 propionate promotes nerve regeneration and repair. Nature 2022, 607, 585–592. [Google Scholar] [CrossRef]
- Jing, Y.; Yang, D.; Bai, F.; Zhang, C.; Qin, C.; Li, D.; Wang, L.; Yang, M.; Chen, Z.; Li, J. Melatonin Treatment Alleviates Spinal Cord Injury-Induced Gut Dysbiosis in Mice. J. Neurotrauma 2019, 36, 2646–2664. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.-J.; Liu, C.; Yu, L.-Z.; Zhou, J.-H.; Li, Y.; Xiong, Y.; Guo, A.; Chao, L.-M.; Qu, Q.; Wei, G.-W.; et al. Melatonin Alleviates Neuroinflammation and Metabolic Disorder in DSS-Induced Depression Rats. Oxidative Med. Cell. Longev. 2020, 2020, 1–17. [Google Scholar] [CrossRef]
- Zhang, Z.; Rasmussen, L.; Saraswati, M.; Koehler, R.C.; Robertson, C.L.; Kannan, S. Traumatic Injury Leads to Inflammation and Altered Tryptophan Metabolism in the Juvenile Rabbit Brain. J. Neurotrauma 2019, 36, 74–86. [Google Scholar] [CrossRef]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar] [CrossRef]
- Krukowski, K.; Nolan, A.; Becker, M.; Picard, K.; Vernoux, N.; Frias, E.S.; Feng, X.; Tremblay, M.-E.; Rosi, S. Novel microglia-mediated mechanisms underlying synaptic loss and cognitive impairment after traumatic brain injury. Brain Behav. Immun. 2021, 98, 122–135. [Google Scholar] [CrossRef]
- Soriano, S.; Curry, K.; Wang, Q.; Chow, E.; Treangen, T.J.; Villapol, S. Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls. Int. J. Mol. Sci. 2022, 23, 4476. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Z.; Wang, S.; Wu, C.; Cao, Y.; Gu, Q.; Zhu, Y.; Zhang, W.; Hu, W. Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype. Nutrients 2022, 14, 3467. https://doi.org/10.3390/nu14173467
Zheng Z, Wang S, Wu C, Cao Y, Gu Q, Zhu Y, Zhang W, Hu W. Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype. Nutrients. 2022; 14(17):3467. https://doi.org/10.3390/nu14173467
Chicago/Turabian StyleZheng, Zhipeng, Shuai Wang, Chenghao Wu, Yang Cao, Qiao Gu, Ying Zhu, Wei Zhang, and Wei Hu. 2022. "Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype" Nutrients 14, no. 17: 3467. https://doi.org/10.3390/nu14173467
APA StyleZheng, Z., Wang, S., Wu, C., Cao, Y., Gu, Q., Zhu, Y., Zhang, W., & Hu, W. (2022). Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype. Nutrients, 14(17), 3467. https://doi.org/10.3390/nu14173467