
Vitamin D and Diseases of Mineral Homeostasis: A Cyp24a1 R396W Humanized Preclinical Model of Infantile Hypercalcemia Type 1

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of the R396W Knock-in Strain
2.2. Genotyping
2.3. Survival Analysis
2.4. Serum Biochemistry
2.5. Microcomputed Tomography of Whole Kidneys
2.6. Gene Expression Monitoring
2.7. Intramedullary Rodded Tibial Osteotomy
2.8. Three-Point Bending Assays
2.9. Statistical Analysis
3. Results
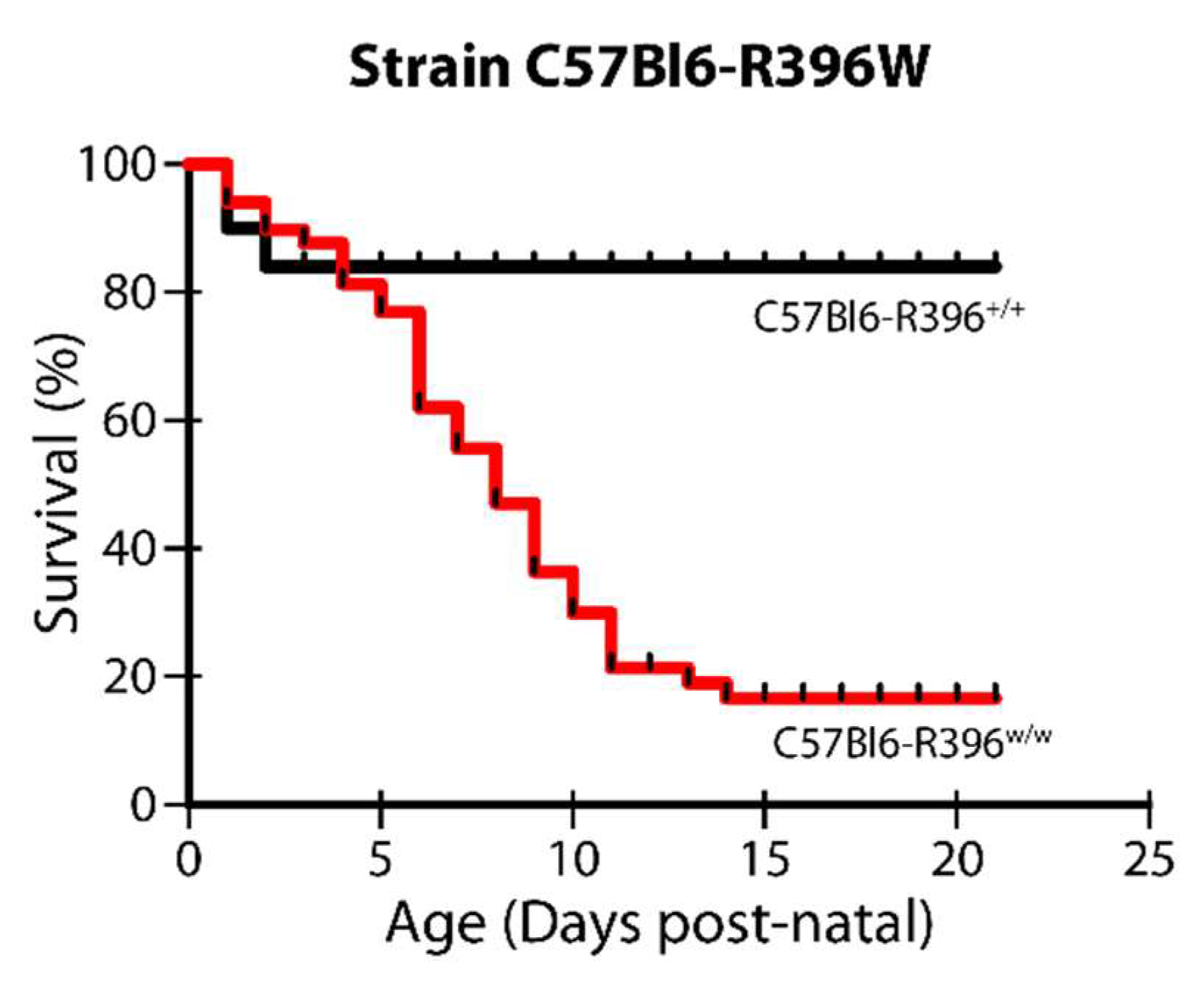
3.1. Severe Postnatal Lethality in C57Bl6-R396w/w Mutant Animals
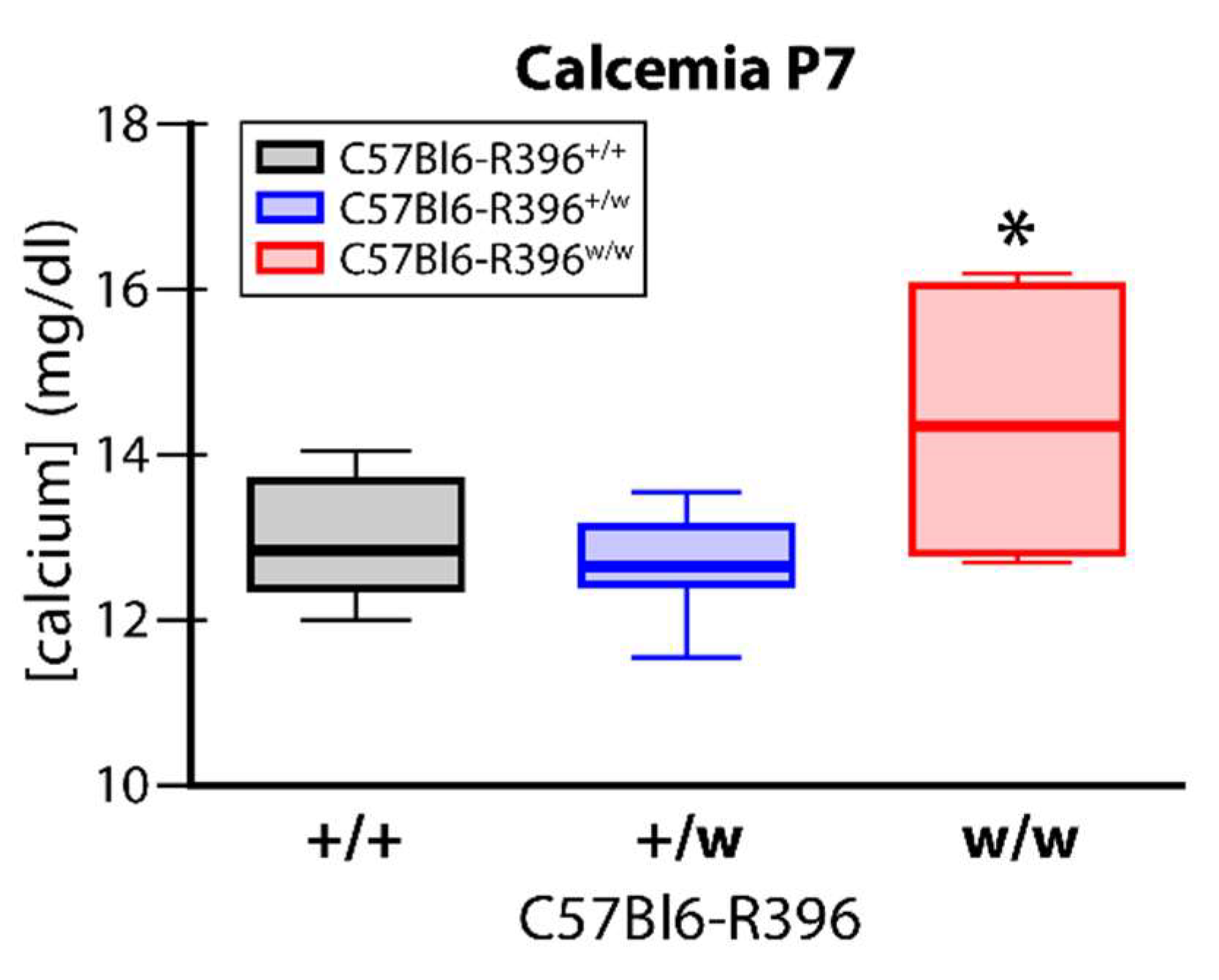
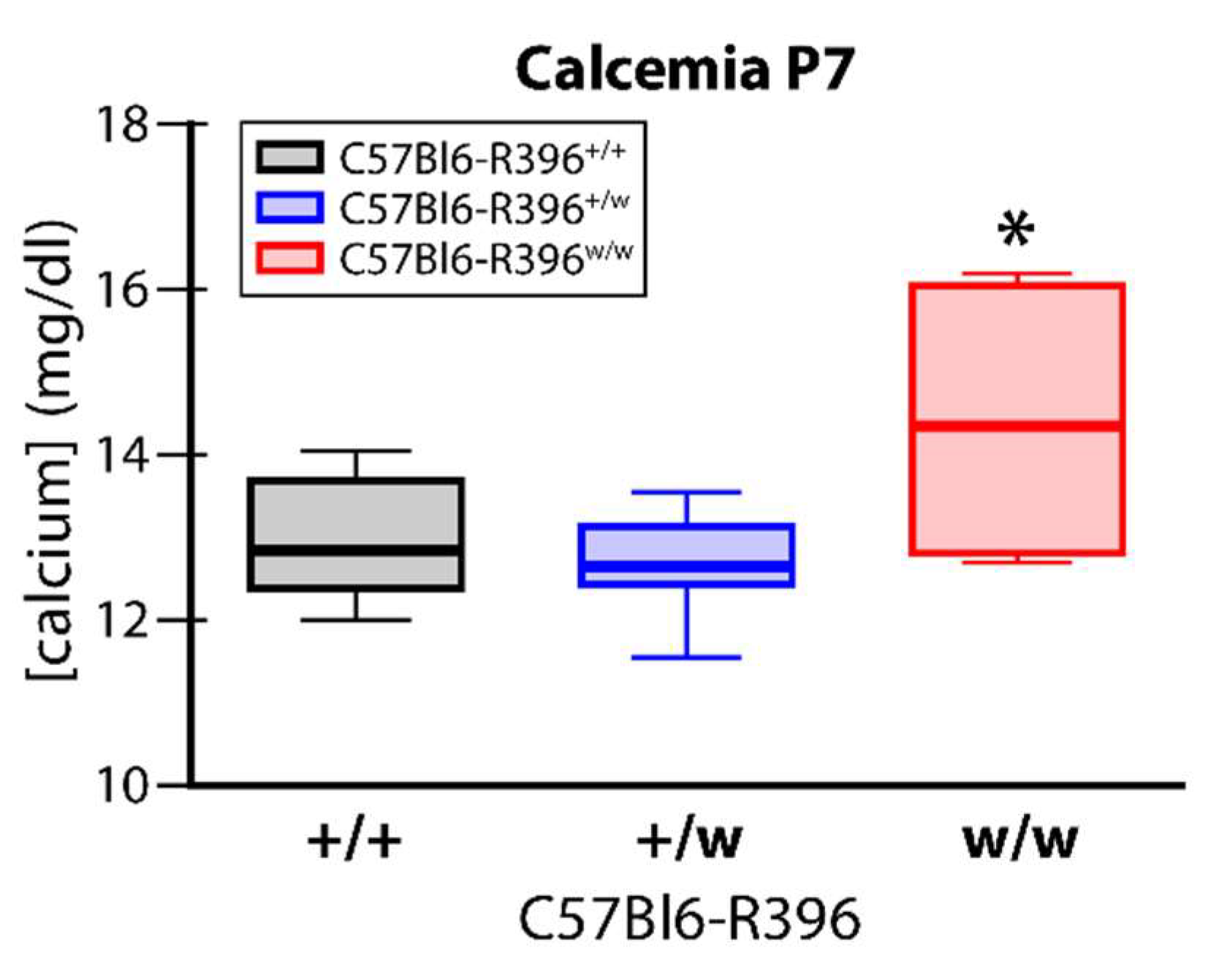
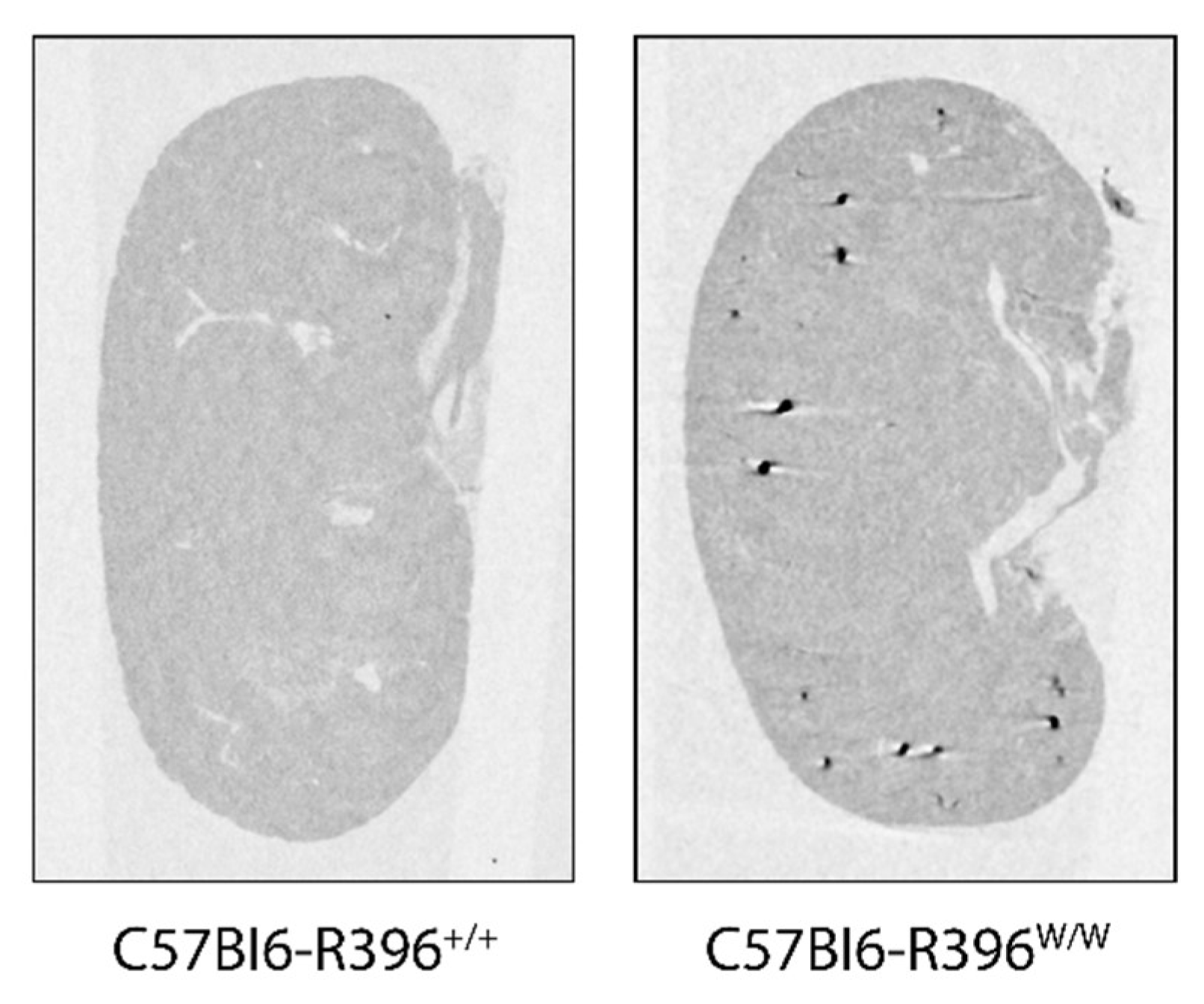
3.2. Early Postnatal Hypercalcemia in C57Bl6-R396w/w Mutant Animals
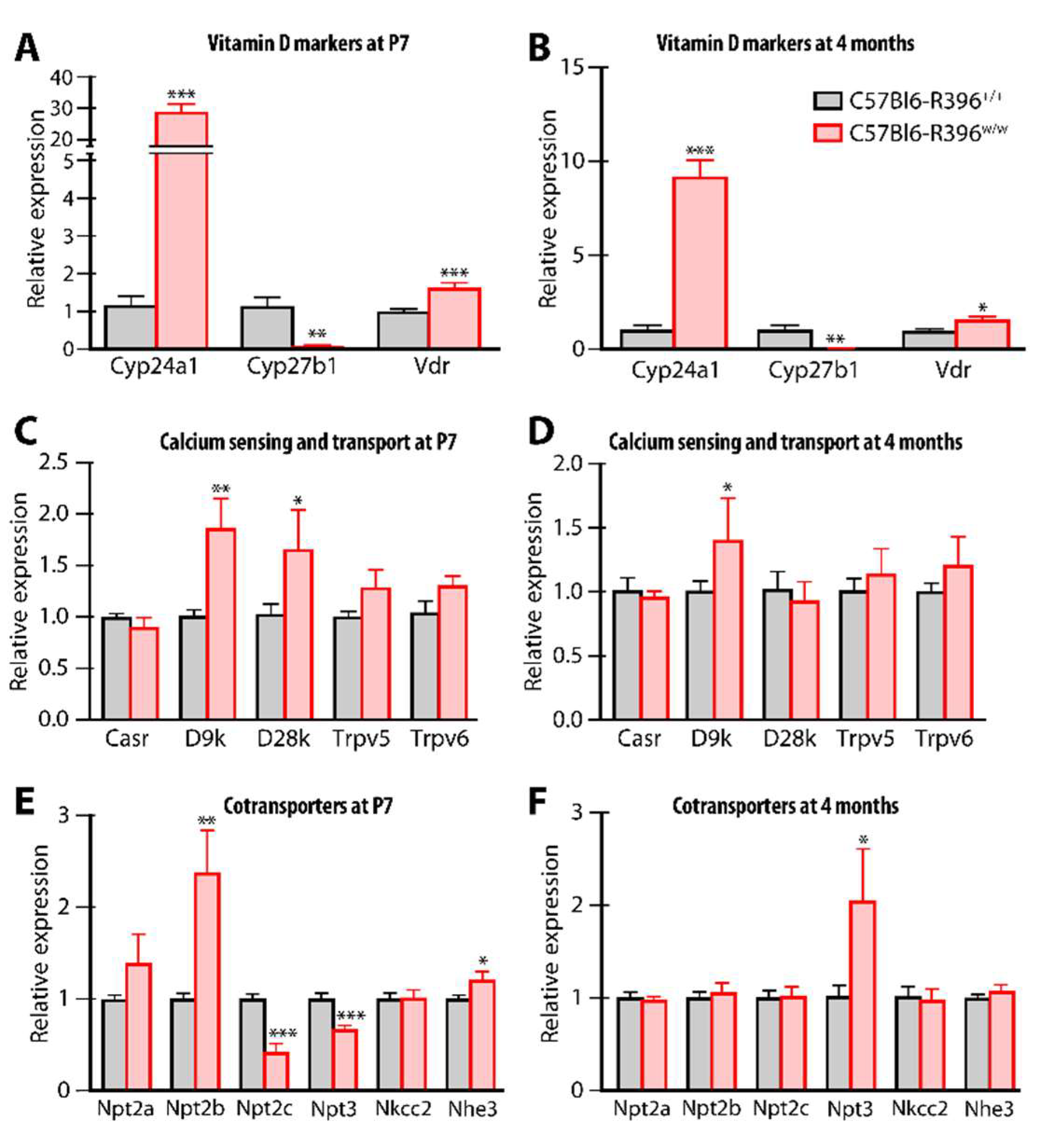
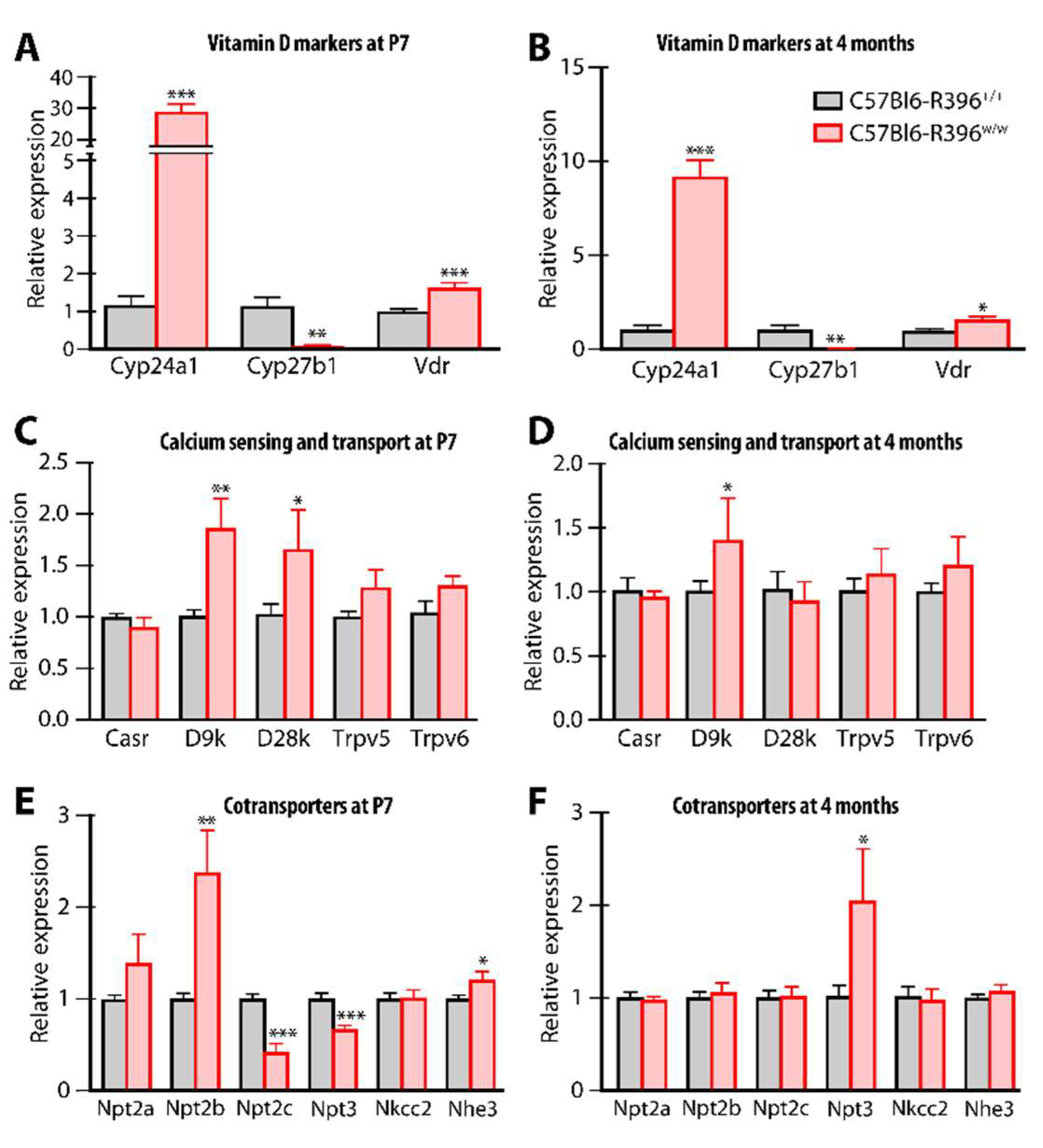
3.3. Gene Expression Monitoring
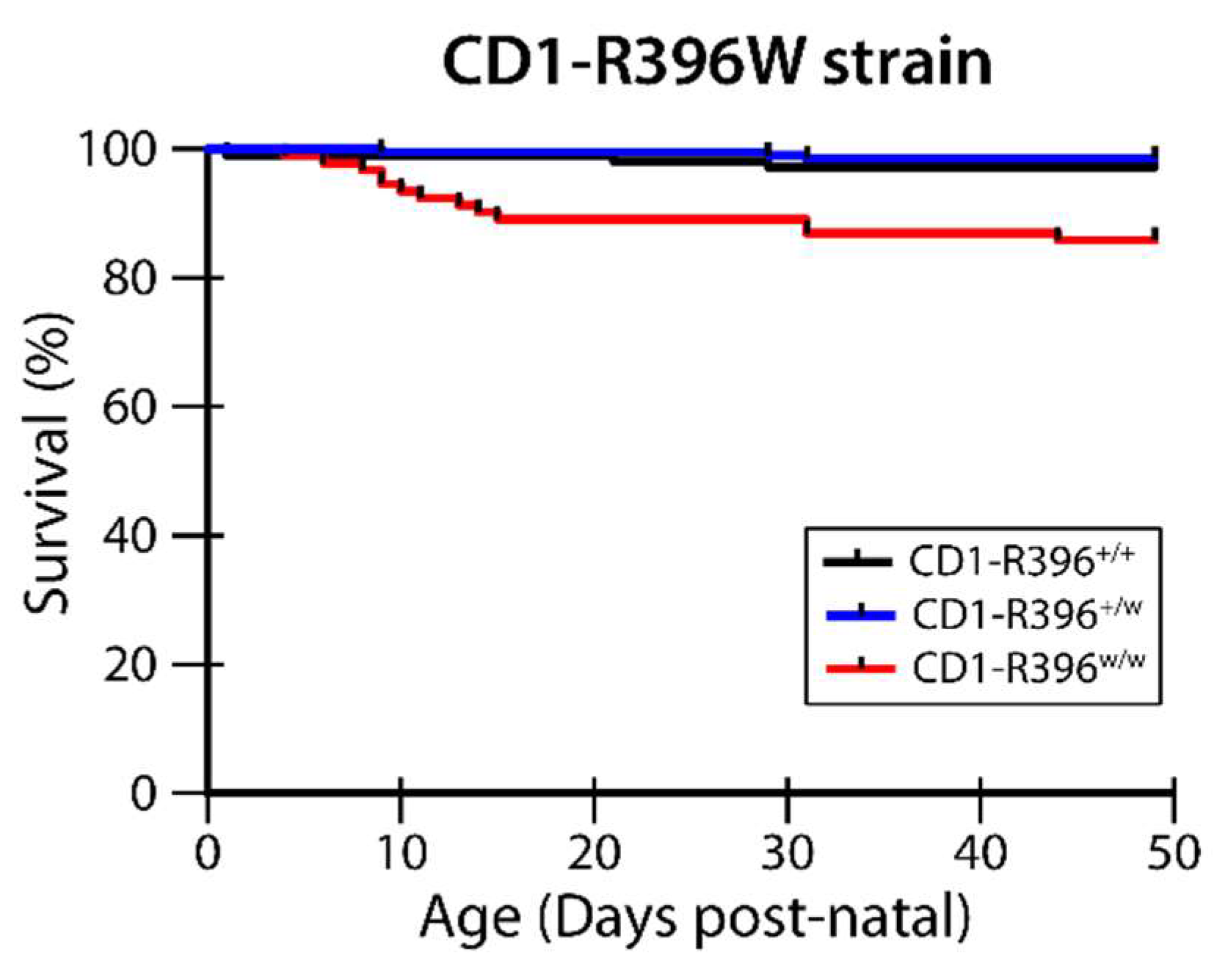
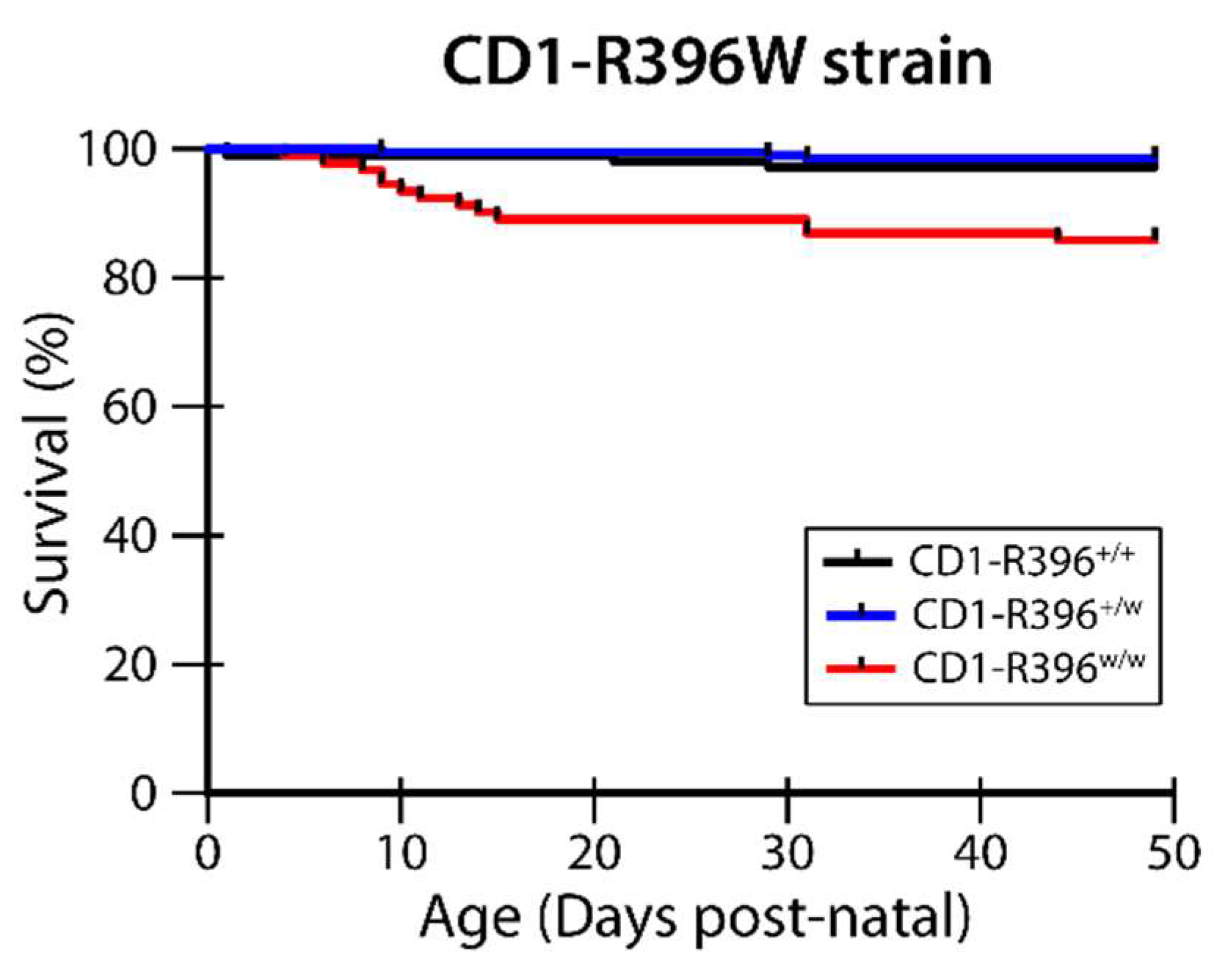
3.4. CD1-R396W Strain
3.5. Vitamin D Metabolites
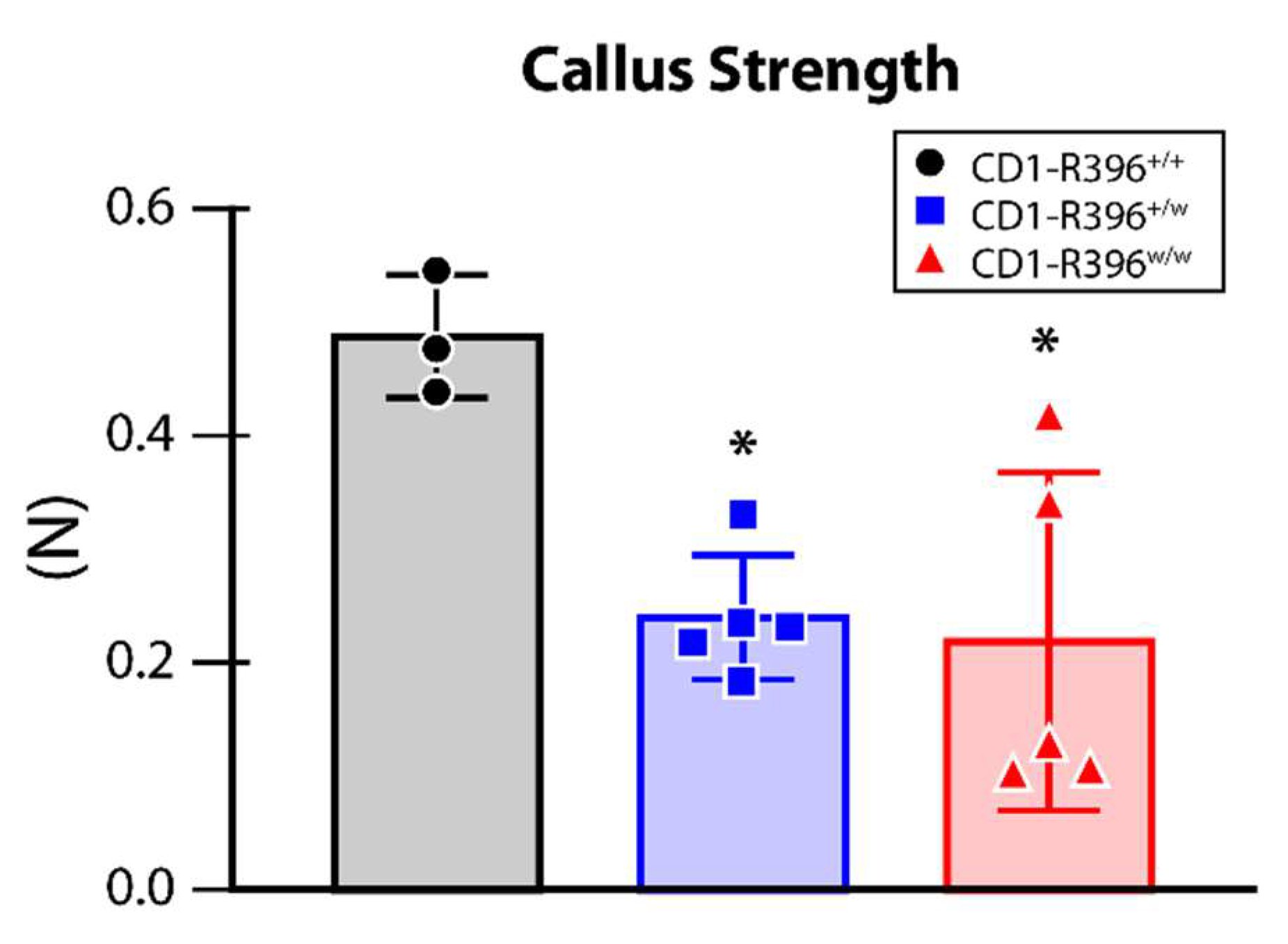
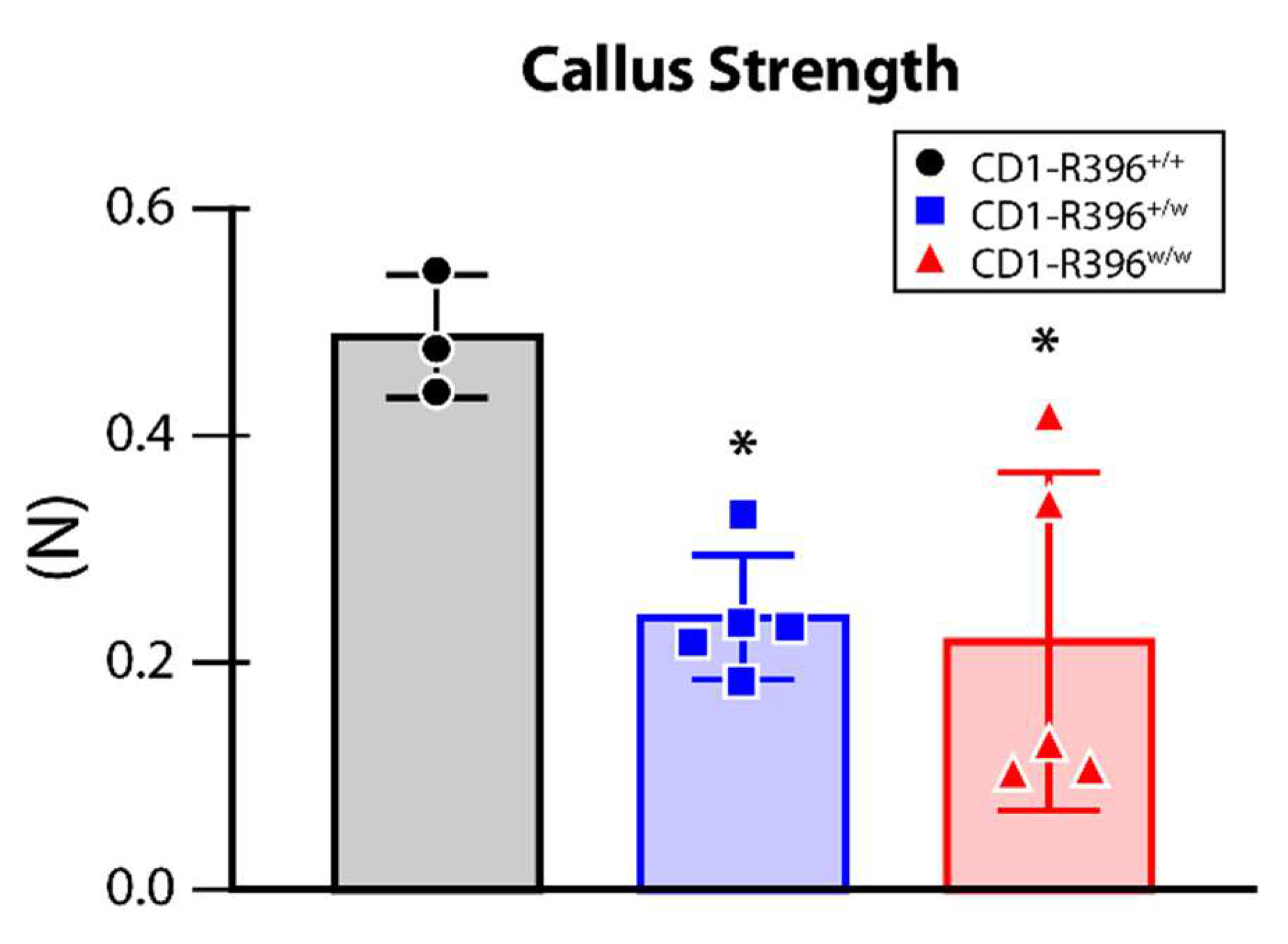
3.6. Impaired Bone Fracture Healing in CD1-R396w/w Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lightwood, R.; Stapleton, T. Idiopathic hypercalcaemia in infants. Lancet 1953, 265, 255–256. [Google Scholar] [CrossRef]
- Stapleton, T.; Macdonald, W.B.; Lightwood, R. The pathogenesis of idiopathic hypercalcemia in infancy. Am. J. Clin. Nutr. 1957, 5, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Gut, J.; Kutilek, S. Idiopathic infantile hypercalcaemia in 5-month old girl. Prague Med. Rep. 2011, 112, 124–131. [Google Scholar] [PubMed]
- Ferraro, P.M.; Minucci, A.; Primiano, A.; De Paolis, E.; Gervasoni, J.; Persichilli, S.; Naticchia, A.; Capoluongo, E.; Gambaro, G. A novel CYP24A1 genotype associated to a clinical picture of hypercalcemia, nephrolithiasis and low bone mass. Urolithiasis 2017, 45, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Figueres, M.L.; Linglart, A.; Bienaime, F.; Allain-Launay, E.; Roussey-Kessler, G.; Ryckewaert, A.; Kottler, M.L.; Hourmant, M. Kidney function and influence of sunlight exposure in patients with impaired 24-hydroxylation of vitamin D due to CYP24A1 mutations. Am. J. Kidney Dis. 2015, 65, 122–126. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Broking, E.; Fehrenbach, H.; et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 410–421. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Ruminska, J.; Kaufmann, M.; Dursun, I.; Patti, M.; Kranz, B.; Pronicka, E.; Ciara, E.; Akcay, T.; Bulus, D.; et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2A Cause Idiopathic Infantile Hypercalcemia. J. Am. Soc. Nephrol. 2016, 27, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Cappellani, D.; Brancatella, A.; Morganti, R.; Borsari, S.; Baldinotti, F.; Caligo, M.A.; Kaufmann, M.; Jones, G.; Marcocci, C.; Cetani, F. Hypercalcemia due to CYP24A1 mutations: A systematic descriptive review. Eur. J. Endocrinol. 2022, 186, 137–149. [Google Scholar] [CrossRef]
- Hanna, C.; Potretzke, T.A.; Cogal, A.G.; Mkhaimer, Y.G.; Tebben, P.J.; Torres, V.E.; Lieske, J.C.; Harris, P.C.; Sas, D.J.; Milliner, D.S.; et al. High Prevalence of Kidney Cysts in Patients With CYP24A1 Deficiency. Kidney Int. Rep. 2021, 6, 1895–1903. [Google Scholar] [CrossRef]
- Macdonald, C.; Upton, T.; Hunt, P.; Phillips, I.; Kaufmann, M.; Florkowski, C.; Soule, S.; Jones, G. Vitamin D supplementation in pregnancy: A word of caution. Familial hypercalcaemia due to disordered vitamin D metabolism. Ann. Clin. Biochem. 2020, 57, 186–191. [Google Scholar] [CrossRef]
- Molin, A.; Lemoine, S.; Kaufmann, M.; Breton, P.; Nowoczyn, M.; Ballandonne, C.; Coudray, N.; Mittre, H.; Richard, N.; Ryckwaert, A.; et al. Overlapping Phenotypes Associated With CYP24A1, SLC34A1, and SLC34A3 Mutations: A Cohort Study of Patients With Hypersensitivity to Vitamin D. Front. Endocrinol. (Lausanne) 2021, 12, 736240. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Nepton, I.; Jones, G.; Schlingmann, K.; Kaufmann, M.; Zuijdwijk, C.S.; Khatchadourian, K.; Gupta, I.R.; Pacaud, D.; Pinsk, M.N.; Mokashi, A.; et al. CYP24A1 and SLC34A1 Pathogenic Variants Are Uncommon in a Canadian Cohort of Children with Hypercalcemia or Hypercalciuria. Horm. Res. Paediatr. 2021, 94, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shen, J.; Hu, X.; Qiao, Y.; Yang, J.; Shen, Y.; Li, G. CYP24A1 Variants in Two Chinese Patients with Idiopathic Infantile Hypercalcemia. Fetal Pediatr. Pathol. 2019, 38, 44–56. [Google Scholar] [CrossRef]
- De Paolis, E.; Scaglione, G.L.; De Bonis, M.; Minucci, A.; Capoluongo, E. CYP24A1 and SLC34A1 genetic defects associated with idiopathic infantile hypercalcemia: From genotype to phenotype. Clin. Chem. Lab. Med. 2019, 57, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.F.; Shen, L. CYP24A1 mutations in idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 1741. [Google Scholar] [PubMed]
- Nesterova, G.; Malicdan, M.C.; Yasuda, K.; Sakaki, T.; Vilboux, T.; Ciccone, C.; Horst, R.; Huang, Y.; Golas, G.; Introne, W.; et al. 1,25-(OH)2D-24 Hydroxylase (CYP24A1) Deficiency as a Cause of Nephrolithiasis. Clin. J. Am. Soc. Nephrol. 2013, 8, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Gigante, M.; Santangelo, L.; Diella, S.; Caridi, G.; Argentiero, L.; D’Alessandro, M.M.; Martino, M.; Stea, E.D.; Ardissino, G.; Carbone, V.; et al. Mutational Spectrum of CYP24A1 Gene in a Cohort of Italian Patients with Idiopathic Infantile Hypercalcemia. Nephron 2016, 133, 193–204. [Google Scholar] [CrossRef] [PubMed]
- St-Arnaud, R.; Arabian, A.; Travers, R.; Barletta, F.; Raval-Pandya, M.; Chapin, K.; Depovere, J.; Mathieu, C.; Christakos, S.; Demay, M.B.; et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 2000, 141, 2658–2666. [Google Scholar] [CrossRef]
- Masuda, S.; Byford, V.; Arabian, A.; Sakai, Y.; Demay, M.B.; St-Arnaud, R.; Jones, G. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology 2005, 146, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, M.; Gallagher, J.C.; Peacock, M.; Schlingmann, K.P.; Konrad, M.; DeLuca, H.F.; Sigueiro, R.; Lopez, B.; Mourino, A.; Maestro, M.; et al. Clinical utility of simultaneous quantitation of 25-hydroxyvitamin D and 24,25-dihydroxyvitamin D by LC-MS/MS involving derivatization with DMEQ-TAD. J. Clin. Endocrinol. Metab. 2014, 99, 2567–2574. [Google Scholar] [CrossRef]
- Laha, T.J.; Strathmann, F.G.; Wang, Z.; de Boer, I.H.; Thummel, K.E.; Hoofnagle, A.N. Characterizing antibody cross-reactivity for immunoaffinity purification of analytes prior to multiplexed liquid chromatography-tandem mass spectrometry. Clin. Chem. 2012, 58, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, M.; Morse, N.; Molloy, B.J.; Cooper, D.P.; Schlingmann, K.P.; Molin, A.; Kottler, M.L.; Gallagher, J.C.; Armas, L.; Jones, G. Improved Screening Test for Idiopathic Infantile Hypercalcemia Confirms Residual Levels of Serum 24,25-(OH)2 D3 in Affected Patients. J. Bone Miner. Res. 2017, 32, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.B.; Benkusky, N.A.; Kaufmann, M.; Lee, S.M.; Onal, M.; Jones, G.; Pike, J.W. A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J. Biol. Chem. 2017, 292, 17541–17558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martineau, C.; Naja, R.P.; Husseini, A.; Hamade, B.; Kaufmann, M.; Akhouayri, O.; Arabian, A.; Jones, G.; St-Arnaud, R. Optimal bone fracture repair requires 24R,25-dihydroxyvitamin D3 and its effector molecule FAM57B2. J. Clin. Investig. 2018, 128, 3546–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, M.J.; Tadikonda, P.; Werner, E.; Prahl, J.; Yamada, S.; DeLuca, H.F. Human 25-hydroxyvitamin D3-24-hydroxylase, a multicatalytic enzyme. Biochemistry 1996, 35, 8465–8472. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; Braun, T.; Hinuma, S.; Jaenisch, R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell 1992, 71, 383–390. [Google Scholar] [CrossRef]
- Pronicka, E.; Ciara, E.; Halat, P.; Janiec, A.; Wojcik, M.; Rowinska, E.; Rokicki, D.; Pludowski, P.; Wojciechowska, E.; Wierzbicka, A.; et al. Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: Molecular study of 11 historical IIH cases. J. Appl. Genet. 2017, 58, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Pronicka, E.; Rowinska, E.; Kulczycka, H.; Lukaszkiewicz, J.; Lorenc, R.; Janas, R. Persistent hypercalciuria and elevated 25-hydroxyvitamin D3 in children with infantile hypercalcaemia. Pediatr. Nephrol. 1997, 11, 2–6. [Google Scholar] [CrossRef]
- Sawada, N.; Sakaki, T.; Ohta, M.; Inouye, K. Metabolism of vitamin D(3) by human CYP27A1. Biochem. Biophys. Res. Commun. 2000, 273, 977–984. [Google Scholar] [CrossRef]
- Cools, M.; Goemaere, S.; Baetens, D.; Raes, A.; Desloovere, A.; Kaufman, J.M.; De Schepper, J.; Jans, I.; Vanderschueren, D.; Billen, J.; et al. Calcium and bone homeostasis in heterozygous carriers of CYP24A1 mutations: A cross-sectional study. Bone 2015, 81, 89–96. [Google Scholar] [CrossRef]
- Ketha, H.; Kumar, R.; Singh, R.J. LC-MS/MS for Identifying Patients with CYP24A1 Mutations. Clin. Chem. 2016, 62, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molin, A.; Baudoin, R.; Kaufmann, M.; Souberbielle, J.C.; Ryckewaert, A.; Vantyghem, M.C.; Eckart, P.; Bacchetta, J.; Deschenes, G.; Kesler-Roussey, G.; et al. CYP24A1 Mutations in a Cohort of Hypercalcemic Patients: Evidence for a Recessive Trait. J. Clin. Endocrinol. Metab. 2015, 100, E1343–E1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Coman, D.; McTaggart, S.J.; Burke, J.R. Long-term follow-up of patients with idiopathic infantile hypercalcaemia. Pediatr. Nephrol. 2006, 21, 1676–1680. [Google Scholar] [CrossRef]
- Nguyen, M.; Boutignon, H.; Mallet, E.; Linglart, A.; Guillozo, H.; Jehan, F.; Garabedian, M. Infantile hypercalcemia and hypercalciuria: New insights into a vitamin D-dependent mechanism and response to ketoconazole treatment. J. Pediatr. 2010, 157, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Dauber, A.; Nguyen, T.T.; Sochett, E.; Cole, D.E.; Horst, R.; Abrams, S.A.; Carpenter, T.O.; Hirschhorn, J.N. Genetic defect in CYP24A1, the vitamin D 24-hydroxylase gene, in a patient with severe infantile hypercalcemia. J. Clin. Endocrinol. Metab. 2012, 97, E268–E274. [Google Scholar] [CrossRef] [Green Version]
- Streeten, E.A.; Zarbalian, K.; Damcott, C.M. CYP24A1 mutations in idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 1741–1742. [Google Scholar]
- Dinour, D.; Beckerman, P.; Ganon, L.; Tordjman, K.; Eisenstein, Z.; Holtzman, E.J. Loss-of-function mutations of CYP24A1, the vitamin D 24-hydroxylase gene, cause long-standing hypercalciuric nephrolithiasis and nephrocalcinosis. J. Urol. 2013, 190, 552–557. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef]
- Nadeau, J.H. Modifier genes in mice and humans. Nat. Rev. Genet. 2001, 2, 165–174. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Genotype | Sex | 25(OH)D3 (ng/mL) | 24,25(OH)2D3 (ng/mL) | Ratio 25:24,25 | 1,24,25(OH)3D3 (pg/mL) | 25(OH)D3-26,23-lactone (ng/mL) | 1,25(OH)2D3 (pg/mL) |
|---|---|---|---|---|---|---|---|
| Cyp24a1+/− | m | – | – | – | – | – | - |
| f | 17.21 ± 2.36 | 6.68 ± 1.05 | 2.60 ± 0.10 | 72.80 ± 4.20 | 3.84 ± 0.72 | 27.80 ± 4.20 | |
| Cyp24a1−/− | m | – | – | – | – | – | – |
| f | 101.29 ± 20.61 *** | 1.45 ± 0.11 *** | 70.60 ± 15.7 # | n.d. | 0.06 ± 0.02 # | 34.00 ± 6.20 | |
| C57bl6-R396+/+ | m | 19.59 ± 2.90 | 6.71 ± 0.76 | 2.91 ± 0.33 | 57.77 ± 10.88 | 2.76 ± 0.32 | 27.39 ± 3.35 |
| f | 18.59 ± 2.50 | 7.38 ± 0.96 | 2.53 ± 0.25 | 66.53 ± 13.18 | 3.16 ± 0.89 | 21.28 ± 5.96 | |
| C57bl6-R396w/w | m | 110.73 ± 15.40 # | 2.59 ± 0.44 # | 43.10 ± 4.08 # | 17.81 ± 3.53 # | 0.07 ± 0.03 # | 37.06 ± 8.85 |
| f | 106.16 ± 26.73 # | 2.12 ± 0.44 # | 49.71 ± 4.37 # | 11.09 ± 5.35 # | 0.05 ± 0.02 # | 37.17 ± 12.12 ** | |
| CD1-R396+/+ | m | 17.23 ± 3.77 | 6.31 ± 1.89 | 2.94 ± 1.07 | 74.66 ± 34.78 | 2.24 ± 0.70 | 45.83 ± 22.74 |
| f | 18.24 ± 4.14 | 8.80 ± 2.14 | 2.08 ± 0.19 | 55.19 ± 17.50 | 2.56 ± 0.67 | 27.43 ± 9.77 | |
| CD1-R396w/w | m | 81.90 ± 19.39 # | 2.26 ± 0.53 # | 36.58 ± 5.35 # | 15.18 ± 5.84 # | 0.06 ± 0.05 # | 45.12 ± 16.57 |
| f | 99.16 ± 13.59 # | 2.23 ± 0.39 # | 44.90 ± 5.53 # | 7.49 ± 1.95 # | 0.03 ± 0.01 # | 29.24 ± 7.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
St-Arnaud, R.; Arabian, A.; Kavame, D.; Kaufmann, M.; Jones, G. Vitamin D and Diseases of Mineral Homeostasis: A Cyp24a1 R396W Humanized Preclinical Model of Infantile Hypercalcemia Type 1. Nutrients 2022, 14, 3221. https://doi.org/10.3390/nu14153221
St-Arnaud R, Arabian A, Kavame D, Kaufmann M, Jones G. Vitamin D and Diseases of Mineral Homeostasis: A Cyp24a1 R396W Humanized Preclinical Model of Infantile Hypercalcemia Type 1. Nutrients. 2022; 14(15):3221. https://doi.org/10.3390/nu14153221
Chicago/Turabian StyleSt-Arnaud, René, Alice Arabian, Dila Kavame, Martin Kaufmann, and Glenville Jones. 2022. "Vitamin D and Diseases of Mineral Homeostasis: A Cyp24a1 R396W Humanized Preclinical Model of Infantile Hypercalcemia Type 1" Nutrients 14, no. 15: 3221. https://doi.org/10.3390/nu14153221
APA StyleSt-Arnaud, R., Arabian, A., Kavame, D., Kaufmann, M., & Jones, G. (2022). Vitamin D and Diseases of Mineral Homeostasis: A Cyp24a1 R396W Humanized Preclinical Model of Infantile Hypercalcemia Type 1. Nutrients, 14(15), 3221. https://doi.org/10.3390/nu14153221