
Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies

Abstract
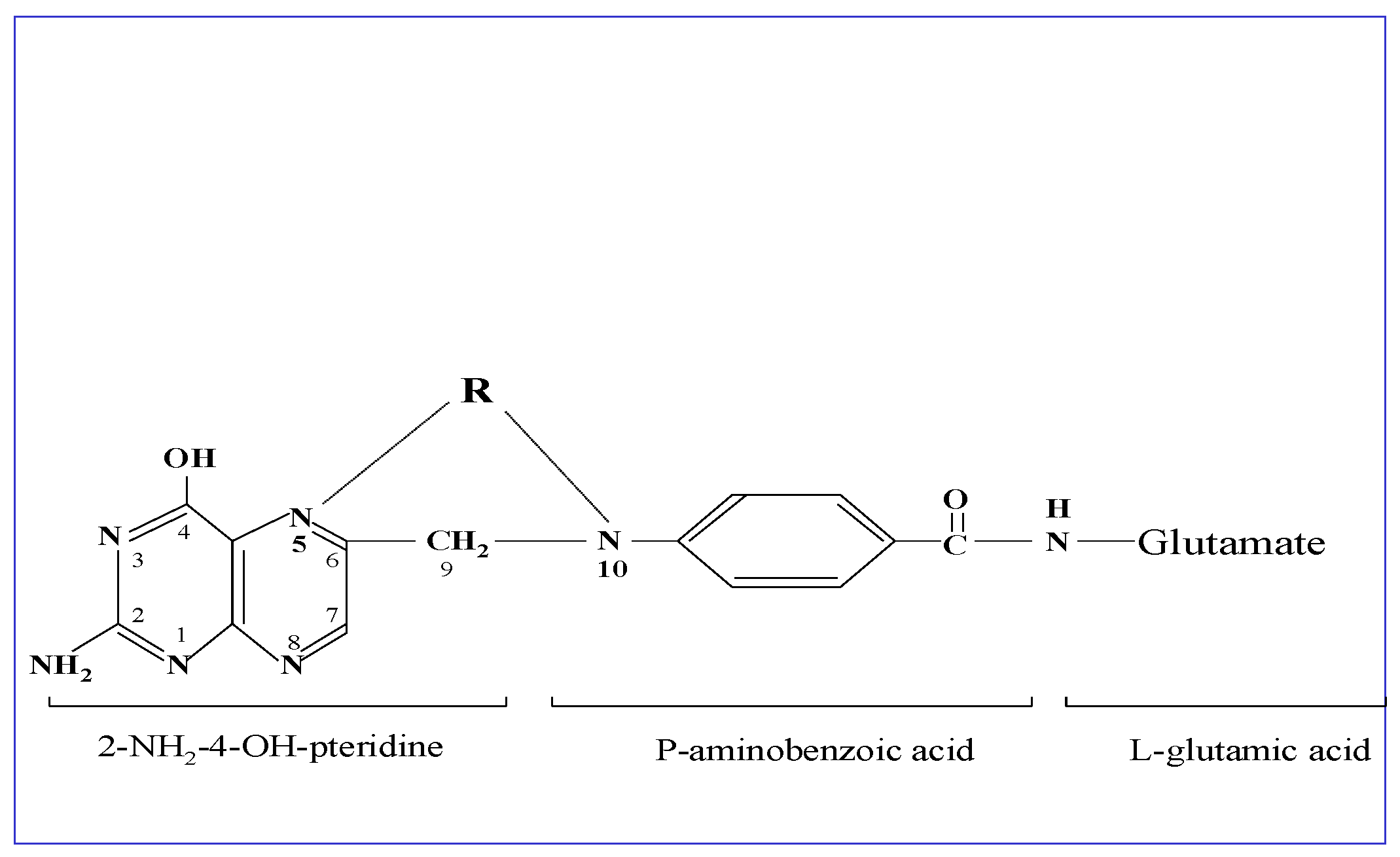
:1. Introduction
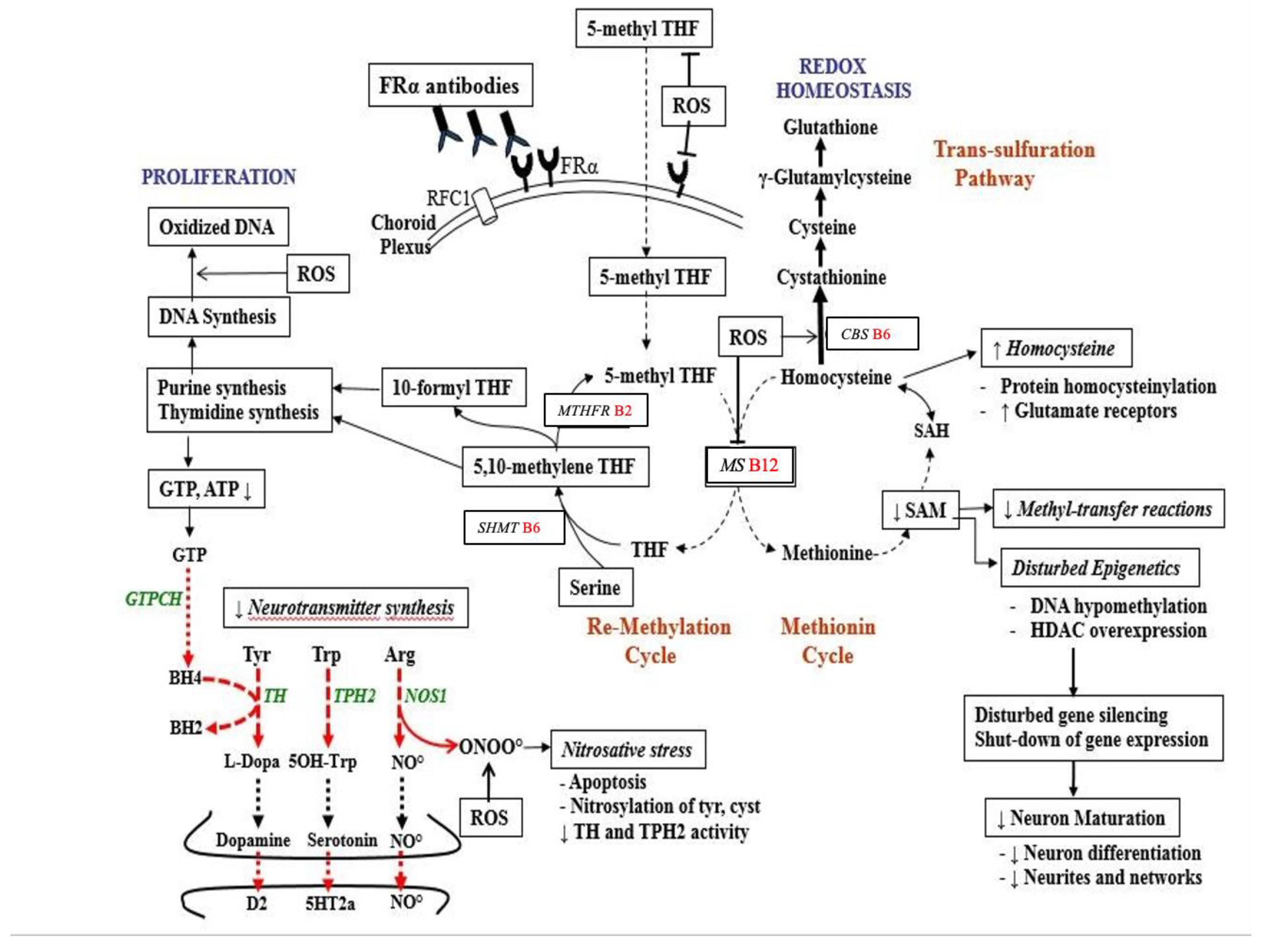
2. Hereditary Folate Malabsorption and Cerebral Folate Deficiency (CFD)
- I.
- Reduced transport of folates across the blood–brain barrier into CSF and across the ependymal into the brain.
- II.
- Reduced folate storage and release from the intracellular folyl-polyglutamate pool.
- III.
- Increased utilization and consumption of reduced folates within the nervous system leading to depletion of the folate pool
- IV.
- Increased catabolism of reduced folates within the nervous system
- V.
- Metabolic conditions affecting folate metabolism within the nervous system
3. Causes of Systemic Folate Deficiency
4. Causes of Cerebral Folate Deficiency (CFD)
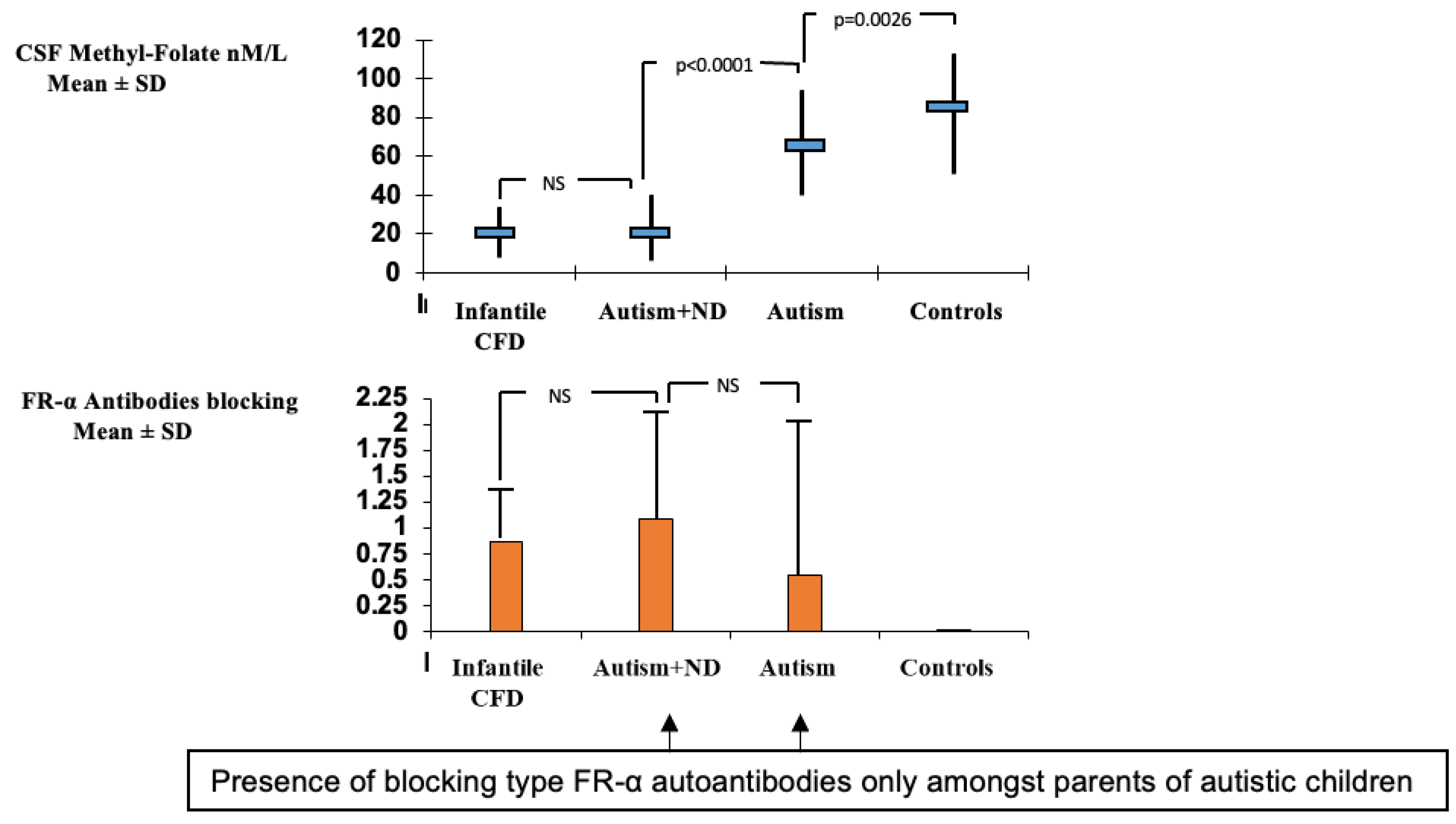
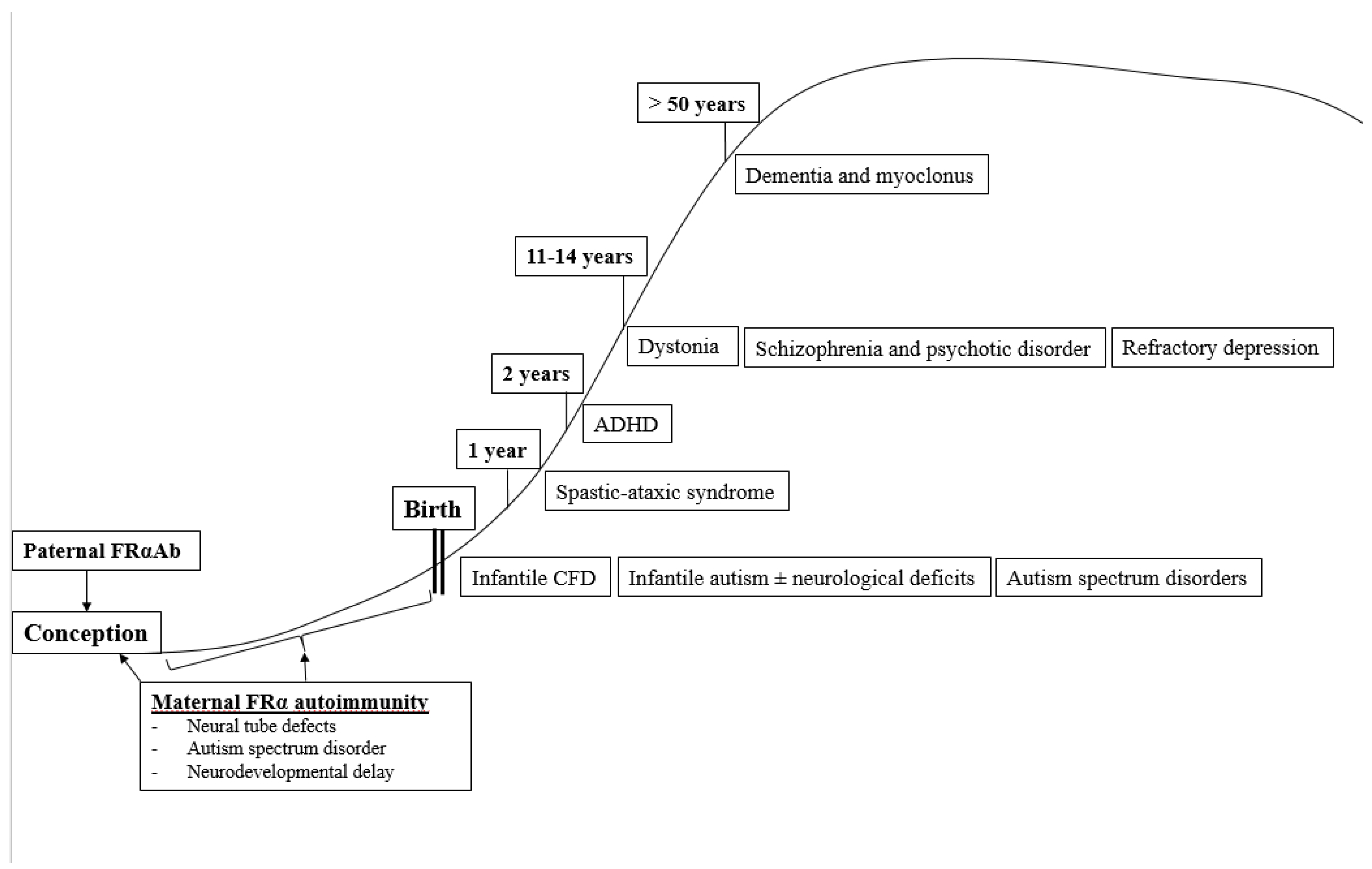
5. Clinical Classification of Different CFD Syndromes Associated with FRα Autoantibodies
6. Diagnostic Investigations
7. Treatment
8. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramaekers, V.T.; Blau, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2004, 46, 843–851. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; Quadros, E.V. The basis for folinic acid treatment in neuro-psychiatric disorders. Biochimie 2016, 126, 79–90. [Google Scholar] [CrossRef]
- Ramaekers, V.; Sequeira, J.M.; Quadros, E.V. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clin. Chem. Lab. Med. 2013, 51, 497–511. [Google Scholar] [CrossRef]
- Grapp, M.; Wrede, A.; Schweizer, M.; Hüwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Bückers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño, E.; Caviedes, L.; Hirsch, S.; Llanos, M.; Iñiguez, G.; Ronco, A.M. Folate Transporters in Placentas from Preterm Newborns and Their Relation to Cord Blood Folate and Vitamin B12 Levels. PLoS ONE 2017, 12, e0170389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E.; Wynne, R.; Rose, S.; Slattery, J.; Delhey, L.; Tippett, M.; Kahler, S.G.; Bennuri, S.C.; Melnyk, S.; Sequeira, J.M.; et al. Thyroid dysfunction in children with autism spectrum disorder is associated with folate receptor α autoimmune disorder. J. Neuroendocrinol. 2017, 29, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weitman, S.D.; Weinberg, A.G.; Coney, L.R.; Zurawski, V.R.; Jennings, D.S.; Kamen, B.A. Cellular localization of the folate receptor: Potential role in drug toxicity and folate homeostasis. Cancer Res. 1992, 52, 6708–6711. [Google Scholar] [PubMed]
- Wills, L. Treatment of “pernicious anaemia of pregnancy” and “tropical anaemia” with special reference to yeast extract as a curative agent. 1931. Natl. Med. J. India 2013, 26, 117–122. [Google Scholar]
- Rosenberg, I.H. A history of the isolation and identification of folic acid (folate). Ann. Nutr. Metab. 2012, 61, 231–235. [Google Scholar] [CrossRef]
- Molloy, A.M.; Kirke, P.N.; Brody, L.C.; Scott, J.M.; Mills, J.L. Effects of folate and vitamin B12 deficiencies during pregnancy on fetal, infant, and child development. Food Nutr. Bull. 2008, 29 (Suppl. 2), S101–S111, discussion S112. [Google Scholar] [CrossRef]
- Liu, X.; Zou, M.; Sun, C.; Wu, L.; Chen, W.X. Prenatal Folic Acid Supplements and Offspring’s Autism Spectrum Disorder: A Meta-analysis and Meta-regression. J. Autism Dev. Disord. 2022, 52, 522–539. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Tancredi, D.J.; Ozonoff, S.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tassone, F.; Hertz-Picciotto, I. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am. J. Clin. Nutr. 2012, 96, 80–89. [Google Scholar] [CrossRef]
- Surén, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzkowsky, P. Congenital malabsorption of folate. Am. J. Med. 1970, 48, 580–583. [Google Scholar] [CrossRef]
- Lubout, C.M.A.; Goorden, S.M.I.; van den Hurk, K.; Jaeger, B.; Jager, N.G.L.; van Koningsbruggen, S.; Chegary, M.; van Karnebeek, C.D.M. Successful Treatment of Hereditary Folate Malabsorption With Intramuscular Folinic Acid. Pediatr. Neurol. 2020, 102, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, V.T.; Segers, K.; Sequeira, J.M.; Koenig, M.; Van Maldergem, L.; Bours, V.; Kornak, U.; Quadros, E.V. Genetic assessment and folate receptor autoantibodies in infantile-onset cerebral folate deficiency (CFD) syndrome. Mol. Genet. Metab. 2018, 124, 87–93. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Häusler, M.; Opladen, T.; Heimann, G.; Blau, N. Psychomotor retardation, spastic paraplegia, cerebellar ataxia and dyskinesia associated with low 5-methyltetrahydrofolate in cerebrospinal fluid: A novel neurometabolic condition responding to folinic acid substitution. Neuropediatrics 2002, 33, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Sahoo, T.; Hyland, K.; Bottiglieri, T.; Peters, S.; Del Gaudio, D.; Roa, B.; Curry, S.; Zhu, H.; Finnell, R.H.; et al. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; West, K.P., Jr.; Black, R.E. The epidemiology of global micronutrient deficiencies. Ann. Nutr. Metab. 2015, 66 (Suppl. 2), 22–33. [Google Scholar] [CrossRef]
- Hoyumpa, A.M. Mechanisms of vitamin deficiencies in alcoholism. Alcohol. Clin. Exp. Res. 1986, 10, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Treasure, J.; Duarte, T.A.; Schmidt, U. Eating disorders. Lancet 2020, 395, 899–911. [Google Scholar] [CrossRef]
- Howard, M.R.; Turnbull, A.J.; Morley, P.; Hollier, P.; Webb, R.; Clarke, A. A prospective study of the prevalence of undiagnosed coeliac disease in laboratory defined iron and folate deficiency. J. Clin. Pathol. 2002, 55, 754–757. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Liu, Y.; Guo, H.; Jabir, M.S.; Liu, X.; Cui, W.; Li, D. Associations between Folate and Vitamin B12 Levels and Inflammatory Bowel Disease: A Meta-Analysis. Nutrients 2017, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Heap, B.J.; Mumford, J.D. Chronic giardiasis with vitamin B12 and folate deficiency presenting with psychiatric symptoms. J. R. Army Med. Corps 1989, 135, 25–26. [Google Scholar] [CrossRef] [Green Version]
- Adan, R.A.H.; van der Beek, E.M.; Buitelaar, J.K.; Cryan, J.F.; Hebebrand, J.; Higgs, S.; Schellekens, H.; Dickson, S.L. Nutritional psychiatry: Towards improving mental health by what you eat. Eur. Neuropsychopharmacol. 2019, 29, 1321–1332. [Google Scholar] [CrossRef]
- Williamson, K.; Kilner, K.; Clibbens, N. A comparison of the nutrient intake of a community-dwelling first-episode psychosis cohort, aged 19–64 years, with data from the UK population. J. Nutr. Sci. 2015, 4, e28. [Google Scholar] [CrossRef] [Green Version]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Villa, D.; Aguilar, M.R.; Rojo, L. Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. Int. J. Mol. Sci. 2019, 20, 4996. [Google Scholar] [CrossRef] [Green Version]
- Klipstein, F.A.; Berlinger, F.C.; Reed, L.J. Folate deficiency associated with drug therapy for tuberculosis. Blood 1967, 29, 697–712. [Google Scholar] [CrossRef]
- Opladen, T.; Blau, N.; Ramaekers, V.T. Effect of antiepileptic drugs and reactive oxygen species on folate receptor 1 (FOLR1)-dependent 5-methyltetrahydrofolate transport. Mol. Genet. Metab. 2010, 101, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Taher, J.; Naranian, T.; Poon, Y.Y.; Merola, A.; Mestre, T.; Suchowersky, O.; Kulasingam, V.; Fasano, A. Vitamins and Infusion of Levodopa-Carbidopa Intestinal Gel. Can. J. Neurol. Sci. 2022, 49, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, C.; Wevers, R.A.; Hyland, K.; Sharma, R.K.; Knust, A.; Hoffman, G.F. The influence of L-dopa on methylation capacity in aromatic L-amino acid decarboxylase deficiency: Biochemical findings in two patients. J. Inherit. Metab. Dis. 2000, 23, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Fattal-Valevski, A.; Bassan, H.; Korman, S.H.; Lerman-Sagie, T.; Gutman, A.; Harel, S. Methylenetetrahydrofolate reductase deficiency: Importance of early diagnosis. J. Child Neurol. 2000, 15, 539–543. [Google Scholar] [CrossRef]
- Kang, S.S.; Wong, P.W.; Susmano, A.; Sora, J.; Norusis, M.; Ruggie, N. Thermolabile methylenetetrahydrofolate reductase: An inherited risk factor for coronary artery disease. Am. J. Hum. Genet. 1991, 48, 536–545. [Google Scholar] [PubMed]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; Den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Banka, S.; Blom, H.J.; Walter, J.; Aziz, M.; Urquhart, J.; Clouthier, C.M.; Rice, G.I.; de Brouwer, A.P.; Hilton, E.; Vassallo, G.; et al. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am. J. Hum. Genet. 2011, 88, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cario, H.; Smith, D.E.; Blom, H.; Blau, N.; Bode, H.; Holzmann, K.; Pannicke, U.; Hopfner, K.P.; Rump, E.M.; Ayric, Z.; et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am. J. Hum. Genet. 2011, 88, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.F.; Christensen, K.E.; Watkins, D.; Raby, B.A.; Renaud, Y.; De la Luna, S.; Estivill, X.; MacKenzie, R.E.; Hudson, T.J.; Rosenblatt, D.S. The molecular basis of glutamate formiminotransferase deficiency. Hum. Mutat. 2003, 22, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Bidla, G.; Watkins, D.; Chéry, C.; Froese, D.S.; Ells, C.; Kerachian, M.; Saskin, A.; Christensen, K.E.; Gilfix, B.M.; Guéant, J.L.; et al. Biochemical analysis of patients with mutations in MTHFD1 and a diagnosis of methylenetetrahydrofolate dehydrogenase 1 deficiency. Mol. Genet. Metab. 2020, 130, 179–182. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, J.M.; Ramaekers, V.T.; Quadros, E.V. The diagnostic utility of folate receptor autoantibodies in blood. Clin. Chem. Lab. Med. 2013, 51, 545–554. [Google Scholar] [CrossRef]
- Bobrowski-Khoury, N.; Ramaekers, V.T.; Sequeira, J.M.; Quadros, E.V. Folate Receptor Alpha Autoantibodies in Autism Spectrum Disorders: Diagnosis, Treatment and Prevention. J. Pers. Med. 2021, 11, 710. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M. Role of folate receptor autoantibodies in infantile autism. Mol. Psychiatry 2013, 18, 270–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaekers, V.T.; Blau, N.; Sequeira, J.M.; Nassogne, M.C.; Quadros, E.V. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics 2007, 38, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaekers, V.T.; Thöny, B.; Sequeira, J.M.; Ansseau, M.; Philippe, P.; Boemer, F.; Bours, V.; Quadros, E.V. Folinic acid treatment for schizophrenia associated with folate receptor autoantibodies. Mol. Genet. Metab. 2014, 113, 307–314. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; Blau, N.; Quadros, E.V. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev. Med. Child Neurol. 2008, 50, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cazorla, A.; Quadros, E.V.; Nascimento, A.; Garcia-Silva, M.T.; Briones, P.; Montoya, J.; Ormazábal, A.; Artuch, R.; Sequeira, J.M.; Blau, N.; et al. Mitochondrial diseases associated with cerebral folate deficiency. Neurology 2008, 70, 1360–1362. [Google Scholar] [CrossRef]
- Quijada-Fraile, P.; O’Callaghan, M.; Martín-Hernández, E.; Montero, R.; Garcia-Cazorla, À.; de Aragón, A.M.; Muchart, J.; Málaga, I.; Pardo, R.; García-Gonzalez, P.; et al. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome. Orphanet. J. Rare Dis. 2014, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, O.; Blau, N.; Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M.; Weissert, M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol. Genet. Metab. 2010, 99, 58–61. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Weis, J.; Sequeira, J.M.; Quadros, E.V.; Blau, N. Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency. Neuropediatrics 2007, 38, 184–187. [Google Scholar] [CrossRef] [Green Version]
- Steinfeld, R.; Grapp, M.; Kraetzner, R.; Dreha-Kulaczewski, S.; Helms, G.; Dechent, P.; Wevers, R.; Grosso, S.; Gärtner, J. Folate receptor alpha defect causes cerebral folate transport deficiency: A treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am. J. Hum. Genet. 2009, 85, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Wolf, A.; Kim, S.E.; Cabrera, R.M.; Wlodarczyk, B.J.; Zhu, H.; Parker, M.; Lin, Y.; Steele, J.W.; Han, X.; et al. CIC de novo loss of function variants contribute to cerebral folate deficiency by downregulating FOLR1 expression. J. Med. Genet. 2021, 58, 484–494. [Google Scholar] [CrossRef]
- Häusler, M.G.; Jaeken, J.; Mönch, E.; Ramaekers, V.T. Phenotypic heterogeneity and adverse effects of serine treatment in 3-phosphoglycerate dehydrogenase deficiency: Report on two siblings. Neuropediatrics 2001, 32, 191–195. [Google Scholar] [CrossRef]
- Rodan, L.H.; Qi, W.; Ducker, G.S.; Demirbas, D.; Laine, R.; Yang, E.; Walker, M.A.; Eichler, F.; Rabinowitz, J.D.; Anselm, I.; et al. 5,10-methenyltetrahydrofolate synthetase deficiency causes a neurometabolic disorder associated with microcephaly, epilepsy, and cerebral hypomyelination. Mol. Genet. Metab. 2018, 125, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Ponzone, A.; Spada, M.; Ferraris, S.; Dianzani, I.; de Sanctis, L. Dihydropteridine reductase deficiency in man: From biology to treatment. Med. Res. Rev. 2004, 24, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Leeming, R.J.; Hall, K. Dihydropteridine Reductase and Folate Metabolism Revisited. Pteridines 2010, 21, 84–85. [Google Scholar] [CrossRef]
- Loens, S.; Chorbadzhieva, E.; Kleimann, A.; Dressler, D.; Schrader, C. Effects of levodopa/carbidopa intestinal gel versus oral levodopa/carbidopa on B vitamin levels and neuropathy. Brain Behav. 2017, 7, e00698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaekers, V.T.; Hansen, S.I.; Holm, J.; Opladen, T.; Senderek, J.; Häusler, M.; Heimann, G.; Fowler, B.; Maiwald, R.; Blau, N. Reduced folate transport to the CNS in female Rett patients. Neurology 2003, 61, 506–515. [Google Scholar] [CrossRef]
- Blau, N.; Bonafé, L.; Krägeloh-Mann, I.; Thöny, B.; Kierat, L.; Häusler, M.; Ramaekers, V. Cerebrospinal fluid pterins and folates in Aicardi-Goutières syndrome: A new phenotype. Neurology 2003, 61, 642–647. [Google Scholar] [CrossRef]
- Deth, R.; Muratore, C.; Benzecry, J.; Power-Charnitsky, V.A.; Waly, M. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology 2008, 29, 190–201. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; Thöny, B.; Quadros, E.V. Oxidative Stress, Folate Receptor Autoimmunity, and CSF Findings in Severe Infantile Autism. Autism Res. Treat. 2020, 2020, 9095284. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Sykes, C.E.; Geddes, T.J.; Jaunarajs, K.L.; Bishop, C. Tryptophan hydroxylase 2 aggregates through disulfide cross-linking upon oxidation: Possible link to serotonin deficits and non-motor symptoms in Parkinson’s disease. J. Neurochem. 2011, 116, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muratore, C.R.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Persico, A.M.; Lintas, C.; De la Monte, S.; Deth, R.C. Age-dependent decrease and alternative splicing of methionine synthase mRNA in human cerebral cortex and an accelerated decrease in autism. PLoS ONE 2013, 8, e56927. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Guéant, J.L.; Namour, F.; Guéant-Rodriguez, R.M.; Daval, J.L. Folate and fetal programming: A play in epigenomics? Trends Endocrinol. Metab. 2013, 24, 279–289. [Google Scholar] [CrossRef]
- Rothenberg, S.P.; Da Costa, M.P.; Sequeira, J.M.; Cracco, J.; Roberts, J.L.; Weedon, J.; Quadros, E.V. Autoantibodies against folate receptors in women with a pregnancy complicated by a neural-tube defect. N. Engl. J. Med. 2004, 350, 134–142. [Google Scholar] [CrossRef]
- Hansen, F.J.; Blau, N. Cerebral folate deficiency: Life-changing supplementation with folinic acid. Mol. Genet. Metab. 2005, 84, 371–373. [Google Scholar] [CrossRef]
- Ho, A.; Michelson, D.; Aaen, G.; Ashwal, S. Cerebral folate deficiency presenting as adolescent catatonic schizophrenia: A case report. J. Child Neurol. 2010, 25, 898–900. [Google Scholar] [CrossRef]
- Masingue, M.; Benoist, J.F.; Roze, E.; Moussa, F.; Sedel, F.; Lubetzki, C.; Nadjar, Y. Cerebral folate deficiency in adults: A heterogeneous potentially treatable condition. J. Neurol. Sci. 2019, 396, 112–118. [Google Scholar] [CrossRef]
- Sadighi, Z.; Butler, I.J.; Koenig, M.K. Adult-onset cerebral folate deficiency. Arch. Neurol. 2012, 69, 778–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurazzi, P.; Kiani, A.K.; Miertus, J.; Paolacci, S.; Barati, S.; Manara, E.; Stuppia, L.; Gurrieri, F.; Bertelli, M. Genetic analysis of intellectual disability and autism. Acta Biomed. 2020, 91, e2020003. [Google Scholar]
- Delmelle, F.; Thöny, B.; Clapuyt, P.; Blau, N.; Nassogne, M.C. Neurological improvement following intravenous high-dose folinic acid for cerebral folate transporter deficiency caused by FOLR-1 mutation. Eur. J. Paediatr. Neurol. 2016, 20, 709–713. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; DiDuca, M.; Vrancken, G.; Thomas, A.; Philippe, C.; Peters, M.; Jadot, A.; Quadros, E.V. Improving Outcome in Infantile Autism with Folate Receptor Autoimmunity and Nutritional Derangements: A Self-Controlled Trial. Autism Res. Treat. 2019, 2019, 7486431. [Google Scholar] [CrossRef] [Green Version]
- Alam, C.; Hoque, M.T.; Finnell, R.H.; Goldman, I.D.; Bendayan, R. Regulation of Reduced Folate Carrier (RFC) by Vitamin D Receptor at the Blood-Brain Barrier. Mol. Pharm. 2017, 14, 3848–3858. [Google Scholar] [CrossRef]
- Vidmar Golja, M.; Šmid, A.; Karas Kuželički, N.; Trontelj, J.; Geršak, K.; Mlinarič-Raščan, I. Folate Insufficiency Due to MTHFR Deficiency Is Bypassed by 5-Methyltetrahydrofolate. J. Clin. Med. 2020, 9, 2836. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.; Morris, A.A.; Walter, J.H. Treatment with Mefolinate (5-Methyltetrahydrofolate), but Not Folic Acid or Folinic Acid, Leads to Measurable 5-Methyltetrahydrofolate in Cerebrospinal Fluid in Methylenetetrahydrofolate Reductase Deficiency. JIMD Rep. 2016, 29, 103–107. [Google Scholar]
- McAuley, E.; McNulty, H.; Hughes, C.; Strain, J.J.; Ward, M. Riboflavin status, MTHFR genotype and blood pressure: Current evidence and implications for personalised nutrition. Proc. Nutr. Soc. 2016, 75, 405–414. [Google Scholar] [CrossRef] [Green Version]
- McNulty, H.; Dowey, L.R.C.; Strain, J.J.; Dunne, A.; Ward, M.; Molloy, A.M.; McAnena, L.B.; Hughes, J.P.; Hannon-Fletcher, M.; Scott, J.M. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation 2006, 113, 74–80. [Google Scholar] [CrossRef] [Green Version]
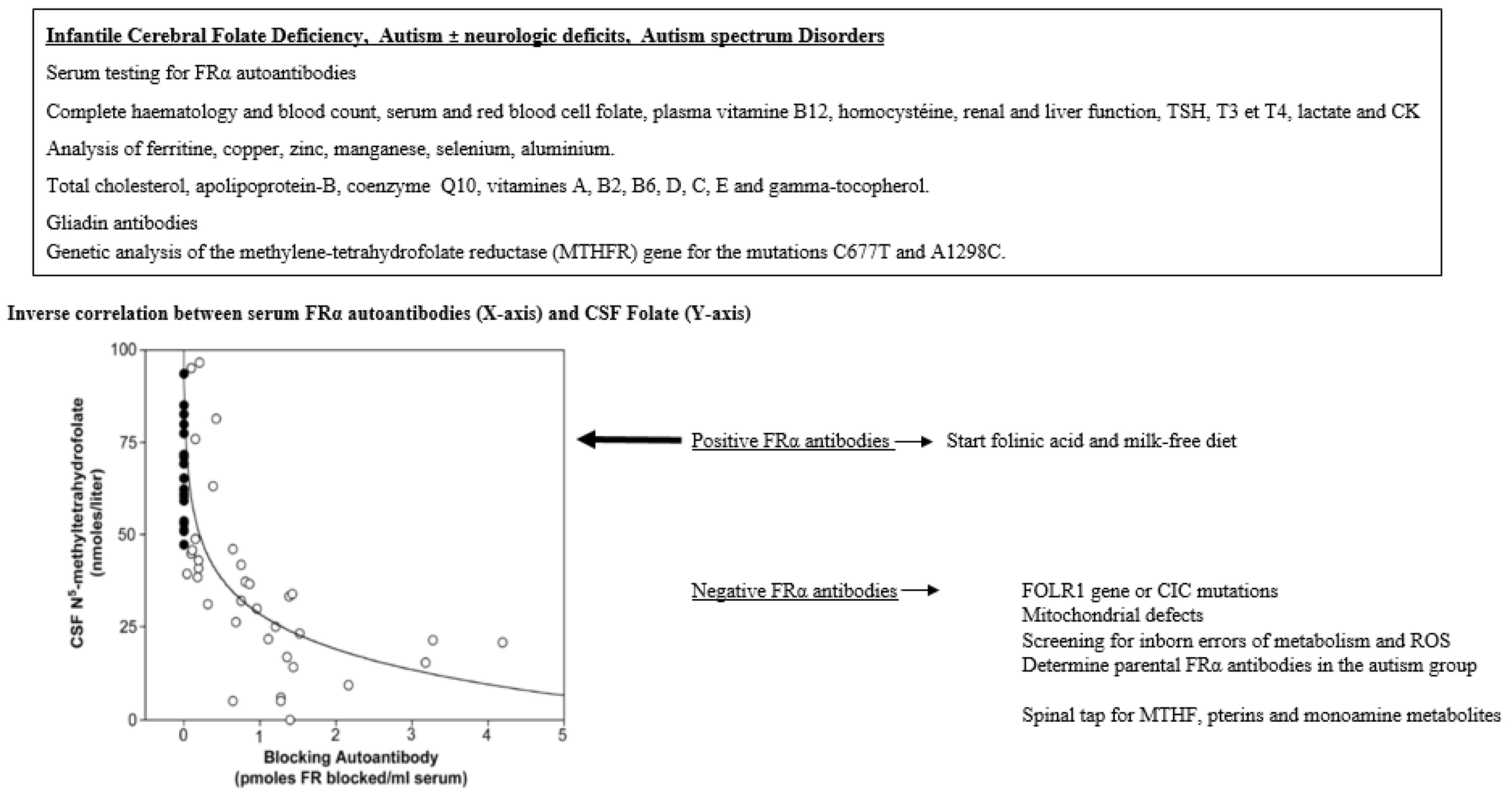


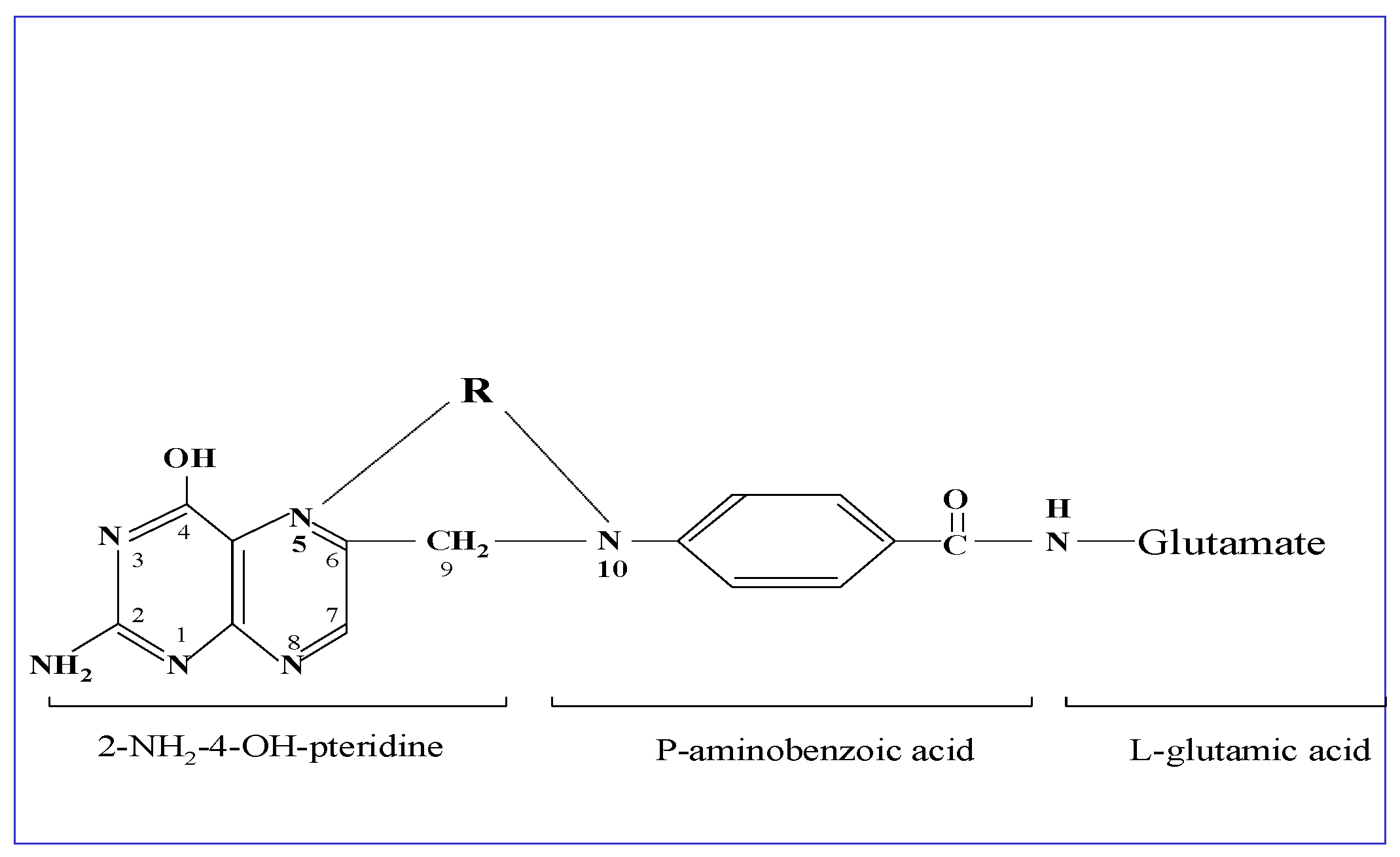{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I. Reduced transport of folates across the blood–brain barrier into CSF and across the ependymal barrier into the brain |
| 1. Disorders of Folate Receptor alpha (FRα). |
|
|
|
|
| 2. Disorders of energy metabolism and ATP production |
|
|
|
| 3. Conditions damaging the transport functions of brain-endothelial vessel walls and choroid epithelial cells |
|
|
|
|
| II. Decreased folate storage due to depletion of intracellular folyl-polyglutamate pool. |
| III. Increased utilization of reduced folates in the brain |
| 1. Hereditary conditions |
|
|
| 2. Iatrogenic conditions |
|
| 3. Infectious, parainfectious and immune-mediated conditions |
|
|
|
| IV. Increased catabolism of reduced folates within the nervous system |
| 1. Conditions associated with oxidative and/or nitrosative stress |
|
| + Glutathione Peroxidase deficiency associated with selenium deficiency |
| + Extra-cellular and intracellular Superoxide Dismutase deficiency secondary to intracellular and/or extracellular manganese deficiency |
| + Ubichinone-10 deficiency states |
| + All conditions associated with vitamin C and E deficiencies |
| 2. Inflammatory processes |
| V. Conditions affecting folate metabolism in the brain |
| 1. Hereditary conditions |
| Enzyme deficiencies |
|
|
|
|
| Depletion of methyl-donor pool glycine, serine, and histidine |
|
|
| 2. Hyper homocysteinemia due to combined deficiencies of vitamin B2, B6, B9 and B12 |
| Classification of Condition | Underlying Mechanism |
|---|---|
| Systemic depletion | |
| Malnutrition | Folate deficient diet or food deprivation |
| Malabsorption | Decreased folate absorption in the jejenum |
| Gluten enteropathy |
| Affects folate absorption |
| Antifolate agents | |
| Blocks RFC1 carrier; inhibits dihydrofolate reductase |
| Mechanism unknown |
| Analogs of para-aminobenzoic acid, interfering with tetrahydrofolate synthesis in sensitive bacteria |
| Interfering with cellular folate uptake |
| Inhibits aromatic amino acid decarboxylase and consequently, SAM and MTHF overconsumption |
| Congenital folate malabsorption | Hereditary factor involving the PCFT gene |
| Inborn errors of metabolism | |
| Depletion of enzymatic product MTHF |
| Defective histidine derived one-carbon transfer to tetrahydrofolate |
| Reduction of the reduced folate pool |
| Reduction of 5-formyl and 10-formyltetrahydrofolate pool |
| Cerebral Folate Deficiency | |
| Infantile-onset CFD | Serum FRα autoantibodies of the blocking and/or binding type |
| Mitochondrial encephalopathies | Decreased active folate transport at choroid plexus |
| Kearns-Sayre syndrome | Decreased active folate transport at choroid plexus |
| Alper’s disease | Decreased active folate transport at choroid plexus |
| CFDS due to FRα dysfunction | FOLR1 gene defects, de novo mutation of Capicua transcriptional repressor (CIC) gene |
| One-carbon pool deficiencies in the CNS | |
| Defective serine synthesis affecting one-carbon pool |
| Inborn errors of metabolism | |
| - Methenyltetrahydrofolate synthase deficiency | Defective conversion of 5-formyl-THF to 5–10-methenyl-THF |
| - Dihydropteridine reductase deficiency | Diminished conversion of dihydrofolate to THF |
| - Aromatic L-amino acid decarboxylase def. | Overconsumption of 5MTHF and SAM |
| Rett syndrome | FRα pseudo gene expression due to MECP2 defect |
| Variant of Aicardi-Goutières syndrome | Variable CFD of unknown origin |
| Reactive Oxygen Species | MTHF instability and dysfunction of FRα and RFC1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramaekers, V.T.; Quadros, E.V. Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies. Nutrients 2022, 14, 3096. https://doi.org/10.3390/nu14153096
Ramaekers VT, Quadros EV. Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies. Nutrients. 2022; 14(15):3096. https://doi.org/10.3390/nu14153096
Chicago/Turabian StyleRamaekers, Vincent Th., and Edward V. Quadros. 2022. "Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies" Nutrients 14, no. 15: 3096. https://doi.org/10.3390/nu14153096
APA StyleRamaekers, V. T., & Quadros, E. V. (2022). Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies. Nutrients, 14(15), 3096. https://doi.org/10.3390/nu14153096