
The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria

Abstract
:
1. Introduction
2. Bioavailability of Polyphenols
3. Mitochondria and Kidneys
3.1. Oxidative Phosphorylation (OXPHOS) System
3.2. Mitochondrial Biogenesis
3.3. Mitochondrial Dynamics
3.4. Mitophagy
4. Kidney and Mitochondria
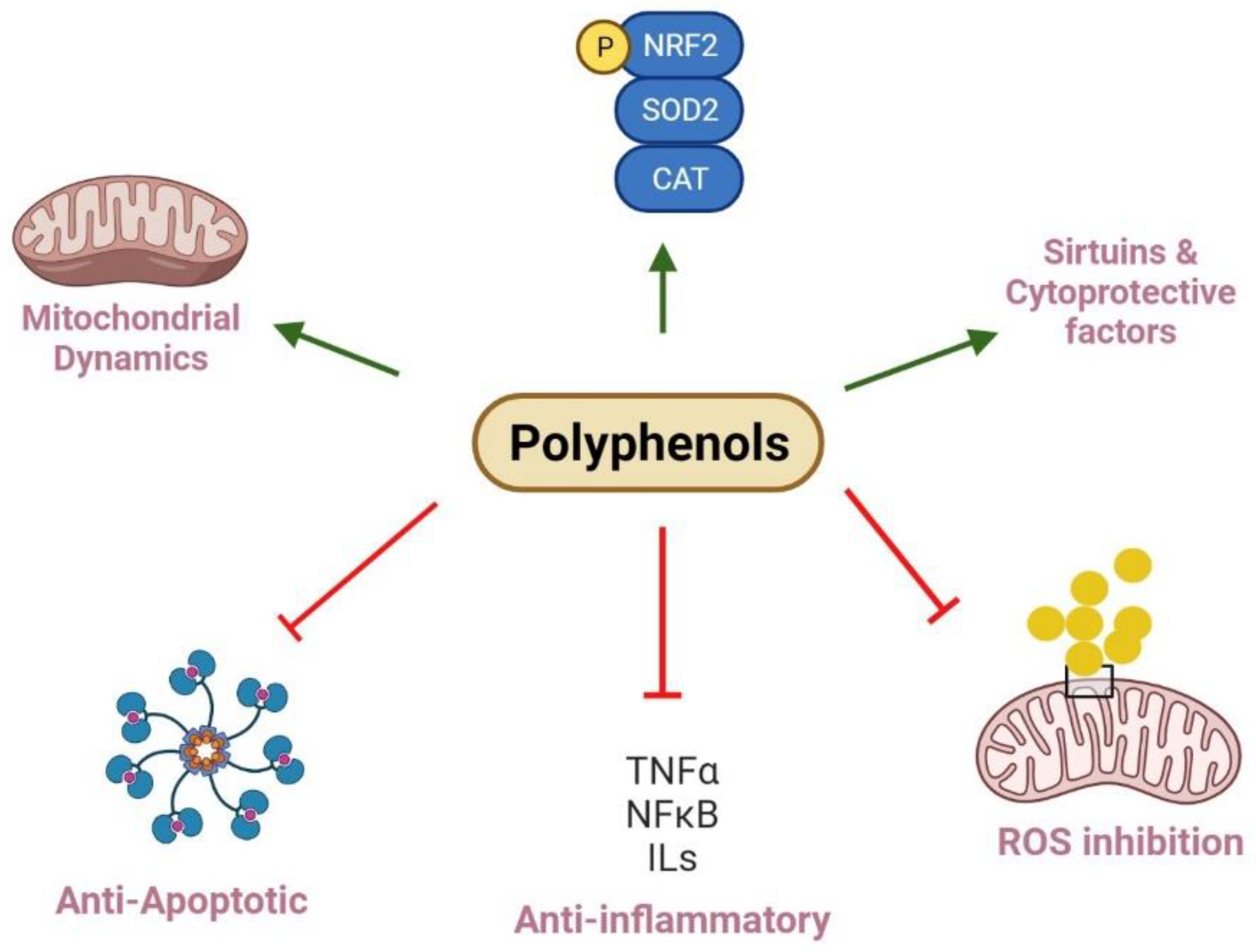
5. Antioxidants and Kidney Diseases
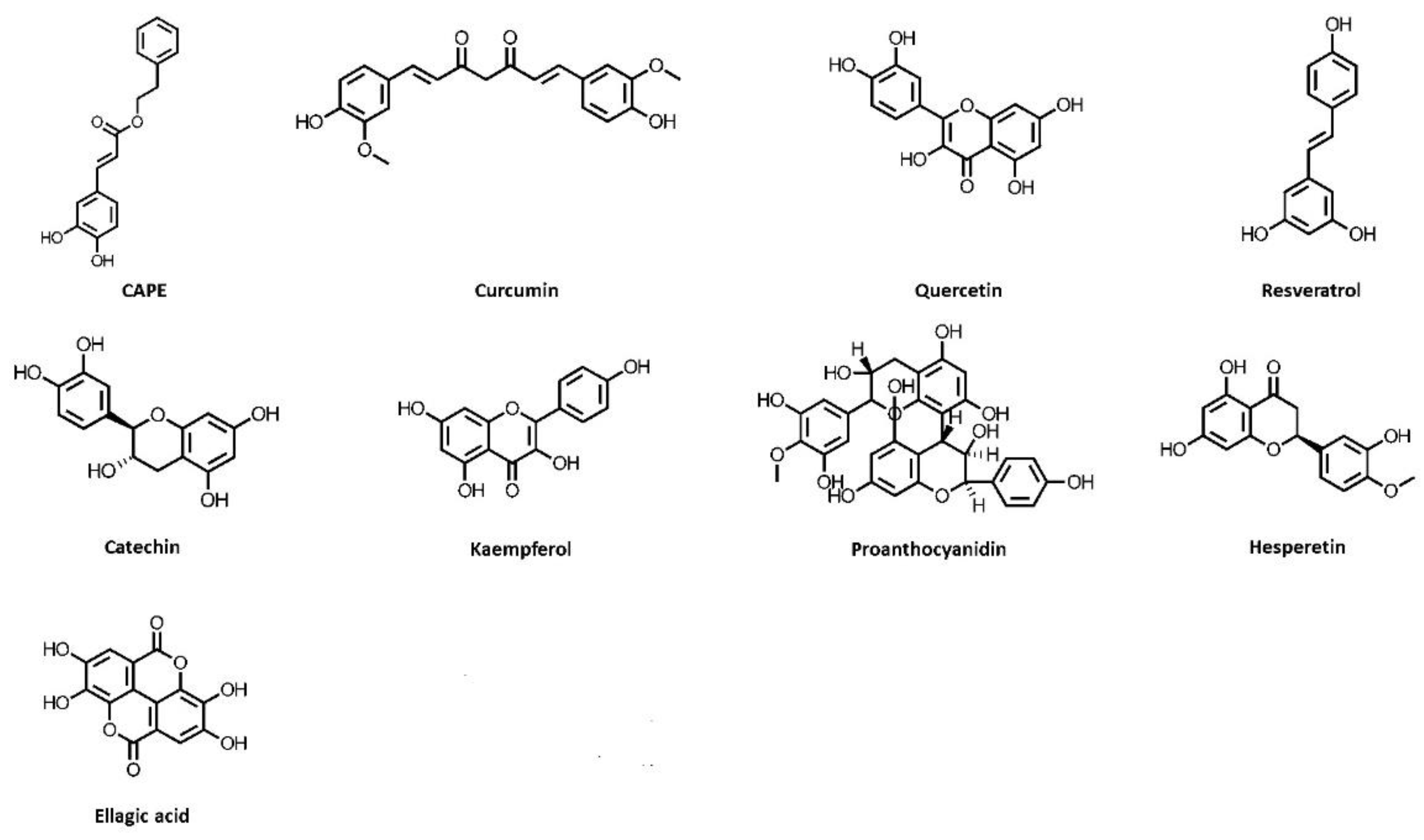
5.1. Caffeic Acid Phenethyl Ester
5.2. Curcumin
5.3. Quercetin
5.4. Resveratrol
5.5. Catechin
5.6. Kaempferol
5.7. Grape Seed Proanthocyanidin
5.8. Hesperetin
5.9. Ellagic Acid
6. Discussion and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vart, P.; Grams, M.E. Measuring and Assessing Kidney Function. Semin. Nephrol. 2016, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Hartogh, D.J.D.; Tsiani, E. Health Benefits of Resveratrol in Kidney Disease: Evidence from In Vitro and In Vivo Studies. Nutrient 2019, 11, 1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M. Mitochondria–Power Players in Kidney Function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.P.; Zeidel, M.L. Homeostasis, the Milieu Intérieur, and the Wisdom of the Nephron. Clin. J. Am. Soc. Nephrol. 2014, 9, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.-H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; A Kellum, J.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.; Monsalve, M.; Ramos, A.; Sanchez-Niño, M.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Duann, P.; Lin, P.-H. Mitochondria Damage and Kidney Disease. Mitochondrial Dyn. Cardiovasc. Med. 2017, 982, 529–551. [Google Scholar] [CrossRef]
- Mafra, D.; Gidlund, E.-K.; Borges, N.A.; Magliano, D.C.; Lindholm, B.; Stenvinkel, P.; Von Walden, F. Bioactive food and exercise in chronic kidney disease: Targeting the mitochondria. Eur. J. Clin. Investig. 2018, 48, e13020. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.; Xie, X.; Du, M.; Bu, Y.; Wu, N.; Yang, H.; Tian, C.; Xu, F.; Xiang, S.; Zhang, P.; et al. Green Tea Polyphenols Ameliorate the Early Renal Damage Induced by a High-Fat Diet via Ketogenesis/SIRT3 Pathway. Oxidative Med. Cell. Longev. 2017, 2017, 9032792. [Google Scholar] [CrossRef] [Green Version]
- Boeing, H.; Bechthold, A.; Bub, A.; Ellinger, S.; Haller, D.; Kroke, A.; Leschik-Bonnet, E.; Müller, M.J.; Oberritter, H.; Schulze, M.; et al. Critical review: Vegetables and fruit in the prevention of chronic diseases. Eur. J. Nutr. 2012, 51, 637–663. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, A.; Zhao, L.; Wang, C.; Nadeem, M.; Raza, A.; Ali, N.; Shah, A.A. Management of hyperuricemia through dietary polyphenols as a natural medicament: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2017, 59, 1433–1455. [Google Scholar] [CrossRef]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef]
- Adekunle, I.A.; Imafidon, C.E.; Oladele, A.A.; Ayoka, A.O. Ginger polyphenols attenuate cyclosporine-induced disturbances in kidney function: Potential application in adjuvant transplant therapy. Pathophysiology 2018, 25, 101–115. [Google Scholar] [CrossRef]
- Tovar-Palacio, C.; Noriega, L.G.; Mercado, A. Potential of Polyphenols to Restore SIRT1 and NAD+ Metabolism in Renal Disease. Nutrients 2022, 14, 653. [Google Scholar] [CrossRef]
- Bendokas, V.; Skemiene, K.; Trumbeckaite, S.; Stanys, V.; Passamonti, S.; Borutaite, V.; Liobikas, J. Anthocyanins: From Plant Pigments to Health Benefits at Mitochondrial Level: Reviews; Critical Reviews in Food Science and Nutrition; Taylor & Francis: Philadelphia, PA, USA, 2020; Volume 60. [Google Scholar]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly) phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects Against Chronic Diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.; Chen, L. Polyphenols and bioavailability: An update. Crit. Rev. Food Sci. Nutr. 2019, 59, 2040–2051. [Google Scholar] [CrossRef]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E.; Chowrimootoo, G.; Choudhury, R.; Debnam, E.S.; Srai, S.K.; Rice-Evans, C. The small intestine can both absorb and glucuronidate luminal flavonoids. FEBS Lett. 1999, 458, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Santhakumar, A.B.; Battino, M.; Alvarez-Suarez, J.M. Dietary polyphenols: Structures, bioavailability and protective effects against atherosclerosis. Food Chem. Toxicol. 2018, 113, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Leonarduzzi, G.; Testa, G.; Sottero, B.; Gamba, P.; Poli, G. Design and development of nanovehicle-based delivery systems for preventive or therapeutic supplementation with flavonoids. Curr. Med. Chem. 2010, 17, 74–95. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Bowey, E.; Adlercreutz, H.; Rowland, I. Metabolism of isoflavones and lignans by the gut microflora: A study in germ-free and human flora associated rats. Food Chem. Toxicol. 2003, 41, 631–636. [Google Scholar] [CrossRef]
- Naven, R.T.; Swiss, R.; Klug-McLeod, J.; Will, Y.; Greene, N. The Development of Structure-Activity Relationships for Mitochondrial Dysfunction: Uncoupling of Oxidative Phosphorylation. Toxicol. Sci. 2012, 131, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.F.; Revel, J.S.; Maier, C.S. Mitochondria-Centric Review of Polyphenol Bioactivity in Cancer Models. Antioxid. Redox Signal. 2018, 29, 1589–1611. [Google Scholar] [CrossRef]
- Spycher, S.; Smejtek, P.; Netzeva, T.I.; Escher, B.I. Toward a Class-Independent Quantitative Structure−Activity Relationship Model for Uncouplers of Oxidative Phosphorylation. Chem. Res. Toxicol. 2008, 21, 911–927. [Google Scholar] [CrossRef]
- Velderrain-Rodríguez, G.R.; Palafox-Carlos, H.; Wall-Medrano, A.; Ayala-Zavala, J.F.; Chen, C.-Y.O.; Robles-Sánchez, M.; Astiazaran-García, H.; Alvarez-Parrilla, E.; González-Aguilar, G.A. Phenolic compounds: Their journey after intake. Food Funct. 2014, 5, 189–197. [Google Scholar] [CrossRef]
- Hussain, M.B.; Hassan, S.; Waheed, M.; Javed, A.; Farooq, M.A.; Tahir, A. Bioavailability and Metabolic Pathway of Phenolic Compounds. 5. In Plant Physiological Aspects of Phenolic Compounds; Marcos, S.-H., Rosario, G.-M., Mariana, P.-T., Eds.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Barchiesi, A.; Bazzani, V.; Tolotto, V.; Elancheliyan, P.; Wasilewski, M.; Chacinska, A.; Vascotto, C. Mitochondrial Oxidative Stress Induces Rapid Intermembrane Space/Matrix Translocation of Apurinic/Apyrimidinic Endonuclease 1 Protein through TIM23 Complex. J. Mol. Biol. 2020, 432, 166713. [Google Scholar] [CrossRef]
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol improves mitochondrial function in the remnant kidney from 5/6 nephrectomized rats. Acta Histochem. 2017, 119, 392–399. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2018, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Nistala, R.; Whaley-Connell, A.; Sowers, J.R. Redox Control of Renal Function and Hypertension. Antioxid. Redox Signal. 2008, 10, 2047–2089. [Google Scholar] [CrossRef] [Green Version]
- Rensvold, J.W.; Ong, S.-E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and Protein Profiling Identifies Iron as a Key Regulator of Mitochondrial Biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Kang, C.-H.; Wang, S.-G.; Lee, H.-M. α-Lipoic acid regulates lipid metabolism through induction of sirtuin 1 (SIRT1) and activation of AMP-activated protein kinase. Diabetologia 2012, 55, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2013, 25, 138–145. [Google Scholar] [CrossRef]
- Chodari, L.; Aytemir, M.D.; Vahedi, P.; Alipour, M.; Vahed, S.Z.; Khatibi, S.M.H.; Ahmadian, E.; Ardalan, M.; Eftekhari, A. Targeting Mitochondrial Biogenesis with Polyphenol Compounds. Oxidative Med. Cell. Longev. 2021, 2021, 4946711. [Google Scholar] [CrossRef]
- Chambers, J.M.; Wingert, R.A. PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef]
- Layal, K.; Perdhana, I.S.; Louisa, M.; Estuningtyas, A.; Soetikno, V. The effects of quercetin on oxidative stress and fibrosis markers in chronic kidney disease rat model. Med. J. Indones. 2017, 26, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Parikh, S.M. Mitochondrial Metabolism in Acute Kidney Injury. Semin. Nephrol. 2020, 40, 101–113. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhuohua, Z.; Liu, J.; Liu, Z.; Chengyuan, T.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Bravi, C.A.; Vertosick, E.; Benfante, N.; Tin, A.; Sjoberg, D.; Hakimi, A.A.; Touijer, K.; Montorsi, F.; Eastham, J.; Russo, P.; et al. Impact of Acute Kidney Injury and Its Duration on Long-term Renal Function After Partial Nephrectomy. Eur. Urol. 2019, 76, 398–403. [Google Scholar] [CrossRef]
- Forni, L.G.; Darmon, M.; Ostermann, M.; Straaten, H.M.O.-V.; Pettilä, V.; Prowle, J.; Schetz, M.; Joannidis, M. Renal recovery after acute kidney injury. Intensiv. Care Med. 2017, 43, 855–866. [Google Scholar] [CrossRef]
- Scammell, M.K.; Sennett, C.M.; Petropoulos, Z.; Kamal, J.; Kaufman, J.S. Environmental and Occupational Exposures in Kidney Disease. Semin. Nephrol. 2019, 39, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Sanders, A.P.; Saland, J.M.; Wright, R.O.; Arora, M. Environmental exposures and pediatric kidney function and disease: A systematic review. Environ. Res. 2017, 158, 625–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, S.; Gassa, A.D.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amani, H.; Habibey, R.; Shokri, F.; Hajmiresmail, S.J.; Akhavan, O.; Mashaghi, A.; Pazoki-Toroudi, H. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 2019, 9, 6044. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, Z.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Liu, X.; Wang, L. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J. 2019, 34, 835–852. [Google Scholar] [CrossRef] [Green Version]
- Kamarauskaite, J.; Baniene, R.; Trumbeckas, D.; Strazdauskas, A.; Trumbeckaite, S. Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants 2021, 10, 747. [Google Scholar] [CrossRef]
- Trumbeckaite, S.; Pauziene, N.; Trumbeckas, D.; Jievaltas, M.; Baniene, R. Caffeic Acid Phenethyl Ester Reduces Ischemia-Induced Kidney Mitochondrial Injury in Rats. Oxidative Med. Cell. Longev. 2017, 2017, 1697018. [Google Scholar] [CrossRef]
- Liu, Q.; Liang, X.; Liang, M.; Qin, R.; Qin, F.; Wang, X. Ellagic Acid Ameliorates Renal Ischemic-Reperfusion Injury Through NOX4/JAK/STAT Signaling Pathway. Inflammation 2019, 43, 298–309. [Google Scholar] [CrossRef]
- Roede, J.R.; Jones, D.P. Reactive species and mitochondrial dysfunction: Mechanistic significance of 4-hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.J.F.; Low, I.C.C.; Pervaiz, S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion 2014, 19, 39–48. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Liu, X.; Wang, M.; Chen, H.; Chen, Z.; Qiu, T. Oxymatrine ameliorates renal ischemia-reperfusion injury from oxidative stress through Nrf2/HO-1 pathway. Acta Cir. Bras. 2015, 30, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, K.S.; Prince, P.S.M. Caffeic acid protects rat heart mitochondria against isoproterenol-induced oxidative damage. Cell Stress Chaperones 2010, 15, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef] [Green Version]
- Robert, F.F.; Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.-S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Chen, W.; Hu, R.; Feng, H. The Injury and Therapy of Reactive Oxygen Species in Intracerebral Hemorrhage Looking at Mitochondria. Oxidative Med. Cell. Longev. 2016, 2016, 2592935. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, C.; Ge, J.; Lv, M.-W.; Talukder, M.; Guo, K.; Li, Y.-H.; Li, J.-L. Ameliorative effects of resveratrol against cadmium-induced nephrotoxicity via modulating nuclear xenobiotic receptor response and PINK1/Parkin-mediated Mitophagy. Food Funct. 2020, 11, 1856–1868. [Google Scholar] [CrossRef]
- Liu, L.; Tao, R.; Huang, J.; He, X.; Qu, L.; Jin, Y.; Zhang, S.; Fu, Z. Hepatic oxidative stress and inflammatory responses with cadmium exposure in male mice. Environ. Toxicol. Pharmacol. 2015, 39, 229–236. [Google Scholar] [CrossRef]
- Karaca, S.; Eraslan, G. The Effects of Flaxseed Oil on Cadmium-Induced Oxidative Stress in Rats. Biol. Trace Elem. Res. 2013, 155, 423–430. [Google Scholar] [CrossRef]
- Cannino, G.; Ferruggia, E.; Rinaldi, A.M. Proteins participating to the post-transcriptional regulation of the mitochondrial cytochrome c oxidase subunit IV via elements located in the 3′UTR. Mitochondrion 2009, 9, 471–480. [Google Scholar] [CrossRef]
- Xu, S.; Pi, H.; Zhang, L.; Zhang, N.; Li, Y.; Zhang, H.; Tang, J.; Li, H.; Feng, M.; Deng, P.; et al. Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J. Pineal Res. 2016, 60, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Pham, D.T.N.; Kim, Y.-M. Alternative strategies for the application of aminoglycoside antibiotics against the biofilm-forming human pathogenic bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 1955–1976. [Google Scholar] [CrossRef]
- Walker, P.D.; Shah, S.V. Gentamicin enhanced production of hydrogen peroxide by renal cortical mitochondria. Am. J. Physiol. Physiol. 1987, 253, C495–C499. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepand, M.R.; Ghahremani, M.H.; Razavi-Azarkhiavi, K.; Aghsami, M.; Rajabi, J.; Keshavarz-Bahaghighat, H.; Soodi, M. Ellagic acid confers protection against gentamicin-induced oxidative damage, mitochondrial dysfunction and apoptosis-related nephrotoxicity. J. Pharm. Pharmacol. 2016, 68, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhong, X.; Yuan, H.; Guo, Y.; Song, D.; Qi, F.; Zhu, Z.; Wang, X.; Guo, Z. Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinum (iv) prodrugs. Chem. Sci. 2020, 11, 3829–3835. [Google Scholar] [CrossRef]
- Waseem, M.; Kaushik, P.; Parvez, S. Mitochondria-Mediated mitigatory role of curcumin in cisplatin-induced nephrotoxicity. Cell Biochem. Funct. 2013, 31, 678–684. [Google Scholar] [CrossRef]
- Kumar, M.; Dahiya, V.; Kasala, E.R.; Bodduluru, L.N.; Lahkar, M. The renoprotective activity of hesperetin in cisplatin induced nephrotoxicity in rats: Molecular and biochemical evidence. Biomed. Pharmacother. 2017, 89, 1207–1215. [Google Scholar] [CrossRef]
- Szewczyk, A. Mitochondria as a Pharmacological Target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.J.; Kim, D.H.; Jung, Y.J.; Kang, K.P.; Lee, A.S.; Lee, S.; Kim, W.; Davaatseren, M.; Hwang, J.-T.; Kim, H.-J.; et al. Genistein protects the kidney from cisplatin-induced injury. Kidney Int. 2008, 74, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Beltrán, C.E.; Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Tapia, E.; García-Torres, I.; Pedraza-Chaverri, J.; Pacher, P. Sulforaphane, a natural constituent of broccoli, prevents cell death and inflammation in nephropathy. J. Nutr. Biochem. 2012, 23, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, W.; Sun, X.; Wang, Y.; Zhou, M. Kaempferol ameliorates Cisplatin induced nephrotoxicity by modulating oxidative stress, inflammation and apoptosis via ERK and NF-κB pathways. AMB Express 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Ciarcia, R.; Damiano, S.; Florio, A.; Spagnuolo, M.; Zacchia, E.; Squillacioti, C.; Mirabella, N.; Florio, S.; Pagnini, U.; Garofano, T.; et al. The Protective Effect of Apocynin on Cyclosporine A-Induced Hypertension and Nephrotoxicity in Rats. J. Cell. Biochem. 2015, 116, 1848–1856. [Google Scholar] [CrossRef]
- Tedesco, D.; Haragsim, L. Cyclosporine: A Review. J. Transplant. 2012, 2012, 230386. [Google Scholar] [CrossRef] [Green Version]
- Niemann, C.U.; Saeed, M.; Akbari, H.; Jacobsen, W.; Benet, L.Z.; Christians, U.; Serkova, N.; Saeed, M. Close Association Between the Reduction in Myocardial Energy Metabolism and Infarct Size: Dose-Response Assessment of Cyclosporine. J. Pharmacol. Exp. Ther. 2002, 302, 1123–1128. [Google Scholar] [CrossRef] [Green Version]
- Serkova, N.; Jacobsen, W.; Niemann, C.U.; Litt, L.; Benet, L.Z.; Leibfritz, D.; Christians, U. Sirolimus, but not the structurally related RAD (everolimus), enhances the negative effects of cyclosporine on mitochondrial metabolism in the rat brain. J. Cereb. Blood Flow Metab. 2001, 133, 875–885. [Google Scholar] [CrossRef]
- Serkova, N.; Klawitter, J.; Niemann, C.U. Organ-Specific response to inhibition of mitochondrial metabolism by cyclosporine in the rat. Transpl. Int. 2003, 16, 748–755. [Google Scholar] [CrossRef]
- Rehman, H.; Krishnasamy, Y.; Haque, K.; Thurman, R.G.; Lemasters, J.J.; Schnellmann, R.G.; Zhong, Z. Green Tea Polyphenols Stimulate Mitochondrial Biogenesis and Improve Renal Function after Chronic Cyclosporin A Treatment in Rats. PLoS ONE 2013, 8, e65029. [Google Scholar] [CrossRef]
- Moon, D.; Kim, J. Cyclosporin A aggravates hydrogen peroxide-induced cell death in kidney proximal tubule epithelial cells. Anat. Cell Biol. 2019, 52, 312–323. [Google Scholar] [CrossRef]
- Wu, Q.; Li, W.; Zhao, J.; Sun, W.; Yang, Q.; Chen, C.; Xia, P.; Zhu, J.; Zhou, Y.; Huang, G.; et al. Apigenin ameliorates doxorubicin-induced renal injury via inhibition of oxidative stress and inflammation. Biomed. Pharmacother. 2021, 137, 111308. [Google Scholar] [CrossRef]
- Alagal, R.I.; AlFaris, N.A.; Alshammari, G.M.; Altamimi, J.Z.; AlMousa, L.A.; Yahya, M.A. Kaempferol attenuates doxorubicin-mediated nephropathy in rats by activating SIRT1 signaling. J. Funct. Foods 2021, 89, 104918. [Google Scholar] [CrossRef]
- Kocahan, S.; Dogan, Z.; Erdemli, E.; Taskin, E. Protective Effect of Quercetin against Oxidative Stress-Induced Toxicity Associated with Doxorubicin and Cyclophosphamide in Rat Kidney and Liver Tissue. Iran. J. Kidney Dis. 2017, 11, 124–131. [Google Scholar] [PubMed]
- Chénais, B.; Andriollo, M.; Guiraud, P.; Belhoussine, R.; Jeannesson, P. Oxidative stress involvement in chemically induced differentiation of K562 cells. Free. Radic. Biol. Med. 2000, 28, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Sutariya, B.; Saraf, M. α-asarone reduce proteinuria by restoring antioxidant enzymes activities and regulating necrosis factor κB signaling pathway in doxorubicin-induced nephrotic syndrome. Biomed. Pharmacother. 2017, 98, 318–324. [Google Scholar] [CrossRef]
- Hekmat, A.S.; Chenari, A.; Alipanah, H.; Javanmardi, K. Protective effect of alamandine on doxorubicin-induced nephrotoxicity in rats. BMC Pharmacol. Toxicol. 2021, 22, 31. [Google Scholar] [CrossRef]
- Pedrycz, A.; Czerny, K. Immunohistochemical study of proteins linked to apoptosis in rat fetal kidney cells following prepregnancy adriamycin administration in the mother. Acta Histochem. 2008, 110, 519–523. [Google Scholar] [CrossRef]
- Pedrycz, A.; Wieczorski, M.; Czerny, K. Late effects of adriamycin single dose on fetal rat kidney—Ultrastructural assessment. Environ. Toxicol. Pharmacol. 2005, 20, 157–160. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Remuzzi, G.; Parving, H.-H.; Keane, W.F.; Zhang, Z.; Shahinfar, S.; Snapinn, S.; Cooper, M.E.; Mitch, W.E.; Brenner, B.M. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004, 65, 2309–2320. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of Oxidative Stress during Diabetes Mellitus. J. Biomark. 2013, 2013, 378790. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- A Nath, K.; Norby, S.M. Reactive oxygen species and acute renal failure. Am. J. Med. 2000, 109, 665–678. [Google Scholar] [CrossRef]
- Fernandes, S.M.; Cordeiro, P.M.; Watanabe, M.; da Fonseca, C.D.; Vattimo, M.D.F.F. The role of oxidative stress in streptozotocin-induced diabetic nephropathy in rats. Arch. Endocrinol. Metab. 2016, 60, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.; Suchal, K.; Khan, S.I.; Bhatia, J.; Kishore, K.; Dinda, A.K.; Arya, D.S. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-κB-TNF-α and TGF-β1-MAPK-fibronectin pathways. Am. J. Physiol. Physiol. 2017, 313, F414–F422. [Google Scholar] [CrossRef] [Green Version]
- Altamimi, J.Z.; AlFaris, N.A.; Alshammari, G.M.; Alagal, R.I.; Aljabryn, D.H.; Aldera, H.; Alrfaei, B.M.; Alkhateeb, M.A.; Yahya, M.A. Ellagic acid protects against diabetic nephropathy in rats by regulating the transcription and activity of Nrf2. J. Funct. Foods 2021, 79, 104397. [Google Scholar] [CrossRef]
- Abdou, H.M.; Elkader, H.-T.A.E.A. The potential therapeutic effects of Trifolium alexandrinum extract, hesperetin and quercetin against diabetic nephropathy via attenuation of oxidative stress, inflammation, GSK-3β and apoptosis in male rats. Chem. Interact. 2021, 352, 109781. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Cai, X.; Dai, X.; Ding, Y.; Jiang, Y.; Li, Y.; Zhang, Z.; Li, Y. Grape seed proanthocyanidin extracts ameliorate podocyte injury by activating peroxisome proliferator-activated receptor-γ coactivator 1α in low-dose streptozotocin-and high-carbohydrate/high-fat diet-induced diabetic rats. Food Funct. 2014, 5, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Bankova, V.; Trusheva, B.; Popova, M. Caffeic Acid Phenethyl Ester (CAPE)—Natural Sources, Analytical Procedures and Synthetic Approaches. Comptes Rendus Lacademie Bulg. Sci. 2018, 71, 1157–1169. [Google Scholar] [CrossRef]
- Zhang, P.; Tang, Y.; Li, N.-G.; Zhu, Y.; Duan, J.-A. Bioactivity and Chemical Synthesis of Caffeic Acid Phenethyl Ester and Its Derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akyol, S.; Ugurcu, V.; Altuntas, A.; Hasgul, R.; Cakmak, O.; Akyol, O. Caffeic Acid Phenethyl Ester as a Protective Agent against Nephrotoxicity and/or Oxidative Kidney Damage: A Detailed Systematic Review. Sci. World J. 2014, 2014, 561971. [Google Scholar] [CrossRef] [Green Version]
- Erdemli, H.K.; Akyol, S.; Armutcu, F.; Gulec, M.A.; Canbal, M.; Akyol, O. Melatonin and caffeic acid phenethyl ester in the regulation of mitochondrial function and apoptosis: The basis for future medical approaches. Life Sci. 2016, 148, 305–312. [Google Scholar] [CrossRef]
- Ozeren, M.; Sucu, N.; Tamer, L.; Aytacoglu, B.; Bayri, O.; Dondas, A.; Ayaz, L.; Dikmengil, M. Caffeic acid phenethyl ester (CAPE) supplemented St. Thomas’ hospital cardioplegic solution improves the antioxidant defense system of rat myocardium during ischemia-reperfusion injury. Pharmacol. Res. 2005, 52, 258–263. [Google Scholar] [CrossRef]
- Migliori, M.; Cantaluppi, V.; Mannari, C.; Bertelli, A.A.E.; Medica, D.; Quercia, A.D.; Navarro, V.; Scatena, A.; Giovannini, L.; Biancone, L.; et al. Caffeic Acid, a Phenol Found in White Wine, Modulates Endothelial Nitric Oxide Production and Protects from Oxidative Stress-Associated Endothelial Cell Injury. PLoS ONE 2015, 10, e0117530. [Google Scholar] [CrossRef]
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef]
- Lu, M.; Li, H.; Liu, W.; Zhang, X.; Li, L.; Zhou, H. Curcumin attenuates renal interstitial fibrosis by regulating autophagy and retaining mitochondrial function in unilateral ureteral obstruction rats. Basic Clin. Pharmacol. Toxicol. 2020, 128, 594–604. [Google Scholar] [CrossRef]
- Shakeri, A.; Cicero, A.F.G.; Panahi, Y.; Mohajeri, M.; Sahebkar, A. Curcumin: A naturally occurring autophagy modulator. J. Cell. Physiol. 2019, 234, 5643–5654. [Google Scholar] [CrossRef]
- Avila-Rojas, S.H.; Lira-León, A.; Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Pedraza-Chaverri, J. Role of Autophagy on Heavy Metal-Induced Renal Damage and the Protective Effects of Curcumin in Autophagy and Kidney Preservation. Medicina 2019, 55, 360. [Google Scholar] [CrossRef] [Green Version]
- Negrette-Guzmán, M.; García-Niño, W.R.; Tapia, E.; Zazueta, C.; Huerta-Yepez, S.; León-Contreras, J.C.; Hernández-Pando, R.; Aparicio-Trejo, O.E.; Madero, M.; Pedraza-Chaverri, J. Curcumin Attenuates Gentamicin-Induced Kidney Mitochondrial Alterations: Possible Role of a Mitochondrial Biogenesis Mechanism. Evid. Based Complement. Altern. Med. 2015, 2015, 917435. [Google Scholar] [CrossRef]
- Iglesias, D.E.; Cremonini, E.; Oteiza, P.I.; Fraga, C.G. Curcumin Mitigates TNFα-Induced Caco-2 Cell Monolayer Permeabilization Through Modulation of NF-κB, ERK1/2, and JNK Pathways. Mol. Nutr. Food Res. 2022, 66, 2101033. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, S.; Sil, P.C. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem. Toxicol. 2015, 83, 111–124. [Google Scholar] [CrossRef]
- Liu, F.-H.; Ni, W.-J.; Wang, G.-K.; Zhang, J.-J. Protective role of curcumin on renal ischemia reperfusion injury via attenuating the inflammatory mediators and Caspase-3. Cell. Mol. Biol. 2016, 62, 95–99. [Google Scholar] [PubMed]
- Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Briones-Herrera, A.; Medina-Campos, O.N.; Reyes-Fermín, L.M.; Martínez-Klimova, E.; León-Contreras, J.C.; Hernández-Pando, R.; Tapia, E.; Pedraza-Chaverri, J. Alterations in mitochondrial homeostasis in a potassium dichromate model of acute kidney injury and their mitigation by curcumin. Food Chem. Toxicol. 2020, 145, 111774. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 4137–4146. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef]
- Ridzuan, N.R.A.; Rashid, N.A.; Othman, F.; Budin, S.B.; Hussan, F.; Teoh, S.L. Protective Role of Natural Products in Cisplatin-Induced Nephrotoxicity. Mini-Rev. Med. Chem. 2019, 19, 1134–1143. [Google Scholar] [CrossRef]
- Ortega-Domínguez, B.; Aparicio-Trejo, O.E.; García-Arroyo, F.E.; León-Contreras, J.C.; Tapia, E.; Molina-Jijón, E.; Hernández-Pando, R.; Sanchez-Lozada, L.-G.; Barrera-Oviedo, D.; Pedraza-Chaverri, J. Curcumin prevents cisplatin-induced renal alterations in mitochondrial bioenergetics and dynamic. Food Chem. Toxicol. 2017, 107, 373–385. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.-M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Jijón, E.; Aparicio-Trejo, O.E.; Rodriguez-Munoz, R.; León-Contreras, J.C.; Cárdenas-Aguayo, M.D.C.; Medina-Campos, O.N.; Tapia, E.; Sanchez-Lozada, L.-G.; Hernández-Pando, R.; Reyes, J.L.; et al. The nephroprotection exerted by curcumin in maleate-induced renal damage is associated with decreased mitochondrial fission and autophagy. BioFactors 2016, 42, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarenga, L.D.A.; Leal, V.D.O.; Borges, N.A.; de Aguiar, A.S.; Faxén-Irving, G.; Stenvinkel, P.; Lindholm, B.; Mafra, D. Curcumin—A promising nutritional strategy for chronic kidney disease patients. J. Funct. Foods 2017, 40, 715–721. [Google Scholar] [CrossRef]
- Malavolta, M.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M.; Provinciali, M. Pleiotropic Effects of Tocotrienols and Quercetin on Cellular Senescence: Introducing the Perspective of Senolytic Effects of Phytochemicals. Curr. Drug Targets 2016, 17, 447–459. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef] [Green Version]
- Renugadevi, J.; Prabu, S.M. Quercetin protects against oxidative stress-related renal dysfunction by cadmium in rats. Exp. Toxicol. Pathol. 2010, 62, 471–481. [Google Scholar] [CrossRef]
- Ko, C.-C.; Chen, Y.-J.; Chen, C.-T.; Liu, Y.-C.; Cheng, F.-C.; Hsu, K.-C.; Chow, L.-P. Chemical Proteomics Identifies Heterogeneous Nuclear Ribonucleoprotein (hnRNP) A1 as the Molecular Target of Quercetin in Its Anti-cancer Effects in PC-3 Cells. J. Biol. Chem. 2014, 289, 22078–22089. [Google Scholar] [CrossRef] [Green Version]
- Symonowicz, M.; Kolanek, M. Flavonoids and Their Properties to form Chelate Complexes; Lodz University of Technology Repository: Łódź, Poland, 2012. [Google Scholar]
- Padma, V.V.; Baskaran, R.; Roopesh, R.S.; Poornima, P. Quercetin attenuates lindane induced oxidative stress in wistar rats. Mol. Biol. Rep. 2012, 39, 6895–6905. [Google Scholar] [CrossRef]
- Liu, T.; Yang, Q.; Zhang, X.; Qin, R.; Shan, W.; Zhang, H.; Chen, X. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci. 2020, 257, 118116. [Google Scholar] [CrossRef]
- Aoi, W.; Niisato, N.; Miyazaki, H.; Marunaka, Y. Flavonoid-Induced reduction of ENaC expression in the kidney of Dahl salt-sensitive hypertensive rat. Biochem. Biophys. Res. Commun. 2004, 315, 892–896. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Cruz, P.; Kone, B.C. Sirtuin 1 Functionally and Physically Interacts with Disruptor of Telomeric Silencing-1 to Regulate α-ENaC Transcription in Collecting Duct. J. Biol. Chem. 2009, 284, 20917–20926. [Google Scholar] [CrossRef] [Green Version]
- Ashkar, F.; Eftekhari, M.H.; Tanideh, N.; Koohpeyma, F.; Mokhtari, M.; Irajie, C.; Iraji, A. Effect of hydroalcoholic extract of Berberis integerrima and resveratrol on ovarian morphology and biochemical parameters in Letrozole-induced polycystic ovary syndrome rat model: An experimental study. Int. J. Reprod. Biomed. (IJRM) 2020, 18, 637. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Sack, M.N.; Finkel, T. Mitochondrial Metabolism, Sirtuins, and Aging. Cold Spring Harb. Perspect. Biol. 2012, 4, a013102. [Google Scholar] [CrossRef] [Green Version]
- Danz, E.D.B.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free. Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef]
- Jang, I.-A.; Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Effects of resveratrol on the renin-angiotensin system in the aging kidney. Nutrients 2018, 10, 1741. [Google Scholar] [CrossRef] [Green Version]
- Albertoni, G.; Schor, N. Resveratrol plays important role in protective mechanisms in renal disease—Mini-Review. J. Bras. Nefrol. 2015, 37, 106–114. [Google Scholar] [CrossRef]
- Saldanha, J.F.; Leal, V.D.O.; Stenvinkel, P.; Carraro-Eduardo, J.C.; Mafra, D. Resveratrol: Why Is It a Promising Therapy for Chronic Kidney Disease Patients? Oxid. Med. Cell. Longev. 2013, 2013, 963217. [Google Scholar] [CrossRef]
- Kitada, M.; Kume, S.; Imaizumi, N.; Koya, D. Resveratrol Improves Oxidative Stress and Protects Against Diabetic Nephropathy Through Normalization of Mn-SOD Dysfunction in AMPK/SIRT1-Independent Pathway. Diabetes 2011, 60, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Koh, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK–SIRT1–PGC1α axis in db/db mice. Diabetologia 2012, 56, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J. Cell. Physiol. 2018, 234, 5033–5043. [Google Scholar] [CrossRef]
- Grzesik, M.; Naparło, K.; Bartosz, G.; Sadowska-Bartosz, I. Antioxidant properties of catechins: Comparison with other antioxidants. Food Chem. 2018, 241, 480–492. [Google Scholar] [CrossRef]
- Crespy, V.; Williamson, G. A Review of the Health Effects of Green Tea Catechins in In Vivo Animal Models. J. Nutr. 2004, 134, 3431S–3440S. [Google Scholar] [CrossRef]
- Li, X.; Jiang, X.; Sun, J.; Zhu, C.; Li, X.; Tian, L.; Liu, L.; Bai, W. Cytoprotective effects of dietary flavonoids against cadmium-induced toxicity. Ann. N. Y. Acad. Sci. 2017, 1398, 5–19. [Google Scholar] [CrossRef]
- Zhang, T.; Mu, Y.; Yang, M.; Al Maruf, A.; Li, P.; Li, C.; Dai, S.; Lu, J.; Dong, Q. (+)-Catechin prevents methylglyoxal-induced mitochondrial dysfunction and apoptosis in EA. hy926 cells. Arch. Physiol. Biochem. 2016, 123, 121–127. [Google Scholar] [CrossRef]
- Silva Santos, L.F.; Stolfo, A.; Calloni, C.; Salvador, M. Catechin and epicatechin reduce mitochondrial dysfunction and oxidative stress induced by amiodarone in human lung fibroblasts. J. Arrhythmia 2016, 33, 220–225. [Google Scholar] [CrossRef]
- Gheysarzadeh, A.; Yazdanparast, R. STAT5 Reactivation by Catechin Modulates H2O2-Induced Apoptosis Through miR-182/FOXO1 Pathway in SK-N-MC Cells. Cell Biophys. 2014, 71, 649–656. [Google Scholar] [CrossRef]
- Shahid, A.; Ali, R.; Ali, N.; Hasan, S.K.; Bernwal, P.; Afzal, S.M.; Vafa, A.; Sultana, S. Modulatory effects of catechin hydrate against genotoxicity, oxidative stress, inflammation and apoptosis induced by benzo (a) pyrene in mice. Food Chem. Toxicol. 2016, 92, 64–74. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.R.; Nabavi, S.F.; Daglia, M.; Rastrelli, L. Epigallocatechin gallate and mitochondria—A story of life and death. Pharmacol. Res. 2016, 104, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green Tea Epigallocatechin 3-Gallate Accumulates in Mitochondria and Displays a Selective Antiapoptotic Effect Against Inducers of Mitochondrial Oxidative Stress in Neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, B.; Du, F.; Su, X.; Sun, G.; Zhou, G.; Bian, X.; Liu, N. Epigallocatechin-3-Gallate Attenuates Oxidative Stress and Inflammation in Obstructive Nephropathy via NF-κB and Nrf2/HO-1 Signalling Pathway Regulation. Basic Clin. Pharmacol. Toxicol. 2015, 117, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Peng, A. The Green Tea Polyphenol (−)-epigallocatechin-3-gallate and its beneficial roles in chronic kidney disease. J. Transl. Intern. Med. 2016, 4, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yu, J.F.; Zhao, C.G.; Sui, F.X.; Teng, X.; BIN Wu, Y. Therapeutic potential of EGCG on acute renal damage in a rat model of obstructive nephropathy. Mol. Med. Rep. 2013, 7, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [Green Version]
- Sahin, K.; Tuzcu, M.; Gencoglu, H.; Dogukan, A.; Timurkan, M.; Sahin, N.; Aslan, A.; Kucuk, O. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010, 87, 240–245. [Google Scholar] [CrossRef]
- Pan, H.; Chen, J.; Shen, K.; Wang, X.; Wang, P.; Fu, G.; Meng, H.; Wang, Y.; Jin, B. Mitochondrial Modulation by Epigallocatechin 3-Gallate Ameliorates Cisplatin Induced Renal Injury through Decreasing Oxidative/Nitrative Stress, Inflammation and NF-kB in Mice. PLoS ONE 2015, 10, e0124775. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Zuo, X.Z.; Tian, C.; Liang, D.; Yi, W.J.; Chen, Z.; Zhang, P.W.; Ding, S.B.; Ying, C.J. Green tea polyphenols attenuate high-fat diet-induced renal oxidative stress through SIRT3-dependent deacetylation. Biomed. Environ. Sci. 2015, 28, 455–459. [Google Scholar]
- Devi, K.P.; Malar, D.S.; Nabavi, S.F.; Sureda, A.; Xiao, J.; Nabavi, S.M.; Daglia, M. Kaempferol and inflammation: From chemistry to medicine. Pharmacol. Res. 2015, 99, 1–10. [Google Scholar] [CrossRef]
- Calderon-Montaño, J.M.; Burgos-Morón, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A Review on the Dietary Flavonoid Kaempferol. Mini-Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef]
- Ali, A.S.; Almalki, A.S.; Alharthy, B.T. Effect of Kaempferol on Tacrolimus-Induced Nephrotoxicity and Calcineurin B1 Expression Level in Animal Model. J. Exp. Pharmacol. 2020, 12, 397–407. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Shah, Z.A.; Saeed, F.; Imran, A.; Arshad, M.U.; Ahmad, B.; Bawazeer, S.; Atif, M.; Peters, D.G.; et al. Chemo-preventive and therapeutic effect of the dietary flavonoid kaempferol: A comprehensive review. Phytother. Res. 2018, 33, 263–275. [Google Scholar] [CrossRef]
- Luo, W.; Chen, X.; Ye, L.; Chen, X.; Jia, W.; Zhao, Y.; Samorodov, A.V.; Zhang, Y.; Hu, X.; Zhuang, F.; et al. Kaempferol attenuates streptozotocin-induced diabetic nephropathy by downregulating TRAF6 expression: The role of TRAF6 in diabetic nephropathy. J. Ethnopharmacol. 2020, 268, 113553. [Google Scholar] [CrossRef]
- Alshehri, A.S. Kaempferol attenuates diabetic nephropathy in streptozotocin-induced diabetic rats by a hypoglycaemic effect and concomitant activation of the Nrf-2/Ho-1/antioxidants axis. Arch. Physiol. Biochem. 2021, 127, 1–14. [Google Scholar] [CrossRef]
- Devi, S.A.; Chandrasekar, B.S.; Manjula, K.; Ishii, N. Grape seed proanthocyanidin lowers brain oxidative stress in adult and middle-aged rats. Exp. Gerontol. 2011, 46, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, H.; Ramachandran, S.; Waypa, G.B.; Yin, J.-J.; Li, C.-Q.; Han, M.; Huang, H.-H.; Sharp, W.W.; Hoek, T.L.V.; et al. Grape Seed Proanthocyanidins Ameliorate Doxorubicin-Induced Cardiotoxicity. Am. J. Chin. Med. 2010, 38, 569–584. [Google Scholar] [CrossRef]
- Pajuelo, D.; Quesada, H.; Díaz, S.; Fernández-Iglesias, A.; Arola-Arnal, A.; Bladé, C.; Salvado, J.; Arola, L. Chronic dietary supplementation of proanthocyanidins corrects the mitochondrial dysfunction of brown adipose tissue caused by diet-induced obesity in Wistar rats. Br. J. Nutr. 2011, 107, 170–178. [Google Scholar] [CrossRef]
- Cheng, M.; Gao, H.-Q.; Xu, L.; Li, B.-Y.; Zhang, H.; Li, X.-H. Cardioprotective Effects of Grape Seed Proanthocyanidins Extracts in Streptozocin Induced Diabetic Rats. J. Cardiovasc. Pharmacol. 2007, 50, 503–509. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Bai, B.S.; Devaraj, S.N. Grape seed proanthocyanidins ameliorates isoproterenol-induced myocardial injury in rats by stabilizing mitochondrial and lysosomal enzymes: An in vivo study. Life Sci. 2007, 81, 1615–1621. [Google Scholar] [CrossRef]
- Li, X.; Xu, L.; Gao, H.; Li, B.; Cheng, M. Effects of grape seed proanthocyanidins extracts on AGEs and expression of bone morphogenetic protein-7 in diabetic rats. J. Nephrol. 2008, 21, 722–733. [Google Scholar]
- Bao, L.; Zhang, Z.; Dai, X.; Ding, Y.; Jiang, Y.; Li, Y.; Li, Y. Effects of grape seed proanthocyanidin extract on renal injury in type 2 diabetic rats. Mol. Med. Rep. 2014, 11, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Kadye, R.; Kramer, A.H.; Joos-Vandewalle, J.; Parsons, M.; Njengele, Z.; Hoppe, H.; Prinsloo, E. Guardian of the Furnace: Mitochondria, TRAP1, ROS and stem cell maintenance. IUBMB Life 2013, 66, 42–45. [Google Scholar] [CrossRef]
- Rigotti, M.; Cerbaro, A.F.; Silva, I.D.R.D.; Agostini, F.; Branco, C.S.; Moura, S.; Salvador, M. Grape seed proanthocyanidins prevent H2O2-induced mitochondrial dysfunction and apoptosis via SIRT 1 activation in embryonic kidney cells. J. Food Biochem. 2020, 44, e13147. [Google Scholar] [CrossRef]
- Ding, Y.; Li, H.; Li, Y.; Liu, D.; Zhang, L.; Wang, T.; Liu, T.; Ma, L.; De La Puerta, R. Protective Effects of Grape Seed Proanthocyanidins on the Kidneys of Diabetic Rats through the Nrf2 Signalling Pathway. Evid. Based Complement. Altern. Med. 2020, 2020, 5205903. [Google Scholar] [CrossRef]
- Yousef, M.; Saad, A.; El-Shennawy, L. Protective effect of grape seed proanthocyanidin extract against oxidative stress induced by cisplatin in rats. Food Chem. Toxicol. 2009, 47, 1176–1183. [Google Scholar] [CrossRef]
- Wei, R.; Ding, R.; Tang, L.; Wang, Y. Grape Seed Proanthocyanidin Extract Reduces Renal Ischemia/Reperfusion Injuries in Rats. Am. J. Med. Sci. 2012, 343, 452–457. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Cheng, H.; Lv, H.; Cheng, G.; Ci, X. Nrf2-Mediated liver protection by esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3β pathway. Free Radic. Biol. Med. 2016, 101, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Xu, Y.; Tang, L.; Yang, X.; Chen, Z.; Wei, Y.; Shao, X.; Shao, X.; Xin, Z.; Cai, B.; et al. Astragalus Polysaccharide Attenuates Cisplatin-Induced Acute Kidney Injury by Suppressing Oxidative Damage and Mitochondrial Dysfunction. BioMed Res. Int. 2020, 2020, 2851349. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z.; Wang, J.; Zhu, H. Antioxidative effects of hesperetin against lead acetate-induced oxidative stress in rats. Indian J. Pharmacol. 2013, 45, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, B.; Guo, F.; Li, Z.; Qin, G. Involvement of the TGFβ1- ILK-Akt signaling pathway in the effects of hesperidin in type 2 diabetic nephropathy. Biomed. Pharmacother. 2018, 105, 766–772. [Google Scholar] [CrossRef]
- Chen, X.; Wei, W.; Li, Y.; Huang, J.; Ci, X. Hesperetin relieves cisplatin-induced acute kidney injury by mitigating oxidative stress, inflammation and apoptosis. Chem. Interact. 2019, 308, 269–278. [Google Scholar] [CrossRef]
- Zhu, C.; Dong, Y.; Liu, H.; Ren, H.; Cui, Z. Hesperetin protects against H2O2-triggered oxidative damage via upregulation of the Keap1-Nrf2/HO-1 signal pathway in ARPE-19 cells. Biomed. Pharmacother. 2017, 88, 124–133. [Google Scholar] [CrossRef]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2015, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mutanen, M.; Pajari, A.-M.; Päivärinta, E.; Misikangas, M.; Rajakangas, J.; Marttinen, M.; Oikarinen, S. Berries as chemopreventive dietary constituents—A mechanistic approach with the ApcMin/+ mouse. Asia Pac. J. Clin. Nutr. 2008, 17, 123–125. [Google Scholar]
- Iino, T.; Ogawa, Y.; Tashima, K.; Kato, S.; Takeuchi, K. Less damaging effect of whisky in rat stomachs in comparison with pure ethanol: Role of ellagic acid, the nonalcoholic ingredient. Gastroenterology 2001, 120, A150. [Google Scholar] [CrossRef]
- Zhou, B.; Li, Q.; Wang, J.; Chen, P.; Jiang, S. Ellagic acid attenuates streptozocin induced diabetic nephropathy via the regulation of oxidative stress and inflammatory signaling. Food Chem. Toxicol. 2018, 123, 16–27. [Google Scholar] [CrossRef]
- Polce, S.A.; Burke, C.; França, L.M.; Kramer, B.; Paes, A.M.D.A.; Carrillo-Sepulveda, M.A. Ellagic Acid Alleviates Hepatic Oxidative Stress and Insulin Resistance in Diabetic Female Rats. Nutrients 2018, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Kulkarni, V.H.; Chakraborty, M.; Habbu, P.V.; Ray, A. Ellagic acid restored lead-induced nephrotoxicity by anti-inflammatory, anti-apoptotic and free radical scavenging activities. Heliyon 2021, 7, e05921. [Google Scholar] [CrossRef]
- Mohammed, E.T.; Hashem, K.S.; Abdelazem, A.Z.; Foda, F.A.M.A. Prospective Protective Effect of Ellagic Acid as a SIRT1 Activator in Iron Oxide Nanoparticle-Induced Renal Damage in Rats. Biol. Trace Elem. Res. 2020, 198, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Du, L.; Hao, M.; Zhou, X.; Xuan, Q.; Apu, C.; Sun, Y.; Lu, Q.; Yin, X. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed. Pharmacother. 2020, 123, 109732. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.; Gok, O.; Beyaz, S.; Ağca, C.A.; Erman, O.; Zerek, A. Ellagic acid prevents kidney injury and oxidative damage via regulation of Nrf-2/NF-κB signaling in carbon tetrachloride induced rats. Mol. Biol. Rep. 2020, 47, 7959–7970. [Google Scholar] [CrossRef] [PubMed]
- Dizakar, S.A.; Saribas, G.S.; Tekcan, A. Effects of ellagic acid in the testes of streptozotocin induced diabetic rats. Drug Chem. Toxicol. 2021, 44, 1–8. [Google Scholar] [CrossRef]
- Lin, W.; Liu, G.; Kang, X.; Guo, P.; Shang, Y.; Du, R.; Wang, X.; Chen, L.; Yue, R.; Kong, F.; et al. Ellagic acid inhibits high glucose-induced injury in rat mesangial cells via the PI3K/Akt/FOXO3a signaling pathway. Exp. Ther. Med. 2021, 22, 1017. [Google Scholar] [CrossRef]
- Soto-Urquieta, M.G.; López-Briones, S.; Pérez-Vázquez, V.; Saavedra-Molina, A.; A González-Hernández, G.; Ramírez-Emiliano, J. Curcumin restores mitochondrial functions and decreases lipid peroxidation in liver and kidneys of diabetic db/db mice. Biol. Res. 2014, 47, 74. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, Y.; Zou, X.; Zheng, Z.; Zhang, J. Curcumin ameliorates CKD-induced mitochondrial dysfunction and oxidative stress through inhibiting GSK-3β activity. J. Nutr. Biochem. 2020, 83, 108404. [Google Scholar] [CrossRef]
- Fu, B.; Zhao, J.; Peng, W.; Wu, H.; Zhang, Y. Resveratrol rescues cadmium-induced mitochondrial injury by enhancing transcriptional regulation of PGC-1α and SOD2 via the Sirt3/FoxO3a pathway in TCMK-1 cells. Biochem. Biophys. Res. Commun. 2017, 486, 198–204. [Google Scholar] [CrossRef]
- Wang, H.; Guan, Y.; Widlund, A.L.; Becker, L.B.; Baur, J.A.; Reilly, P.M.; Sims, C.A. Resveratrol ameliorates mitochondrial dysfunction but increases the risk of hypoglycemia following hemorrhagic shock. J. Trauma Acute Care Surg. 2014, 77, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxidative Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Wongmekiat, O.; Peerapanyasut, W.; Kobroob, A. Catechin supplementation prevents kidney damage in rats repeatedly exposed to cadmium through mitochondrial protection. Naunyn-Schmiedebergs Arch. Pharmacol. 2018, 391, 385–394. [Google Scholar] [CrossRef]
- Barnett, L.M.A.; Cummings, B.S. Cellular and Molecular Mechanisms of Kidney Toxicity. Semin. Nephrol. 2019, 39, 141–151. [Google Scholar] [CrossRef]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Arduan, A.O.; Sanchez-Niño, M.D. Mitochondria-Targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Bosco, C. Oxidative stress and protective effects of polyphenols: Comparative studies in human and rodent kidney. A review. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2006, 142, 317–327. [Google Scholar] [CrossRef]
- Yeh, W.-J.; Hsia, S.-M.; Lee, W.-H.; Wu, C.-H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2016, 25, 84–92. [Google Scholar] [CrossRef]
- Vargas, F.; Romecín, P.; Guillen, A.I.G.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; García-Estañ, J. Flavonoids in Kidney Health and Disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Virgili, F.; Marino, M. Regulation of cellular signals from nutritional molecules: A specific role for phytochemicals, beyond antioxidant activity. Free Radic. Biol. Med. 2008, 45, 1205–1216. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Substance | Model (Animal or Cell) | Outcomes | References |
|---|---|---|---|
| CAPE (22 mg/kg and 34 mg/kg) | Wistar rat | Protected oxidative phosphorylation of kidney mitochondrial and decreased ROS production at Complex II in ischemia/reperfusion model. | [68] |
| CAPE (pretreated with two doses (22 mg/kg and 34 mg/kg)) | Wistar rats | Ameliorated ischemia-induced renal mitochondrial injury, improved oxidative phosphorylation with complex I-dependent substrate glutamate/malate, increased mitochondria Ca2+ uptake, blocked ischemia-induced caspase-3 activation, and protected kidney cells from necrosis. | [69] |
| Curcumin (200 mg/kg) | Sprague Dawley rats | Attenuated renal fibrosis, inflammatory response, and mitochondrial dysfunction. Inhibited the PI3K/AKT/mTOR pathway. Revealed anti-fibrotic effects mediated through the regulation of autophagy and protection of mitochondrial function. | [131] |
| Curcumin (60 mg/kg) | db/+ mice | Protected kidneys of diabetic mice from hyperglycemia modify oxygen consumption rate and NO synthesis and increasing in TBARS levels in mitochondria. | [219] |
| Curcumin (Pre-treatment with 200 mg/kg) | Wistar rats | Replenished the mitochondrial lipid peroxidase levels with pre-treatment of curcumin. Restored the cisplatin-induced modulatory effects on altered enzymatic and non-enzymatic antioxidants in kidney mitochondria. | [92] |
| Curcumin (400 mg/kg) | Wistar rats | Decreased mitochondrial hydrogen peroxide production, increased the respiration related to oxidative phosphorylation and mitochondrial membrane potential, reduced fission and enhanced fusion, and increased the expression of the PGC1α and TFAM. | [138] |
| Curcumin (200 mg/kg) | Wistar rats | Prevented the increase of mitochondrial Fis1 protein, decreased OPA1 and SIRT3, and increased in the mitophagy associated proteins Parkin and PINK1. | [142] |
| Curcumin (400 mg/kg) | Wistar rats | Attenuated the decrease in activities of respiratory complexes I and IV and induction of calcium-dependent permeability transition in gentamycin-induced mitochondrial alterations. Mediated mitochondrial functions and biogenesis through nuclear factor Nrf2. | [134] |
| Curcumin (diet containing 0.04% (w/w) curcumin) | C57BL/6 mice | Exerted beneficial effects include increasing mitochondrial biogenesis, alleviating mitochondrial dysfunction by increasing ATP levels, activities of mitochondrial electron transport chain complexes and mitochondrial respiration, and suppressing mitochondrial membrane potential. | [220] |
| Quercetin (20 mg/kg for animal, 20 μM for cell) | Sprague Dawley rats Renal tubular epithelial cells | Enhanced mitophagy. The antifibrotic effect was through activation of SIRT1/PINK1/Parkin-mediated mitophagy. | [153] |
| Quercetin (10 mg/kg) | Wistar rats | Ameliorated the cytotoxic effects of doxorubicin and cyclophosphamide on the kidney through the elevation of antioxidant expression and the suppression of lipid peroxidation. Suppressed the accumulation of MDA and increased GPx levels. | [107] |
| Resveratrol (40 mg/kg) | C57BL/6 mice | Improved renal function and inflammation in aging mice. Increased the expression of Nrf2-HO-1-NQO1 signaling and SIRT1-AMPK-PGC1α signaling. | [157] |
| Resveratrol (10 mg/kg) | Mice | Decreased mitochondria ROS generation by enhancing SIRT3 within the upregulation of PGC1 α and SOD2 mitochondria gene expression. Suppressed cadmium-induced apoptosis in mice kidney. | [221] |
| Resveratrol (30 mg/kg) | Long-Evans rats | Restored mitochondrial respiratory capacity and decreased mitochondrial ROS and lipid peroxidation following hemorrhagic shock. Increased SIRT1, PGC1α, SOD2, and CAT expression. | [72] |
| Resveratrol (30 mg/kg) | Long-Evans rats | Restored mitochondrial function and reduced insulin resistance. The anti-glycemic effects of resveratrol mediated by reduced mitochondrial ROS. | [222] |
| Resveratrol (20 mg/kg for animal, 10 μM for cell) | Sprague-Dawley rats Mouse mesangial cell | Upregulated SIRT1 and PGC1α deacetylation contributed to the mitochondrial protective effects of resveratrol. | [34] |
| Resveratrol (50 mg/kg) | Sprague-Dawley rats | Restored SIRT1/3 activity, decreased acetylated SOD2 levels, ameliorated oxidative stress and mitochondrial function of renal cell. | [223] |
| Resveratrol (diet contained resveratrol) | White chickens | Mitigated cadmium-induced oxidative stress and restored the antioxidant enzyme activity. Enhanced the phase I and II detoxification systems to relieve oxidative damage. Ameliorated cadmium-induced mitochondria dysfunction by SIRT3 upregulation and SIRT1, PGC1α, Nrf1, and TFAM transcription restrictions. Attenuated mitochondrial fission and promoted mitochondrial fusion reversed PINK1/Parkin-mediated mitophagy initiation. | [82] |
| Catechin (25, 50, and 100 mg/kg) | Wistar rats | Decreased MDA, NO, and TNF-α while increased SOD and CAT. Protected the kidney against the toxic effect of cadmium through its antioxidant, anti-inflammation, and mitochondrial protection. | [224] |
| EGCG (100 mg/kg for animal, 10 μM for cell) | C57BL/6 mice HK-2 cells | Attenuated cisplatin-induced mitochondrial oxidative stress and mitochondrial damage to electron transport chain activities while improved antioxidant defense enzyme activities in mitochondria. | [181] |
| Kaempferol (200 mg/kg) | Rat | Decreased the renal expression of Bax and cleaved caspase-3 and the production of ROS, MDA, TNF-α, and IL-6. Improved GSH and SOD levels and Bcl2 mRNA. Increased renal mRNA and SIRT1 protein levels that was related to increased acetylation of Nrf2 and NF-κB. | [106] |
| Kaempferol (200 mg/kg) | Balb/C mice | Modulated oxidative stress, inflammation, and apoptosis via ERK and NF-κB pathway. Corrected the levels of renal antioxidants and elevated the nuclear levels of HO-1 and Nrf2 in renal tissues. Attenuated the cisplatin mediated apoptosis via down-regulating the levels of Bax/Bcl2 imbalance and activating caspase-3. | [97] |
| GSPE (125, 250, and 500 mg/kg) | Sprague-Dawley rats | Ameliorated podocyte injury in diabetic nephropathy by activation of AMPK-SIRT1-PGC1α signaling, inhibited oxidative stress and mitochondrial dysfunction in the kidney. | [123] |
| GSPE (100 μM) | HEK-293 cells | Prevented H2O2 induced oxidative damage to proteins and lipids and depletion in SOD activity. Prevented mitochondrial electron transport chain dysfunction, ATP depletion, and apoptosis induced by H2O2. Regulated SIRT 1 and 3 expressions. | [197] |
| GSPE (125 and 250 mg/kg) | Sprague Dawley rats | Decreased renal damage by activating the Nrf2 signaling pathway; consequently, enhanced the antioxidant capacity of the tissue in diabetic rats. | [198] |
| Hesperetin (2.5, 5 and 10 μM) | HK2 cells | Attenuated oxidative-stress-induced apoptosis by reducing ROS levels in cisplatin-treated HK-2 cells. Activated the Nrf2 signaling pathway and regulating its downstream genes, including NQO1 and HO-1. Attenuated the MAPK signaling pathway against inflammation and inhibited the expression of apoptotic proteins to protect kidneys from AKI caused by cisplatin. | [205] |
| Ellagic acid (40 µM) | MCs | Protected mesangial cells from high glucose-induced injury. Inhibited some inflammatory factors and activation of PI3K/Akt signaling pathway. | [218] |
| Ellagic acid (100 mg/kg) | Sprague Dawley rats | Protected gentamicin-induced mitochondrial damage by preventing MMP loss and decreased mitochondrial ROS content, mitochondrial swelling, and cytochrome C release. | [90] |
| Ellagic acid (150, 100, and 50 mg/kg for animals, 100 μg/mL for cells) | Mice NRK52E cells | Ameliorated Streptozotocin induced oxidative renal injury by inhibiting NF-κB pathway. | [211] |
| Ellagic acid (10 mg/kg) | Wistar rats | With antioxidant and anti-apoptotic effects through overexpression of SIRT1 in renal tissues led to the decrease in renal MDA content and P53 protein level and an increase in renal GSH level and CAT activity. | [214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashkar, F.; Bhullar, K.S.; Wu, J. The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria. Nutrients 2022, 14, 3115. https://doi.org/10.3390/nu14153115
Ashkar F, Bhullar KS, Wu J. The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria. Nutrients. 2022; 14(15):3115. https://doi.org/10.3390/nu14153115
Chicago/Turabian StyleAshkar, Fatemeh, Khushwant S. Bhullar, and Jianping Wu. 2022. "The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria" Nutrients 14, no. 15: 3115. https://doi.org/10.3390/nu14153115
APA StyleAshkar, F., Bhullar, K. S., & Wu, J. (2022). The Effect of Polyphenols on Kidney Disease: Targeting Mitochondria. Nutrients, 14(15), 3115. https://doi.org/10.3390/nu14153115